




Immune-mediated necrotising myopathy (IMNM) is a type of idiopathic inflammatory myopathy (IIM) characterized by subacute, proximal and symmetric muscle weakness, with evidence on histological examination of myocyte necrosis, minimal inflammatory cell infiltration and absence of perifascicular atrophy. The detection of antibodies helps distinguish it from other IIMs and connective tissue diseases associated with myositis1 and classify it (Table 1).1,2
Characteristics of the inflammatory myopathies.
| Dermatomyositis/juvenile dermatomyositis | [0,3–5] IMNM | Polymyositis | Overlap myositis | Inclusion body myositis | |||
|---|---|---|---|---|---|---|---|
| Anti-SRP (22%–39%) | Anti-HMGCR (26%–50%) | Antibody-negative (25%–40%) | |||||
| Muscle weakness | Proximal | [0,3–5]Proximal | Proximal | Proximal | Distal (finger flexors, knee extensors) | ||
| Extramuscular manifestations | Cutaneous (heliotrope rash, Gottron papules) | Dysphagia, lung and heart involvement | Infrequent | Frequent | Infrequent (except for dysphagia) | Antisynthetase syndrome: interstitial lung disease, mechanic’s hands, arthritis, Raynaud syndrome. | Infrequent (except for dysphagia) |
| Dysphagia | Associated with connective tissue disorders and malignancy (adulthood) | Scleroderma | |||||
| Systemic lupus erythematosus | |||||||
| Autoantibodies | Mi-2, MDA2, TIF-1γ, NXP2, SAE | SRP | HMGCR | Negative | Nonspecific | Associated with antisynthetase syndrome: Jo-1, PL-7, PL-12, HA, EJ, KS, Zo, OJ. | c-N1A |
| Other: Ku, Ro/SS-S, SS-B, PM/Scl, U-snRNP | |||||||
| Histology | Perimysial inflammation, perifascicular atrophy, MHC class I and complement deposition in capillaries or sarcolemma | [0,3–5]Severe necrosis, MHC class I and complement deposition in capillaries or sarcolemma | Endomysial CD8 + T cells | Perifascicular necrosis, MHC class I and II complement deposition on sarcolemma | Endomysial CD8 + T cells, MHC-I, amyloid, vacuoles, tubulofilaments | ||
Onset in childhood is rare, especially for the antibody-negative subtype, of which only 1 case has been reported.3 Since a standard of care has not been established for the paediatric population, the management is extrapolated from the approach used in adult patients, initiating treatment with steroids and intravenous immunoglobulin (IVIG). Although children tend to have a favourable response to this approach,3 in severe cases it may be necessary to resort to other immunomodulatory drugs, and there is no consensus on which drug should be the first choice.
We present the case of a boy with IMNM refractory to treatment that developed cardiac and later cutaneous manifestations compatible with scleroderma overlap syndrome (SOS).
The patient was a boy aged 10 years that experienced progressive, symmetric and non-fluctuating weakness in the upper and lower extremities and axial hypotonia of 2 months’ duration. He presented with xerosis and hyperpigmentation in the presternal and hypogastric regions and knees, and periocular and perioral hypopigmentation (Fig. 1a–c). The patient reported absence of dysphagia, fever or other symptoms. The physical examination revealed severe muscle weakness, predominantly axial (12/52 points in the Childhood Myositis Assessment Scale [CMAS]). The results of blood testing were: creatinine kinase (CK), 13 798 U/L; aldolase, 319 U/L, lactate dehydrogenase (LDH), 1700 U/L, aspartate transaminase/alanine transaminase (AST/ALT), 509/664 U/L; erythrocyte sedimentation rate (ESR), 8 mm. The patient tested negative for autoantibodies, including anti-SRP and anti-HMGCR. The electromyogram (EMG) revealed predominantly proximal myopathy. The magnetic resonance imaging (MRI) scan revealed extensive symmetric oedema in the shoulder and pelvic girdles (Fig. 1d). There was evidence of dilatation of the distal 2/3 of the oesophagus, with inefficient peristalsis in the oesophageal-gastric-duodenal transit. The cardiac function (left ventricular ejection fraction [LVEF], 72%) and lung function were normal. Examination of a muscle biopsy sample revealed myocyte necrosis and marked overexpression of major histocompatibility complex class I (MHC-I) molecules, without perifascicular atrophy or deposition of membrane attack complex, features compatible with IMNM.
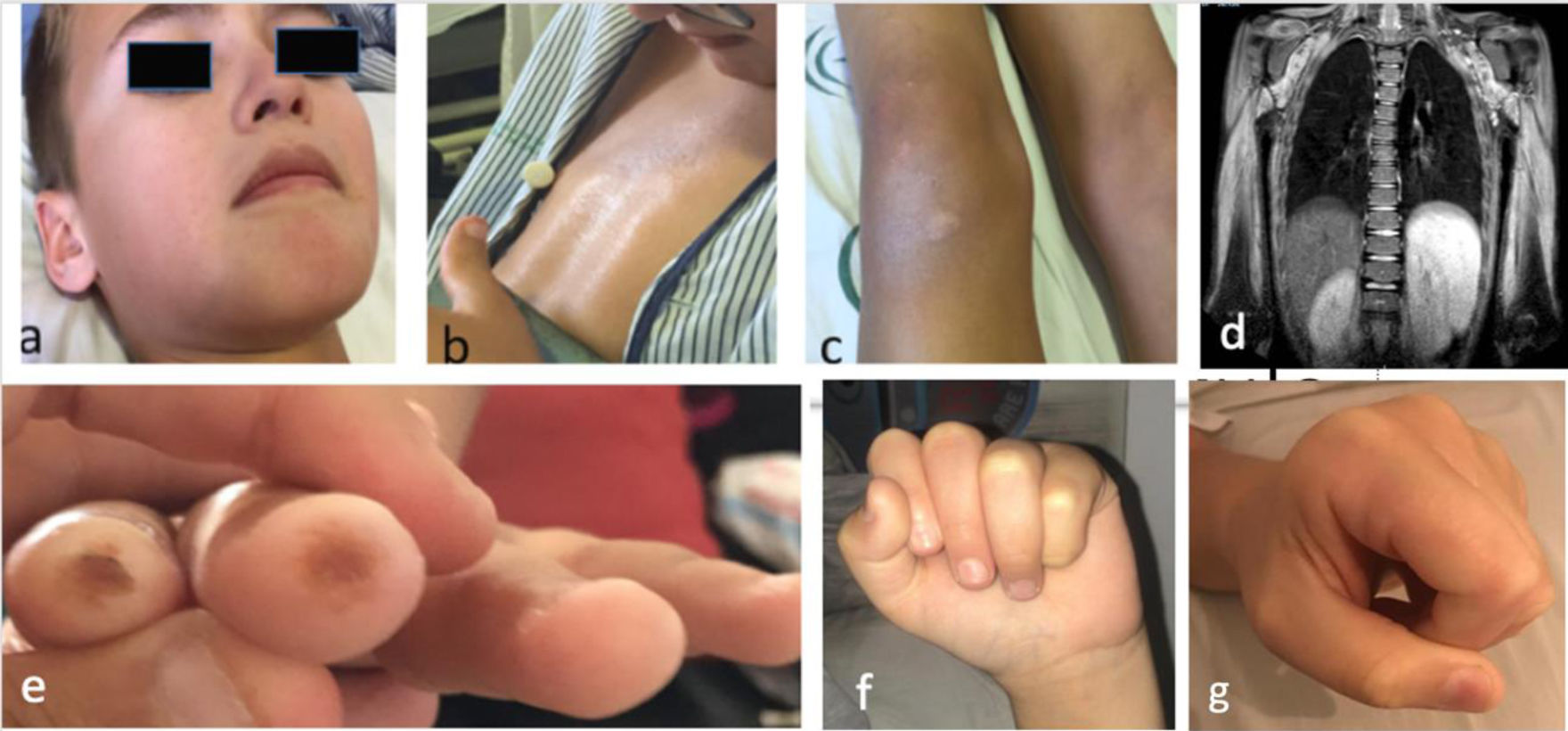
a) Facial hypopigmentation (periocular and perioral); b) Skill with an atrophic, shiny and hyperpigmented appearance at the sternal level; c) Hyperpigmentation and psoriasiform desquamative lesions on knees; d) Coronal view on MRI (extensive involvement of muscles of both shoulders, thorax and proximal arm, bilateral and symmetric, hyperintense signal in STIR sequence indicative of oedema); e) Digital ulcers; f) Scleroderma in hands, difficulty making a fist (frontal view); g) Scleroderma in hands, difficulty making a fist (lateral view).
The patient received methylprednisolone boluses (30 mg/kg/days) for 5 days, IVIG (2 g/kg) and subcutaneous methotrexate (15 mg/m2/week). At discharge, the patient exhibited mild clinical improvement, with a marked descent in CK. Forty-eight hours later, the patient was readmitted due to heart failure with ventricular dysfunction (LVEF, 25%–30%) and mild pericardial effusion (1 cm), a CK level of 2500 U/L, a CK-MB level of 540 ng/mL and a troponin level of 5.94 ng/mL. The electrocardiogram revealed a sinus rhythm with ST segment elevation and small R waves in leads V4 and V5, suggestive of myocardial damage. The patient was admitted to the paediatric intensive care unit (PICU) with cardiogenic shock secondary to myopericarditis and received initial support with milrinone, subsequently switched to dopamine due to hypotension, followed by 1 cycle of levosimendan. The initial regimen was optimised with boluses of methylprednisolone and initiation of tacrolimus (0.1 mg/kg/day), with gradual improvement of myocardial function until it normalised. The extended cardiac workup, performed after cardiac function improved (heart MRI and Holter monitoring) ruled out structural and functional cardiac abnormalities and residual arrhythmia.
After 6 months of medical treatment and rehabilitation, the weakness had resolved (CMAS, 52/52) and the laboratory parameters normalized.
Fifteen months after onset, the patient developed cutaneous ulcers in the fingertips with toughening of the palmar skin (Fig. 1e–g), prompting a new screening for connective tissue disorders (autoantibodies, and heart, lung and digestive tract evaluation) that did not detect any abnormalities. Due to the suspicion of overlap syndrome (scleroderma and IMNM), methotrexate was replaced by mycophenolate, maintaining tacrolimus.
The patient is currently asymptomatic, save for the cutaneous sclerosis, which remains stable.
We think this case was exceptional due to the difficult prognostication and treatment of a disease this rare, especially in the presence of life-threatening complications, in this case cardiac involvement. Although the latter has been described in the context of IMNM, it has been reported in anti-SRP + cases but not in antibody-negative cases. Therefore, it is essential to assess for cardiac involvement at onset and during the follow-up, as it can develop at any point in the course of disease and it is not necessarily accompanied by peripheral muscle weakness.4 In addition, the development of sclerodermatous lesions required reconsideration of the initial diagnosis to contemplate SOS with myopericarditis, given the increased frequency of myocardial involvement described in such cases,4 even in the absence of a compatible autoantibody profile.
Combination therapy with steroids, IVIG and methotrexate is typically used as first-line treatment, although there is no consensus on this matter. However, weakness in IMNM tends to be more persistent and refractory to treatment compared to other IIMs, occasionally requiring combinations of other drugs,5,6 as was the case in our patient. In such cases, tacrolimus, mycophenolate, azathioprine, ciclosporin or cyclophosphamide could be contemplated for second-line treatment, reserving rituximab for patients that respond poorly.6 In case of cardiac involvement, inotropic support (for instance, with levosimendan) can maintain adequate myocardial function while a therapeutic dose of the immunomodulatory drugs is achieved to control the disease. Although the adequate duration of treatment has yet to be established, the severity of the disease warrants contemplating the possibility of maintaining it indefinitely.
Please cite this article as: Martín Pedraz L, Galindo Zavala R, Yun Castilla C, Ortiz Garrido A, Núñez Cuadros E. Miopatía necrosante inmunomediada seronegativa con afectación miocárdica. An Pediatr. 2021;95:470–472.
Previous presentation: The study was presented at the 6th Forum of the Sociedad Española de Reumatología Pediátrica (SERPE); November 2016, Murcia, Spain.

Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals