


Primary adrenal insufficiency (PAI) in children is a rare condition characterized by deficient production of glucocorticoids and/or mineralocorticoids. The clinical manifestations are nonspecific and insidious. Providers need to know about this disorder to be able to make an early diagnosis, as appropriate management can be life-saving.
MethodsWe conducted a multicentre retrospective study including every patient aged less than 18 years given a diagnosis of PAI in the last 30 years at 5 Spanish hospitals.
ObjectivesThe objective was to determine the aetiologies, signs, symptoms and laboratory findings of PAI in the paediatric age group.
ResultsTwenty nine patients received a diagnosis of PAI at a median age of 5.6 years. An aetiological diagnosis was established in 23 patients (79.3%): X-linked adrenoleukodystrophy in 8 (27.6%), autoimmune adrenalitis in 6 (20.7%), X-linked adrenal hypoplasia congenita in 4 (13.8%), adrenocorticotropic hormone (ACTH) resistance syndrome in 2 (6.9%), Pearson syndrome in 2 (6.9%) and Allgrove syndrome in 1 (3.4%). In the remaining 6 patients, no clear aetiology was identified. Sixteen patients (55.2%) had onset with an adrenal crisis. Twenty patients (69%) needed combination therapy (hydrocortisone and fludrocortisone).
ConclusionsAsthenia, hyperpigmentation and hyponatraemia were the most prevalent sign, symptom and electrolyte abnormality at onset of PAI, although their absence does not rule out this disease. The elevation of ACTH persists despite adequate glucocorticoid replacement therapy.
La insuficiencia suprarrenal primaria (ISP) es una enfermedad rara en niños caracterizada por la incapacidad en la producción de glucocorticoides y/o mineralocorticoides. Las manifestaciones clínicas son inespecíficas e insidiosas. Debemos conocer esta enfermedad para poder realizar un diagnóstico precoz, ya que el correcto manejo de la enfermedad puede salvar vidas.
DiseñoSe ha realizado un estudio multicéntrico y retrospectivo, registrándose todos los pacientes diagnosticados de ISP (menores de 18 años) en los últimos 30 años de 5 hospitales españoles.
ObjetivosDeterminar la etiología, signos, síntomas y alteraciones analíticas en la insuficiencia suprarrenal primaria en la edad pediátrica.
ResultadosVeintinueve pacientes fueron diagnosticados de ISP, con una mediana de 5,6 años. Se logró emitir un diagnóstico etiológico en 23 pacientes (79,3%): encontramos 8 casos de adrenoleucodistrofia ligada al cromosoma X (27,6%), 6 adrenalitis autoinmunes (20,7%), 4 hipoplasias suprarrenales congénitas ligadas al X (13,8%), 2 síndromes de resistencia a la ACTH (6,9%), 2 pacientes con síndrome de Pearson (6,9%) y un paciente con síndrome de Allgrove (3,4%). En los otros 6 pacientes, la etiología es desconocida por el momento. Dieciséis pacientes (55,2%) debutaron en forma de crisis adrenal. Veinte pacientes (69%) precisaron tratamiento combinado (hidrocortisona y fludrocortisona).
ConclusionesLa astenia, la hiperpigmentación cutánea y la hiponatremia fueron el síntoma, el signo y la alteración electrolítica más frecuentes al debut, aunque su ausencia, no descarta una ISP. La ACTH (hormona adrenocorticotropa) permanece elevada a pesar de un correcto tratamiento con glucocorticoides.

Primary adrenal insufficiency (PAI) is a condition in which the capacity of adrenal glands to produce adrenocortical hormones (glucocorticoids and mineralocorticoids) is impaired.
It is a rare condition, with a prevalence of 82–144 cases per million inhabitants, an incidence of 4–6 cases per million inhabitants and a predominance of the female sex,1–3 and its incidence in children is unknown.
The most frequent cause of PAI present at birth is congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. There are few case studies in children, like the one we present here, that analyse rare aetiologies, such as autoimmune forms, corticotropic resistance or other genetic disorders. To our knowledge, only one such study has been conducted before in Spain.4
Autoimmune adrenalitis is the most frequent cause of PAI in adults, accounting for 80% of cases, with a clear predominance of the female sex.5 It is much less frequent in the paediatric population, in which it accounts for only 10%–15% of cases.5
It can present in isolation or in association with other diseases such as hypoparathyroidism, mucocutaneous candidiasis or other autoimmune diseases as part of an autoimmune pluriglandular syndrome (APS).
X-linked adrenoleukodystrophy (X-ALD) is a peroxisomal disorder with an incidence of 1 in 14 700 births.6 It is caused by changes in the ABCD1 gene (Xq28) that encodes a transmembrane protein (ALDP) involved in the transport of very long chain fatty acids (VLCFAs) from the cytosol to the peroxisome, where they are degraded. The accumulation of VLCFAs compromises the structural integrity of the myelin sheath, which causes neurological manifestations, and causes damage to the adrenal cortex and hypogonadism as VLCFAs accumulate in Leydig cells.
Congenital adrenal hypoplasia is caused by changes in the NR0B1 gene, located in the Xp21.2 region, which encodes the DAX1 protein. It is a rare disorder with an incidence of less than 1 in 12 500 births.7 It manifests with primary adrenal insufficiency, delayed puberty and impaired spermatogenesis in adulthood.
This disorder should be suspected in male patients once CAH and X-ALD have been ruled out, especially if there is a family history of infertility in males.
Familial glucocorticoid deficiency (FGD) or ACTH resistance syndrome is a rare disease of unknown prevalence with an autosomal recessive pattern of inheritance. It manifests with isolated glucocorticoid deficiency due to a lack of response to ACTH by cells in the zona fasciculata of the adrenal cortex. It is due to defects in the ACTH receptor (MC2R) or its accessory protein (MRAP) necessary for transmitting the ACTH signal into the cell.8 It should be suspected in patients with recurrent hypoglycaemia and mucocutaneous hyperpigmentation. A family history of consanguinity or unexplained premature death supports the diagnosis.
Pearson syndrome is a rare disease caused by a deletion in mitochondrial DNA. Its incidence is 1 per million births.9 It is characterised by sideroblastic anaemia associated with neutropenia and thrombopenia, exocrine pancreatic dysfunction and renal tubulopathy. It may be associated with diabetes, primary adrenal insufficiency, hypoparathyroidism and growth delay.
Allgrove syndrome (AS), also known as triple A syndrome, is characterised by alacrima, achalasia and adrenal insufficiency secondary to ACTH resistance. It is associated with peripheral motor and sensory neuropathy, dysautonomia, optic atrophy and ataxia. Its prevalence is unknown. It is an autosomal recessive disorder caused by changes in the AAAS gene (12q13.13) encoding the ALADIN protein.10
In many cases, the clinical manifestations resulting from combined cortisol and mineralocorticoid deficiency develop gradually, and the nonspecificity of symptoms complicates the diagnosis.
Glucocorticoid deficiency manifests with asthenia, anorexia and a tendency toward hypoglycaemia due to the impact of the lack of cortisol on energy metabolism (decreased gluconeogenesis and glycogenolysis); it can also cause orthostatic hypotension due to decreased sensitivity to catecholamines and gastrointestinal symptoms due to an increase in hydrochloric acid and pepsin in the intestinal lumen. Weight loss is common.
The decrease in cortisol leads to an increase in corticotropin-releasing hormone (CRH), ACTH and other peptides derived from pro-opiomelanocortin, such as β-lipotropin, which has melanocyte-stimulating activity, causing hyperpigmentation of the skin and mucous membranes, a feature characteristic of Addison disease.
Mineralocorticoid deficiency is responsible for salt loss due to hypoaldosderonism, which in turn results in hypovolaemia, hyponatraemia with increased natriuresis, hyperkalaemia and metabolic acidosis. Salt craving is typical. Blood tests will show low aldosterone levels with a compensatory increase in renin.
Its suspicion is of vital importance, as any type of stress (infections, trauma, surgery, etc) can trigger an acute episode of adrenal insufficiency, referred to as an adrenal crisis, manifesting with hypovolaemic shock with hyponatraemia, hyperkalaemia, acidosis and hypoglycaemia.
The estimated incidence of adrenal crises in Europe is of 6–8 cases per 100 inhabitants per year, of which 70% occur in before age 10 years.11,12 The mortality rate is 0.5 deaths per 100 patients per year.2 Thus, it is a serious condition requiring urgent medical attention.
We consider this study to be of great interest, given the low prevalence in the paediatric population, the high level of suspicion required for diagnosis and the high mortality rate of adrenal crises.
ObjectivesThe general objective was to identify the causes of primary adrenal insufficiency in paediatric patients. The specific objectives were to obtain epidemiological data on the different aetiologies, to gather data on the symptoms, signs and analytical findings present at onset, to determine the frequency of onset in the form of an adrenal crisis, to assess the value of ACTH measurement in treatment monitoring and to establish the number of patients that required combination therapy (glucocorticoids and mineralocorticoids).
Patients and methodsPatients. We conducted a multicentre and descriptive case study including all patients who received a diagnosis of PAI during childhood in the past 30 years (from January 1990 to December 2020) in 5 Spanish hospitals.
The sample included all patients with suspected and subsequently confirmed PAI with onset from birth to age 18 years. Their medical history had to be compatible with PAI and include elevated serum ACTH levels and/or decreased cortisol levels.
We excluded all deceased patients for whom we were unable to collect data, cases of congenital adrenal hyperplasia and cases with a iatrogenic, neoplastic, infectious and/or traumatic aetiology.
Methods. We collected data from health records (paper and/or electronic format).
The data analysis was performed with the software SPSS version 25.0 (IBM, NY, USA). We expressed results as frequencies and percentages (qualitative variables) or as median and range (quantitative variables). We used the χ2 test or the Fisher exact test to compare proportions. We compared quantitative or ordinal variables in different groups by means of the Kruskal-Wallis test and the Mann-Whitney U test. P values of less than 0.05 were considered statistically significant.
Ethical considerations. The project was approved by the local clinical research ethics committee.
ResultsCauses of adrenal insufficiency and epidemiologyThe final sample consisted of 29 patients, 22 (75%) male and 7 (25%) female. An aetiological diagnosis was made in 23 (79.3%): we identified 8 cases of X-linked adrenoleukodystrophy (27.6%), 6 of autoimmune adrenalitis (20.7%), 4 of congenital adrenal hypoplasia secondary to DAX1 protein abnormalities (13.8%), 2 of ACTH resistance syndrome (6.9%), 2 of Pearson syndrome (6.9%) and 1 of Allgrove syndrome (3.4%).
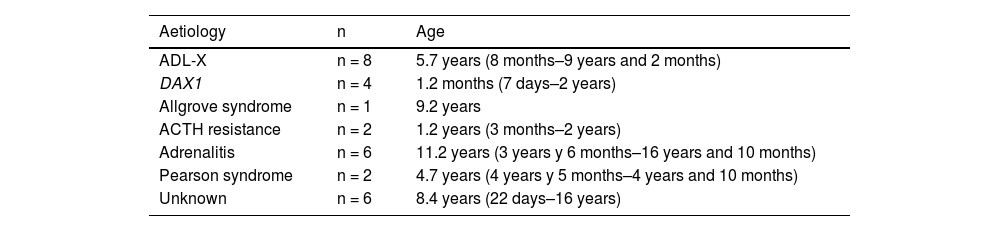
The age at diagnosis ranged from seven days to 16 years, with a median of 5.6 years. Table 1 shows the age of the patients at onset based on the aetiology.
Age at onset in patients by aetiology of PAI.
| Aetiology | n | Age |
|---|---|---|
| ADL-X | n = 8 | 5.7 years (8 months–9 years and 2 months) |
| DAX1 | n = 4 | 1.2 months (7 days–2 years) |
| Allgrove syndrome | n = 1 | 9.2 years |
| ACTH resistance | n = 2 | 1.2 years (3 months–2 years) |
| Adrenalitis | n = 6 | 11.2 years (3 years y 6 months–16 years and 10 months) |
| Pearson syndrome | n = 2 | 4.7 years (4 years y 5 months–4 years and 10 months) |
| Unknown | n = 6 | 8.4 years (22 days–16 years) |
ACTH, adrenocorticotropic hormone; DAX1, congenital adrenal hypoplasia; PAI, primary adrenal insufficiency; X-ADL, X-linked adrenoleukodystrophy.
Three female and three male patients received a diagnosis of autoimmune adrenalitis, with detection of adrenal cortical autoantibodies in all.
Four of them (67%) had onset with an adrenal crisis, while the other two had a more insidious onset, with one seeking care for mucocutaneous candidiasis and pubertal delay and the other for weight loss and asthenia.
An associated autoimmune disorder was identified in 66.7% of these patients, with detection of thyroid, pituitary and ovarian autoantibodies.
Half of the patients currently have adrenal insufficiency in isolation, while in the other 3 it is present as part of an autoimmune polyglandular syndrome (APS); 2 of them have associated primary hypothyroidism.
X-linked adrenoleukodystrophy (X-ADL)We identified 8 males with X-ADL, 2 of whom were siblings and 2 of whom had a sibling who had died of the same disease.
In this series, 87.5% of the cases were diagnosed as a result of progressive neurological manifestations, most frequently behavioural changes, detected in 5 patients (62.5%), followed by gait and language disturbances, found in 3 (37.5%). Twenty-five percent of the patients had visual or hearing loss or hypotonia. In one patient, the diagnosis was made following an episode of hyponatraemic dehydration (adrenal crisis).
The plasma concentration of very-long-chain fatty acids (VLCFAs) was elevated in all cases.
Genetic testing was carried out in 50% of the patients, confirming the presence of changes in the ABCD1 gene. The identified pathogenic variants were c.521A>G (p.Tyr174Cys) and c.901-3C>G.
Two of the patients died during the follow-up.
Congenital adrenal hypoplasiaWe identified 4 male patients with this diagnosis. As can be seen in Table 1, these were the patients with the earliest onset, which occurred as early as the neonatal period in 2 of them.
In all 4, molecular diagnostic techniques confirmed the presence of changes in the NR0B1 gene, which encodes the DAX1 protein. The identified pathogenic variants were c.707G>A (p.Trp236X), c.773C>A (p.Ala258Asp) and c.315G>A (p.Trp105X).
Two of the patients received a diagnosis of hypogonadotropic hypogonadism during the follow-up and were treated with intramuscular testosterone.
Familial glucocorticoid deficiency (FGD) or ACTH resistance syndromeDiagnosed in two female patients. In one, the aetiology was confirmed when genetic testing detected a compound heterozygous variant in the ACTH receptor gene (MC2R). The identified changes were MC2R:c.320A>G (p.Asp107Gly) and MC2R:c.433C>T (p.Arg145Cys).
In the other patient, genetic testing was not conducted but FGD was suspected due to parental consanguinity and the absence of mineralocorticoid deficiency.
It is worth noting that both patients had onset with seizures secondary to severe hypoglycaemia.
Pearson syndromeThere were 2 male patients who, in the course of their underlying disease, developed pancytopenia, exocrine pancreatic insufficiency, tubulopathy, postnatal growth retardation and psychomotor retardation. One developed non-autoimmune primary hypothyroidism, and it is worth noting that both patients were diagnosed with growth hormone deficiency.
Allgrove syndromeThe only patient was a boy who had onset with seizures secondary to severe hypoglycaemia. Before receiving the diagnosis of PAI, he had been diagnosed with achalasia, alacrima and neurologic abnormalities: abnormal visual evoked potentials (VEPs) and brainstem auditory evoked potentials (BAEPs) and distal spinal muscular atrophy (DSMA). In the presence of this characteristic triad, he received the diagnosis of Allgrove syndrome, although it was not confirmed by genetic testing.
Symptoms, signs and laboratory findings present at onsetAsthenia was the most frequent symptom, reported by 16 patients (55%), followed by vomiting and weight loss, reported by 14 patients (48.3%), and anorexia, reported by 11 (37.9%).
Four patients presented with seizures at onset, secondary to hypoglycaemia in 3 of them (with blood glucose levels of 30, 14 and 8 mg/dL) and to severe hyponatraemia in the remaining patient (with a blood sodium level of 104 mEq/L).
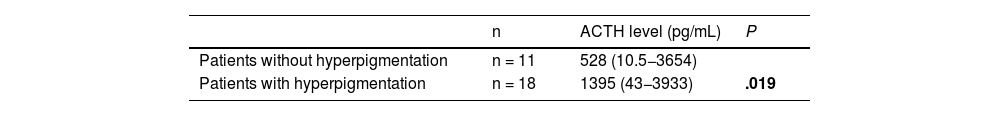
Hyperpigmentation was the most prevalent clinical feature, present in 18 patients (62.1%). Adrenocorticotropic hormone values at diagnosis were higher in patients with hyperpigmentation (Table 2).
Hyponatraemia was the most frequent electrolyte disturbance, present in 19 patients (65.5%). The median blood sodium level was 129.5 mEq/L (103–141).
Hyperkalaemia was found in only 7 patients (24.1%). The median blood potassium was 5 mEq/L (3.8–7.8).
When it came to blood glucose levels, 10 patients (34.5%) had levels of less than 60 mg/dL, of who 6 had levels of less than 50 mg/dL. The median blood glucose level was 71.5 mg/dL (8–111).
The serum ACTH level was elevated at onset in most cases (93.1%). The median ACTH level was 1100 pg/mL (10.5–3933). We found no differences in the ACTH levels in relation to the different aetiologies.
As for cortisol levels, values of less than 5 µg/dL were detected in 17 patients (58.6%), values of 5–9 µg/dL in 7 (24.1%) and values greater than 9 µg/dL in 5 (17.2%). All patients with cortisol levels greater than 5 µg/dL had elevated ACTH levels.
The data failed to demonstrate a negative correlation between cortisol levels and ACTH levels at diagnosis.
Aldosterone levels were decreased in 17 patients (58.6%); however, the plasma renin activity (PRA) was elevated in only 11 cases (37.9%).
Onset with an adrenal crisisFrom the clinical point of view, the onset was acute (adrenal crisis) in 16 cases (55.2%), although hypotension was only detected in 11.
Table 3 shows the patients who had onset with an adrenal crisis based on the aetiology.
Percentage of patients who had onset with an adrenal crisis by aetiology of PAI.
| ADL-X | DAX1 | Allgrove syndrome | ACTH resistance | Autoimmune adrenalitis | Pearson syndrome | Unknown aetiology |
|---|---|---|---|---|---|---|
| n = 8 | n = 4 | n = 1 | n = 2 | n = 6 | n = 2 | n = 6 |
| 1 (12.5%) | 4 (100%) | 1 (100%) | 2 (100%) | 4 (66.7%) | 0% | 4 (66.7%) |
ACTH, adrenocorticotropic hormone; DAX1, congenital adrenal hypoplasia; PAI, primary adrenal insufficiency; X-ADL, X-linked adrenoleukodystrophy.
We did not find significant differences in the presence of adrenal crises based on the age at diagnosis.
Of the patients who had onset with an adrenal crisis, 6 (37.5%) exhibited one of the biochemical abnormalities of the adrenal crisis triad (hyponatraemia, hyperkalaemia and hypoglycaemia), 7 patients (43.8%) two of them, and only 3 patients (18.7%) all three.
Monitoring of ACTH levels during follow-upACTH levels fluctuated during follow-up, normalising or even becoming transiently suppressed in 69% of cases. The last measurement was elevated in 66% of patients. We found no differences in this parameter based on the aetiology.
TreatmentTwenty patients (69%) received combination therapy with hydrocortisone and 9-α-fluorohydrocortisone, while 9 (31%) received glucocorticoids as monotherapy (the two patients with ACTH resistance syndrome, the two patients with Pearson syndrome and five of the patients with adrenoleukodystrophy).
DiscussionThe most frequent causes of PAI in our study were X-linked adrenoleukodystrophy, autoimmune adrenalitis and congenital adrenal hypoplasia. In our series, X-linked adrenoleukodystrophy predominated, in agreement with a recently published article13; However, other studies continue to find that the most frequent type of aetiology is the autoimmune aetiology.14–16
We ought to highlight that congenital adrenal hypoplasia was the aetiology with the earliest onset (median age at diagnosis, 1.2 months), while the oldest age at diagnosis corresponded to autoimmune adrenalitis (median age at diagnosis, 11.2 years), which was consistent with the previous literature.7,15
The male/female ratio was 1/0.3, and the male predominance was consistent with other paediatric case series.4,13,14
In our study, asthenia, hyperpigmentation and hyponatraemia were the most frequent symptom, sign and electrolyte disturbance, respectively, although their absence does not rule out PAI.
Cutaneous hyperpigmentation is a cardinal sign that suggests the diagnosis, but it is not always present. Published studies show that hyperpigmentation is present in 90% of patients,17–19 although the frequency was lower in our series (62.1%). Hyperpigmentation can be very subtle at onset or in the first 6 weeks of life,17 which could explain its absence in 3 of our patients, who had onset at 7, 22 and 24 days post birth.
Hyponatraemia was present in 65.5% of patients, although this feature can be even more frequent (90%).20,21
Hyperkalaemia was infrequent in our sample (24.1%) compared to other published series, in which it was found in up to 50% of the cases.16,20 Its absence may be explained in patients with profuse vomiting, since the loss of hydrochloric acid causes metabolic alkalosis and, as a result, hypokalaemia, although we did not find an association between potassium levels and the presence of vomiting in our sample.
In this case series, 55.2% of the patients had onset with an acute adrenal crisis. In the paediatric population, adrenal crisis is defined as an acute deterioration in health status with haemodynamic changes (arterial hypotension, slow capillary refill or increased heart rate for age) or severe electrolyte disturbances (hyponatraemia or hyperkalaemia) or hypoglycaemia not attributable to any other cause. The diagnosis is confirmed when the disturbances are reversed after glucocorticoid administration.22
As this series demonstrates, arterial hypotension is not required for diagnosis of adrenal crisis, as more than half of our patients had onset with adrenal crisis but hypotension was only detected in 37.9%. Similarly, we should not wait for blood glucose, sodium and potassium levels to all be abnormal before suspecting an adrenal crisis.
The ACTH level is not a suitable marker for the purpose of treatment monitoring. Values of ACTH should remain elevated, as illustrated by our sample; if levels are within the normal range, the patient may be overtreated.3,17 Similarly, treatment should not be modified based on the degree of hyperpigmentation.17 The corticoid dose should be adjusted based on the assessment of weight gain, growth and clinical manifestations.
In this series, 69% of patients required combination therapy (glucocorticoid + mineralocorticoid). The rest of the patients did not show mineralocorticoid axis involvement, which can be explained by the fact that the zona glomerulosa of the adrenal cortex is usually less affected than the zona fasciculata and the zona reticularis.6
In conclusion, clinicians must be vigilant to recognise the signs and symptoms of primary adrenal insufficiency in order to achieve an early diagnosis and prevent the development of adrenal crises. Furthermore, knowledge of the different causes of PAI allows the clinician to narrow down the aetiological diagnosis.
FundingThis research did not receive any external funding.

