

Congenital hypogonadotropic hypogonadism (CHH) can present alone or in association with anosmia or other congenital malformations. More than 30 genes have been identified as being involved in the pathogenesis of CHH with different patterns of inheritance, and the increasing availability of next generation sequencing (NGS) has increased the diagnostic yield.
MethodsWe analysed the diagnostic yield of NGS in patients with CHH using the clinical exome filtered with virtual panels. We also assessed whether designing panels based on the presence/absence of microsmia increased the diagnostic yield.
ResultsThe use of a 34-gene virtual panel confirmed the diagnosis of CHH in 5 out of 9 patients (55%) patients. In 2 out of 9 (22%), the findings were inconclusive. Applying the presence/absence of microsmia criterion to choose genes for analysis did not improve the diagnostic yield.
ConclusionsThe approach to the genetic study of patients with CHH varies depending on the resources of each healthcare facility, so the sensitivity of testing may vary substantially depending on whether panels, clinical exome sequencing or whole exome sequencing (WES) are used. The analysis of all genes related to CHH regardless of the presence/absence of microsmia seems to be the best approach.
El hipogonadismo hipogonadotropo congénito (HHC) puede presentarse de manera aislada o acompañado de anosmia o de malformaciones congénitas. Más de 30 genes han sido implicados en la patogénesis de HHC; además, se han descrito varios patrones de herencia asociados a esta entidad. La creciente disponibilidad de técnicas de secuenciación masiva (NGS) ha permitido que aumente el rendimiento diagnóstico del estudio de esta patología.
Pacientes y métodosEvaluamos el rendimiento diagnóstico del estudio mediante NGS de pacientes con HHC, usando la secuenciación del exoma clínico filtrado por paneles virtuales. Además, se analizó si el diseño de estos paneles, basándose en la presencia/ausencia de microsmia/anosmia aumentaban este rendimiento diagnóstico.
ResultadosUsando un panel virtual compuesto de 34 genes pudimos confirmar el diagnóstico de HHC en cinco de nueve pacientes (55%). En dos de nueve individuos (22%) estudiados se obtuvieron resultados no concluyentes. La ausencia/presencia de microsmia para la elección de genes a estudiar no mejora el rendimiento diagnóstico.
ConclusionesEl abordaje del estudio genético de pacientes con HHC puede variar en función de las técnicas disponibles en cada centro, por lo que la sensibilidad del test utilizado variará, dependiendo si se utiliza secuenciación de paneles, exoma clínico o exoma completo. El análisis de todos los genes relacionados con HHC independientemente de la presencia/ausencia de microsmia pareciera el abordaje con mejor rendimiento.
The term congenital hypogonadotropic hypogonadism (CHH) refers to deficiencies in the production, secretion, or activity of gonadotropin-releasing hormone (GnRH). It encompasses a broad clinical spectrum, including pubertal delay and/or infertility. It can present alone or in association with other features such as anosmia or microsmia, palate defects, hearing impairment, ear anomalies, renal malformations and/or skeletal anomalies, among others.1 The incidence of CHH is of approximately 1 per 30 000 male individuals, and it is 3 –5 times higher in men than in women.2–4
The pathophysiology of CHH is classically divided into 2 major forms. The first involves abnormal differentiation, development, or migration of GnRH-producing neurons during foetal life, preventing the correct placement of these neurons in the hypothalamus and also affecting olfactory neurons. This form corresponds to Kallmann syndrome (KS).5 In KS, the common pathophysiological feature is fibroblast growth factor signalling, with abnormalities in the proteins involved in this pathway, such as anosmin 1 and prokineticin, resulting in changes in GnRH neuron migration.2,5–7 The second form involves changes in the GnRH pathway at different levels (synthesis, release, actions on gonadotropic pituitary cells), and corresponds to the normosmic CHH presentation. The pathophysiology of CHH may sometimes involve a combination of genetic abnormalities that simultaneously affect both neuronal development and the hypothalamic-pituitary-gonadal axis.5–7
More than 30 genes have been identified to be associated with the pathogenesis of CHH and several patterns of inheritance have been described, including autosomal dominant, autosomal recessive, X-linked and sporadic cases product of de novo variants.1,6,8 In addition, there is evidence of oligogenic transmission in up to 20% of cases.1,6,8
As next generation sequencing (NGS) has become more affordable and thus more widely available, the diagnosis of this disease has increased enormously.1,7 At least 30 loci account for roughly 50% of cases, although variants in certain loci are exceedingly rare and tend to be identified in complex syndromes.5
Genetic diagnosis is now affordable in most settings thanks to advances in NGS technology and bioinformatics. Due to the genetic heterogeneity of CHH, diagnosis formerly required a tedious “gene by gene” approach, which was expensive and had a low diagnostic yield. Therefore, in this study, we evaluate the diagnostic yield of NGS in patients with CHH by means of clinical exome capture and filtering by a virtual panel. We also assessed whether designing virtual panels based on the presence or absence of microsmia/anosmia was cost-effective.
Patients and methodsWe conducted a descriptive study of patients with CHH managed in the outpatient paediatric endocrinology clinic between 2014 and 2019.
Inclusion criteria:- -
Absent or incomplete puberty.
- -
Girls: absence of progressive thelarche after age 13 years and/or absence of menarche after more than 4 years of progressive thelarche and/or absence of menarche at age 16 years (primary amenorrhea).
- -
Boys: absence of onset of testicular growth or testicular volume of less than 4 mL (measured with a Prader orchidometer) by age 14 years, or arrested development within 5 years after reaching a testicular volume of 4 mL or greater;
- -
- -
Prepubertal response in the luteinizing hormone-releasing hormone (LHRH) stimulation test: LH peak of less than 5 IU/L. Inhibin B values below the 5th percentile;
- -
Absence of abnormalities in another hormonal axis;
- -
Constitutional delay of growth and puberty ruled out based on personal and family history and after confirming normal progress of puberty during the follow-up8,9;
- -
Functional hypogonadotropic hypogonadism associated with chronic disease ruled out based on medical history and laboratory testing including a complete blood count, acute phase reactant serum levels and serological testing for coeliac disease8,9;
- -
Intracranial disease ruled out based on the findings of cranial magnetic resonance imaging; and
- -
Patient/parental consent to participation in the study.
- -
We collected data on the following demographic and clinical variables: age, sex, family history, anthropometric measurements, Tanner stage and associated symptoms. We also recorded the results of the University of Pennsylvania Smell Identification Test (UPSIT) (Sensonics Inc.; Haddon Height, NJ, USA). The test includes 40 microcapsules of standardized odours in a “scratch and sniff” booklet, and patients are asked to identify each odour by picking 1 out of 4 answer choices provided. The UPSIT score is interpreted based on normative data for age and sex described in the UPSIT manual. A score of less than 30 points corresponds to microsmia and a score of less than 18 to anosmia.10,11
Genetic analysisWe obtained samples of genomic DNA from peripheral blood lymphocytes with an automatic DNA extractor (EZ1 DNA Blood kit-EZ1 Advanced XL, QIAGEN, Germany) following the directions of the manufacturer. We measured the concentration of DNA and assessed the quality of samples by means of spectrophotometry (NanoDrop® ND-1000 spectrophotometer, Wilmington DE, USA). Library preparation, sequencing, and data analysis were performed following validated protocols. The clinical exome was captured with the Clinical Exome Solution®️ kit (SOPHiA Genetics) and the sequencing performed with the NextSeq 500 system (Illumina).
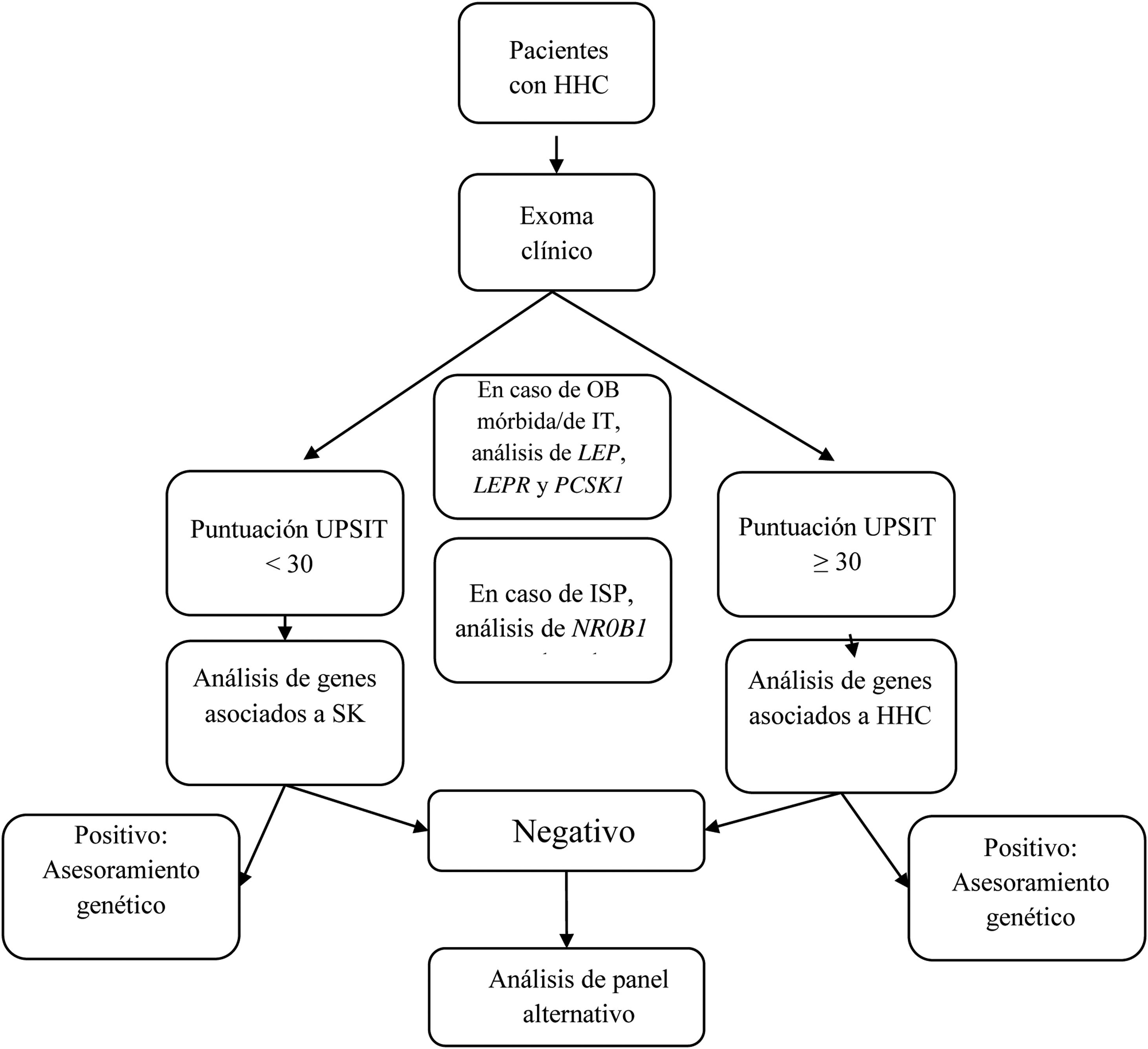
We designed a virtual panel of 34 genes out of those included in the capture kit and associated with CHH with and without microsmia/anosmia based on the European Consensus Statement published in 2015 by Bohem et al.1 and other reviews2,5–8,12–16 (Table 1). Although our primary objective was to assess the diagnostic yield of genetic tests guided by the presence of anosmia/microsmia, we only included some genes associated with syndromic presentations in the evaluation of patients with a compatible phenotype. Our analysis consisted of the following steps (see Fig. 1):
- 1.
If the patient presented with morbid or early-onset obesity, we included the genes LEP, LEPR, and PCSK1;
- 2.
If the patient had primary adrenal insufficiency, we included gene NR0B1(DAX1);
- 3.
In patients presenting with a KS phenotype, we analysed the following genes: ANOS1, AXL, CHD7, FGF8, FGFR1, HESX1, NSMF, PNPLA6, POLR3B, PROK2, PROKR2, SEMA3A, SEMA3E, SEMA7A, SMCHD1, SOX9, SOX10, SPRY4, WDR11, RAB18, RAB3GAP1, and RAB3GAP2;
- 4.
In patients with CHH without microsmia/anosmia, we analysed the following genes: GNRH1, GNRHR, KISS1, KISS1R, TAC3, TACR3, FSHB and LHB; and
- 5.
In patients with KS that had a negative result, we also analysed the genes included in step 4, and in patients with CHH without microsmia/anosmia and a negative result, we also analysed the genes included in step 3.
Genes included in the virtual panel for Kallmann syndrome and for CHH without anosmia.
| Genes associated with Kallmann syndrome and “syndromic presentation” | Genes associated with CHH without anosmia | |
|---|---|---|
| POLR3B (NM_018082.5) | SOX10 (NM_006941) | FSHB (NM_000510.2) |
| LEP (NM_00230.2) | ANOS1 KAL1 (NM_000216) | GNRH1(NM_000825) |
| NR0B1/DAX1 (NM_00074.4) | AXL (NM_001699) | GNRHR (NM_000406.2) |
| LEPR (NM_002303) | SEMA3E (NM_012431.3) | KISS1 (NM_002256) |
| PCSK1 (NM_000439) | FGF8 (NM_033163.3) | KISS1R (NM_032551) |
| SOX9 (NM_000346.3) | FGFR1 (NM_001174063) | LHB (NM_000894.2) |
| PNPLA6 (NM_006702) | SEMA7A (NM_003612.4) | TAC3 (NM_013251.3) |
| CHD7 (NM_017780) | HESX1 (NM_003865) | TACR3 (NM_001059) |
| RAB18 (NM_021252.4) | NSMF (NELF) (NM_001130969) | |
| RAB3GAP1 (NM_012233) | PROK2 (NM_001126128.1) | |
| RAB3GAP2 (NM_012414.3) | SPRY4 (NM_030964.3) | |
| SEMA3A (NM_006080) | WDR11(NM_018117.11) | |
| SMCHD1 (NM_015295.2) | PROKR2 (NM_144773) | |

Algorithm for genetic analysis guided by the presence of absence of anosmia/microsmia.
CHH, congenital hypogonadotropic hypogonadism; EO, early-onset; KS, Kallmann syndrome; OB, obesity; PAI, primary adrenal insufficiency; UPSIT, University of Pennsylvania Smell Identification Test.
We used the SOPHiA DDM platform (SOPHiA Genetics) for variant analysis. We used conventional Sanger sequencing (Genetic analyzer 3130, Applied Bio-systems, Foster City, CA, USA) to confirm suspected pathogenic variants. When available, we also obtained the genotypes of family members by Sanger sequencing. We classified the variants following the recommendations of the American College of Medical Genetics (ACMG).17
Recruitment of participantsThe study was reviewed and approved by the ethics committee of our hospital (code EO 11-2015, date of approval, March 26, 2015) and conducted in adherence to the principles of the Declaration of Helsinki. Participants signed a written informed consent form following a thorough explanation of the nature of the study procedures. Participants did not receive any financial compensation.
The collection of samples is preserved in the biobank of the hospital.
ResultsThe study included 9 male patients that underwent genetic testing for diagnosis with the exome approach. Five (56%) exhibited a KS phenotype, 3 (33%) CHH without olfactory impairment and 1 (11%) features of CHARGE syndrome. Table 2 presents the clinical and genetic data for these patients.
Description of the genotype and phenotype of patients under study.
| Patient | Gene | Variant, zygosity and origin | Classification | Testicular volume (mL) left/right. | Peak (P) or Basal(B) LH (IU/L) | Testosterone (ng/mL) |inhibin B (pg/mL) | UPSIT score | Height (z-score) at diagnosis |
|---|---|---|---|---|---|---|---|
| Locus | Age at examination (years) | Additional phenotypic features | |||||
| 1 | SOX10 | NM_006941: c.929dupT | Likely pathogenic | 1/2 | <0.07(B)|<0.4|NM | 8/40 | +0.07. |
| 22q13.1 | NP_008872: p.Ser311Glufs*91 | 12 | Deafness, microphallus | ||||
| Heterozygous, UDO | |||||||
| 2 | SOX10 | NM_006941: c.473A>C | Likely pathogenic | 5/5 | <0.07(B)|<0.4|NM | 36/40 | −1. |
| 22q13.1 | NP_008872: p.Glu158Ala | 15.5 | None | ||||
| Heterozygous, UDO | |||||||
| 3 | – | – | 2/absent | 0.47(P)|<0.4|NM | 15/40 | −2.12. | |
| 16 | Cryptorchidism | ||||||
| 4 | ANOS1 | NM_000216.4: c.932_941delTCACTATAGT | Pathogenic | 2/2 | 0.8(P)|0.39|17 | 7/40 | −1.46. |
| Xp22.31 | NP_000207: p.Val311Leufs*25 | 15.7 | Bilateral cryptorchidism, olfactory bulb agenesis | ||||
| Hemizygous, de novo | |||||||
| 5 | SEMA3A | NM_006080: c.1303G>A | VUS | 2/2 | 0.7(P)|0.29|10 | 32/40 | −1.06. |
| 7q21.11 | NP_006071: p.Val435Ile | 14 | Microphallus, cryptorchidism | ||||
| Heterozygous, UDO | |||||||
| 6 | – | – | – | 2/3 | 2.1(P)|0.2|12 | 36/40 | +0.07. |
| 19 | None | ||||||
| 7 | FGFR1 | NM_001174063: c.1196delT | Likely pathogenic | 2/2 | <0.07(B)|1.77|NM | 14/40 | + 1.23. |
| 8p11.23 | NP_001167534.1p.Met399Argfs*37 | 18 | Facial dysmorphic features, cryptorchidism | ||||
| Heterozygous, UDO | |||||||
| 8 | NSMF | NM_001130969: c.419G>T | VUS | 3/3 | 0.4 (B)|0.1|NM | 26/40 | −1.89. |
| 9q34.3 | NP_001124441: p.Arg140Leu | 22 | Cryptorchidism | ||||
| Heterozygous, UDO | |||||||
| CHD7 | NM_017780: c.4226T>A | VUS | |||||
| 8q12.2 | NP_060250: p.Val1409Glu | ||||||
| Heterozygous, UDO | |||||||
| 9 | CHD7 | NM_017780.3: c.7160C>A | Pathogenic | 2/2 | 0.7 (B)|0.11|NM | NA | −2.7. |
| 8q12.2 | NP_060250.2: p.Ser2387 | 16 | Optic nerve dysplasia, iris coloboma, mitral insufficiency, cryptorchidism, deafness, ASD | ||||
| Heterozygous, de novo |
ASD, autism spectrum disorder; LH, luteinizing hormone; NA, not available due to lack of collaboration (patient with ASD); NM, not measured; UDO, undetermined origin; VUS, variant of uncertain significance.
Using the virtual panel of 34 genes designed for the study, we detected pathogenic or likely pathogenic variants that explained the phenotype in 5 of the 9 patients (55%). In another 2 patients, testing detected variants of unknown significance that did not fully explain the phenotype, while no variants were detected in the remaining 2. Every relevant variant was detected with the virtual panel designed for KS. We classified the detected variants following the criteria of the American College of Medical Genetics and Genomics (ACMG).17
Below, we present detailed information on each of the variants detected in the patients under study:
SOX10: Patient 1 was a boy aged 12 years that presented with a testicular volume of less than 4 mL, microphallus, indetectable basal gonadotropins, microsmia and sensorineural hearing loss. Testing in this patient detected a pathogenic variant in SOX10, a gene that belongs to the SOX transcription factor family. Members of this family are involved in several multiorgan developmental processes. SOX10 encodes a transcription factor expressed by GnRH neuron precursors.5 Loss-of-function variants in the heterozygous state are associated with KS with incomplete penetrance,5 and these patients may also present sensorineural hearing loss,5,7 iris hypopigmentation and wisps/patches of white hair.5 The variant that we found in patient 1, c.929dupT; p.Ser311Glufs*91, duplicates the thymine at position 930, resulting in a frameshift that produces a premature stop codon creating a truncated protein or could result in nonsense-mediated mRNA decay (NMD).18 This variant has not been described before (Clinvar19, Varsome20, gnomAD21). Unfortunately, parental DNA was unavailable. We applied the ACMG guidelines for the interpretation of variants17 and classified this variant as likely pathogenic.
Patient 2 was a boy aged 17 years with incomplete puberty (arrested testicular volume of 5 mL, the same volume measured in the initial evaluation at age 15.5 years, see Table 2) and a hormone profile compatible with hypogonadotropic hypogonadism, without evidence of olfactory abnormalities in the UPSIT test and an unclear family history of delayed puberty in the father. Genetic testing detected a heterozygous missense variant in SOX10, c.473A>C. This is a novel variant, and in silico predictions using different tools classified it as deleterious. The variant was located in the HMG-box DNA-binding domain, where other missense variants have been classified as pathogenic; 38 out of 44 (86%) missense variants in gene SOX10 that are not variants of unknown significance (VUS) are pathogenic, exceeding the threshold of 51.0%, and 97 out of 152 (63.8%) clinically reported variants in gene SOX10 are pathogenic, exceeding the threshold of 12.0%.19 We classified this variant as likely pathogenic based on the ACMG criteria.17 Parental DNA was not available in this case, either.
ANOS1: In patient 4, a boy aged 15.7 years presenting with features of the KS phenotype (Table 2), testing detected the variant c.932_941delTCACTATAGT in ANOS1. This is a frameshift mutation that changes a valine to a leucine in position 311 and the amino acid sequence that follows, leading to a premature stop codon that may create a truncated protein or result in NMD.18 This variant has not been described before (Clinvar,19 Varsome,20 gnomAD21). Since other frameshift variants have been described as pathogenic, we concluded that this variant is pathogenic based on the ACMG guidelines.17 The study of maternal DNA showed that this was a de novo variant. ANOS1 was the first gene found to be associated with KS22 and follows an X-linked pattern of inheritance.
FGFR1: In patient 7, a boy aged 18 years with a testicular volume of less than 4 mL and undetectable basal LH levels presenting with the KS phenotype, we found a frameshift variant in the FGFR1 gene, c.1196delT. This variant produces a premature stop codon and a truncated protein without a protein-kinase domain, or it may result in NMD.18 It has not been described before (Clinvar19, GnomAD21, Varsome20). The patient had cryptorchidism, CHH and dysmorphic facial features. Applying the ACMG guidelines,17 we classified this variant as likely pathogenic. FGFR1 was the first gene known to cause an autosomal dominant (AD) form of KS19–21 through loss-of-function variants with incomplete penetrance.1,5,12 We ought to underscore that pathogenic variants of the FGFR1 gene may give rise to the KS or the normosmic CHH phenotypes5,7 and account for up to 10% of KS cases and 7% of normosmic CHH cases.6 Some patients have syndromic presentations with skeletal abnormalities, ectrodactyly, cleft palate, and severe CHH. It is worth noting that these manifestations are related to biallelic FGFR1 pathogenic variants.5
NSMF: Patient number 8 was a man aged 22 years that presented with the KS phenotype (Table 2) and carried the novel missense variant c.419G>T, which we interpreted as a VUS.17 This patient also had a missense variant in the CHD7 gene, c.4226T>A, which is not included in population databases or pathogenic variant databases. This variant is in the helicase domain, and in silico predictors classified it as deleterious. Parental samples were not available.
CHD7 has not been associated with the oligogenic pattern of inheritance to date,23 so we cannot be sure whether there is an association between the NSMF variant and the variant found in CHD7. Unfortunately, parental samples were unavailable to test the segregation pattern of the variants.
CHD7: In addition to the variant detected in patient 8, in patient 9, a boy aged 16 years with a phenotype compatible with CHARGE syndrome (Table 2), we found the nonsense variant c.7160C>A; p.Ser2387*. This variant results in a premature stop codon and a truncated protein or possibly NMD.18 It has been described as pathogenic by Janssen et al. in patients with CHARGE syndrome.24 The analysis of parental DNA revealed that it was a de novo variant.
SEMA3A: In patient 5, a boy aged 14 years that presented with cryptorchidism and microphallus, we found the missense variant c.1303G>A. The GnomAD database reports a population frequency for this variant of 0.014.21 We concluded that it was a VUS because this gene is also associated with an oligogenic inheritance pattern and we did not detect any other variant in genes known to be involved in oligogenic CHH.5,7
DiscussionThe diagnostic yield of our approach was 55%, which was consistent with the previous literature.1,5,7,12 In addition, we identified 2 patients with novel pathogenic variants in genes associated with the oligogenic form of CHH, suggesting that the presence of a pathogenic variant in one gene is not enough to cause the disease. Also, it is reasonable to hypothesise that common variants categorised as benign in these genes or in regulatory or intronic regions could be the additional factor required to cause CHH.
In our study, all pathogenic and likely pathogenic variants and VUS were detected with the virtual panel designed for KS. This may be explained by a higher frequency of changes in genes associated with KS being compared to changes in genes associated with normosmic CHH,12 in addition to the phenotypic overlap between genes involved in these two phenotypes,1,7 even in patients without microsmia/anosmia. Thus, given the revolution in genomic analysis that we are currently experiencing, in which many laboratories use the clinical exome or the whole exome filtered by virtual panels, it would make sense to design a virtual panel that offers the highest sensitivity but remains focused on the primary purpose of the panel, despite the broad possibilities offered by virtual panels, to decrease the likelihood of secondary findings.
In 2 patients we found VUS in genes associated with the oligogenic pattern of inheritance, but we did not identify the second variant required for this pattern, so our results were inconclusive. The genes known to be associated with oligogenic inheritance of CHH are the following2,5–7,14–16,25–28:
- •
FGF8/FGFR1 pathway: FGF8, FGFR1, ANOS1, FGF17, KLB, IL17RD, HS6ST1, NSMF, SPRY4, FLRT3 and DUSP6;
- •
Migration of neurons from the nasal placode to the hypothalamus: PROR2, PROKR2, SEMA3A, SEMA3E and SEMA7A; and
- •
GnRH expression and function: WDR11, GNRHR, KISS1R, TAC3, and TACR3.
There are limitations to our study. First, we used a clinical exome capture kit, the Clinical Exome Solution version 2 of SOPHiA GENETICS, that includes 4493 genes but does not include every gene associated with CHH. The 15 relevant genes that are not included in this kit are CCDC141, DUSP6, FEZF1, FGF17, FLRT3, HS6ST1, IL17RD, KLB, IGSF10, DMXL2, OTUD4, POLR3A, RNF216, STUB1, and TBC1D20. Out of these genes, we must highlight IL17RD and FGF17, which account for 3% of CHH cases.15 In addition, parental DNA samples were not available in most cases. Lastly, the sample was small.
Nonetheless, the diagnostic yield in our study was consistent with other research conducted worldwide.1,4,7,28,29 In our sample, all relevant genetic findings were identified through the virtual panel designed to study genes associated with KS. Therefore, in patients with normosmic CHH, the analysis of the alternative virtual panel was crucial to diagnosis. In our opinion, this is related to the different phenotypic expression of some genes related to CHH as regards the sense of smell.2,5,7 This fact led us reflect on the usefulness of WES filtered by two different virtual panels taking into account the presence or absence of olfactory disturbances. It may be more practical to design a single virtual panel that includes all genes related to KS and normosmic CHH.
In our opinion, WES filtered by a virtual panel of genes related to CHH is a very sensitive test and offers the possibility of reassessment at any time. However, use of a virtual panel has the following drawbacks: (a) from an ethics standpoint, the issue of secondary findings must be discussed with patients and/or guardians during pre-test consultation; and (b) this kind of test is not widely available yet. Furthermore, pre-designed panels lack some of the genes identified most recently and do not allow for reanalysis of the data, although they are available in most laboratories.
In conclusion, the genetic study of well-characterized patients with CHH that present with KS or normosmic CHH is useful and should be performed in all patients with this disease. Nevertheless, the sensitivity of testing may vary substantially depending on whether a gene panel, the clinical exome, or the whole exome (WES) is analysed.
FundingThis study was supported by the Department of Genomic Medicine of the Universidad Autónoma de Madrid-Fundación Jiménez Díaz (file 081800).
Conflict of interestThe authors declare no conflicts of interest.
We are very grateful to Oliver Shaw for his advice in drafting and organizing the English version of the manuscript. We thank the children who participated in the study and their families.
Previous presentation: Partial data presented as an oral communication “Aproximación al diagnóstico molecular del hipogonadismo hipogonadotropo aislado con y sin alteración del olfato”, XLI Congress of the Sociedad Española de Endocrinología Pediátrica; May 24–26, 2019.

