
El hipogonadismo hipogonadotropo congénito (HHC) puede presentarse de manera aislada o acompañado de anosmia o de malformaciones congénitas. Más de 30 genes han sido implicados en la patogénesis de HHC; además, se han descrito varios patrones de herencia asociados a esta entidad. La creciente disponibilidad de técnicas de secuenciación masiva (NGS) ha permitido que aumente el rendimiento diagnóstico del estudio de esta patología.
Pacientes y métodosEvaluamos el rendimiento diagnóstico del estudio mediante NGS de pacientes con HHC, usando la secuenciación del exoma clínico filtrado por paneles virtuales. Además, se analizó si el diseño de estos paneles, basándose en la presencia/ausencia de microsmia/anosmia aumentaban este rendimiento diagnóstico.
ResultadosUsando un panel virtual compuesto de 34 genes pudimos confirmar el diagnóstico de HHC en cinco de nueve pacientes (55%). En dos de nueve individuos (22%) estudiados se obtuvieron resultados no concluyentes. La ausencia/presencia de microsmia para la elección de genes a estudiar no mejora el rendimiento diagnóstico.
ConclusionesEl abordaje del estudio genético de pacientes con HHC puede variar en función de las técnicas disponibles en cada centro, por lo que la sensibilidad del test utilizado variará, dependiendo si se utiliza secuenciación de paneles, exoma clínico o exoma completo. El análisis de todos los genes relacionados con HHC independientemente de la presencia/ausencia de microsmia pareciera el abordaje con mejor rendimiento.
Congenital hypogonadotropic hypogonadism (CHH) can present alone or in association with anosmia or other congenital malformations. More than 30 genes have been identified as being involved in the pathogenesis of CHH with different patterns of inheritance, and the increasing availability of next generation sequencing (NGS) has increased the diagnostic yield.
MethodsWe analysed the diagnostic yield of NGS in patients with CHH using the clinical exome filtered with virtual panels. We also assessed whether designing panels based on the presence/absence of microsmia increased the diagnostic yield.
ResultsThe use of a 34-gene virtual panel confirmed the diagnosis of CHH in 5 out of 9 patients (55%). In 2 out of 9 (22%), the findings were inconclusive. Applying the presence/absence of microsmia criterion to choose genes for analysis did not improve the diagnostic yield.
ConclusionsThe approach to the genetic study of patients with CHH varies depending on the resources of each healthcare facility, so the sensitivity of testing may vary substantially depending on whether panels, clinical exome sequencing or whole exome sequencing (WES) are used. The analysis of every genes related to CHH regardless of the presence/absence of microsmia seems to be the best approach.
El término hipogonadismo hipogonadotropo congénito (HHC) hace referencia a deficiencias en la producción, secreción o actividad de la hormona liberadora de gonadotropina (GnRH). Su espectro clínico es amplio, incluyendo el retraso puberal y/o la infertilidad. Puede aparecer aislado o asociado a otras alteraciones, como la anosmia o hiposmia, defectos del paladar, déficits auditivos, malformaciones de pabellón auricular, malformaciones renales y/o anomalías esqueléticas1. Su incidencia es de aproximadamente un caso por 30.000 varones y es de tres a cinco veces mayor en hombres que en mujeres2–4.
La HHC tradicionalmente se clasifica según su fisiopatología en dos formas principales. La primera implica la diferenciación, desarrollo o migración alterada de las neuronas productoras de hormona liberadora de gonadotropinas (GnRH) durante el desarrollo fetal que impide el emplazamiento correcto de estas neuronas en el hipotálamo, afectando también a las neuronas olfatorias. Esta presentación corresponde al síndrome de Kallmann (SK)5. El denominador común en la fisiopatología del SK es la afectación de la vía de señalización de los factores de crecimiento de fibroblastos, con alteraciones en las proteínas implicadas, como la anosmina 1 y la procineticina, que producen alteraciones en la migración de las neuronas productoras de GnRH2,5–7. La otra forma implica alteraciones en la síntesis, liberación y/o acción de GnRH y corresponde a la presentación de HHC con olfato normal. La fisiopatología de la HHC puede implicar una combinación de alteraciones genéticas que afectan simultáneamente al desarrollo neuronal y el eje gonadotropo5–7.
Se han identificado más de 30 genes implicados en la etiopatogenia del HHC con distintos patrones de herencia: autosómico dominante, autosómico recesivo, ligada al cromosoma X y casos esporádicos producidos por variantes de novo1,6,8. También hay evidencia de herencia oligogénica en hasta el 20% de los casos1,6,8.
Con la reducción en el coste de las técnicas de secuenciación de nueva generación (NGS) y, por tanto, su mayor disponibilidad, el diagnóstico del HHC ha aumentado considerablemente1,7. Al menos 30 loci son responsables de aproximadamente el 50% de los casos, aunque los cambios en ciertos loci son muy infrecuentes y suelen detectarse en síndromes complejos5.
Actualmente, el diagnóstico genético es asequible en casi todos los entornos gracias a los avances en la tecnología NGS y en el posterior análisis bioinformático. Debido a la heterogeneidad genética del HHC, en el pasado, el diagnóstico requería una aproximación tediosa «gen por gen», con elevados costes y una baja rentabilidad diagnóstica. En consecuencia, el objetivo del estudio fue evaluar el rendimiento diagnóstico de la NGS en pacientes con HHC mediante la secuenciación del exoma clínico y su filtrado por paneles virtuales. También se analizó si el diseño de estos paneles, basándose en la presencia o ausencia de hiposmia/anosmia, resultaba coste-efectivo.
Pacientes y métodosEstudio descriptivo de pacientes con HHC seguidos en la Unidad de Endocrinología Infantil y el Servicio de Endocrinología del Hospital Universitario Fundación Jiménez Díaz entre el 2014 y el 2019.
Criterios de inclusión:- -
Mujeres: ausencia de telarquia progresiva a partir de los 13 años y/o ausencia de menarquia tras cuatro años de telarquia progresiva y/o ausencia de menarquia a los 16 años (amenorrea primaria).
- -
Varones: volumen testicular inferior a 4 mL (medido mediante orquidómetro de Prader) a los 14 años o detención del desarrollo puberal tras haberse alcanzado un volumen ≥ 4mL en los cinco años siguientes;
- -
Respuesta prepuberal en la prueba de estimulación con hormona liberadora de hormona luteinizante (test LHRH): pico de LH<5 UI/L. Valores de inhibina B por debajo del percentil 5;
- -
Ausencia de afectación de otros ejes hormonales;
- -
Tras descartar retraso constitucional del crecimiento y pubertad, teniendo en cuenta la historia clínica y los antecedentes familiares junto con la constatación de adecuado desarrollo puberal durante el seguimiento en consultas8,9;
- -
Tras descartar hipogonadismo hipogonadotropo funcional asociado a patología crónica mediante información clínica y tras realizar pruebas de laboratorio que incluyen hemograma, reactantes de fase aguda y anticuerpos para la enfermedad celíaca entre otros.
- -
Patología endocraneal descartada mediante resonancia magnética craneal; y
- -
Consentimiento a participar del paciente/padre o tutor.
Se recogieron los siguientes datos demográficos y clínicos: edad, sexo, antecedentes familiares, antropometría, estadio de Tanner y manifestaciones clínicas asociadas. También se recogieron los resultados del University of Pennsylvania Smell Identification Test (UPSIT) (Sensonics Inc; Haddon Height, NJ, EE. UU.) para valoración de alteraciones del olfato. La prueba incluye 40 microcápsulas de olores estandarizados en un cuaderno de «rascar y oler», y el paciente tiene que identificar cada olor, escogiendo una respuesta entre cuatro opciones. La puntuación del UPSIT se interpreta según datos de normalidad para el sexo y la edad incluidos en el manual del UPSIT. Los resultados inferiores a 30 puntos son indicativos de hiposmia y los inferiores a 18 puntos revelan anosmia10,11.
Análisis genéticoLas muestras de ADN genómico se extrajeron de linfocitos de sangre periférica con un extractor de ADN automático (EZ1 DNA Blood kit-EZ1 Advanced XL, QIAGEN, Alemania) de acuerdo con las instrucciones del fabricante. La concentración y pureza del ADN se evaluaron mediante espectrofotometría (espectrofotómetro NanoDrop® ND-1000, Wilmington, DE, EE. UU.). La construcción de librerías, secuenciación y análisis de datos se llevaron a cabo mediante protocolos validados. La captura del exoma clínico se realizó con el kit Clinical Exome Solution®? (SOPHiA Genetics) y la secuenciación con el sistema NextSeq 500 (Illumina).
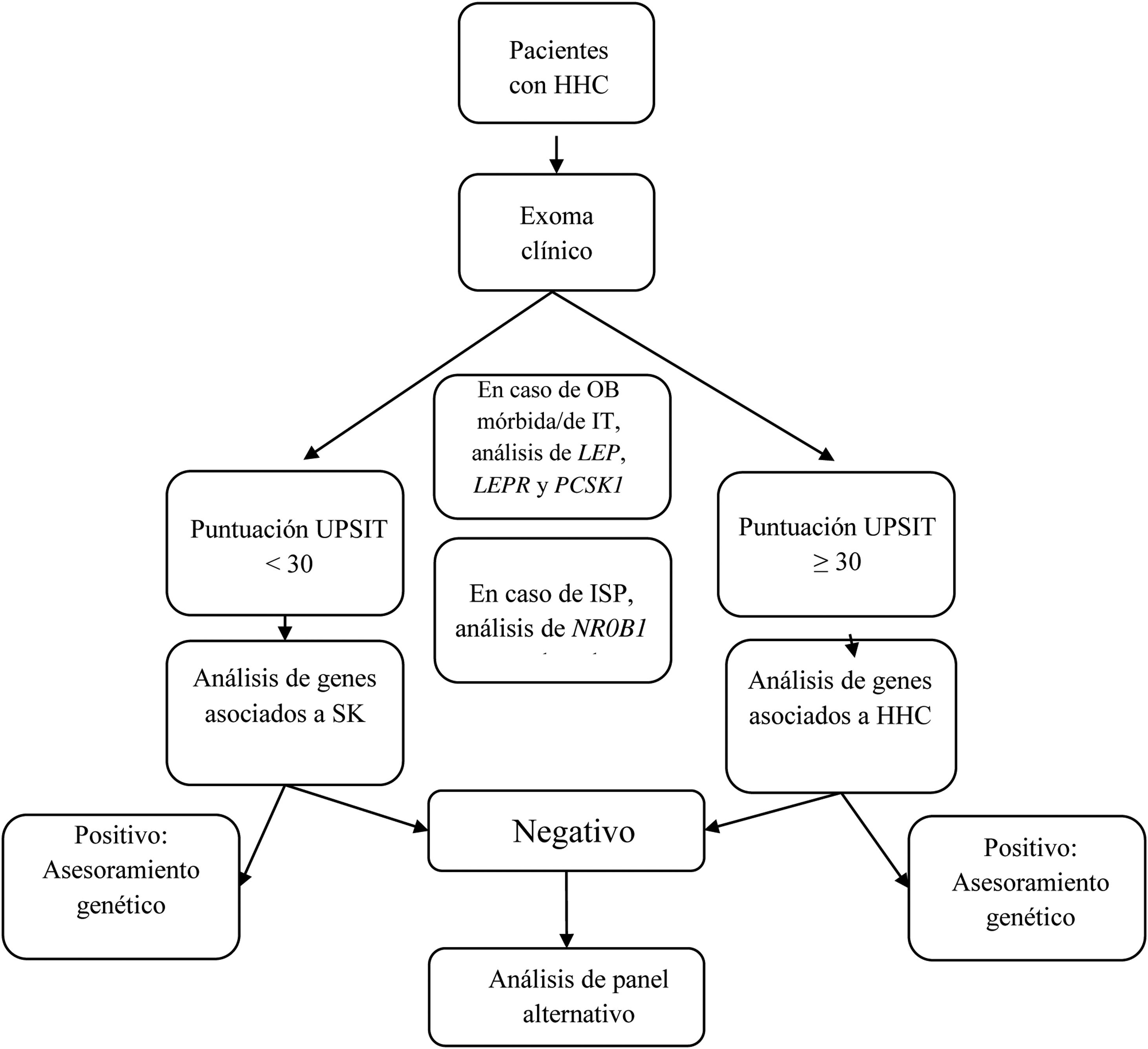
Se diseñó un panel virtual con 34 de los genes incluidos en el kit de captura, genes relacionados con el HHC con y sin hiposmia/anosmia asociada, según el documento de consenso europeo publicado en 2015 por Boehm et al.1 y otras revisiones2,5–8,12–16 (tabla 1). Aunque el objetivo principal era evaluar el rendimiento diagnóstico del estudio genético guiado por la presencia o ausencia de hiposmia/anosmia, el estudio solo incluyó algunos genes asociados a patología sindrómica en pacientes con fenotipo compatible. El análisis genético incluyó los siguientes pasos (fig. 1):
- 1.
Si el paciente presentaba obesidad mórbida o de inicio precoz, el análisis incluía los genes LEP, LEPR, and PCSK1;
- 2.
Si el paciente tenía insuficiencia suprarrenal primaria, se incluía el gen NR0B1(DAX1);
- 3.
En pacientes con fenotipo compatible con SK, se analizaron los siguientes genes: ANOS1, AXL, CHD7, FGF8, FGFR1, HESX1, NSMF, PNPLA6, POLR3B, PROK2, PROKR2, SEMA3A, SEMA3E, SEMA7A, SMCHD1, SOX9, SOX10, SPRY4, WDR11, RAB18, RAB3GAP1, and RAB3GAP2;
- 4.
En pacientes con HHC sin hiposmia/anosmia asociada, se analizaron los siguientes genes: GNRH1, GNRHR, KISS1, KISS1R, TAC3, TACR3, FSHB and LHB; y
- 5.
En pacientes con SK con resultado negativo, también se analizaron los genes incluidos en el paso 4, y en pacientes con HHC sin hiposmia/anosmia con resultado negativo, también se examinaron los genes incluidos en el paso 3.
Genes incluidos en el panel virtual del síndrome de Kallman y el HHC sin anosmia
| Genes asociados al síndrome de Kallmann y genes asociados a distintos cuadros sindrómicos | Genes asociados al HHC sin anosmia | |
|---|---|---|
| POLR3B (NM_018082.5) | SOX10 (NM_006941) | FSHB (NM_000510.2) |
| LEP (NM_00230.2) | ANOS1 KAL1 (NM_000216) | GNRH1(NM_000825) |
| NR0B1/DAX1 (NM_00074.4) | AXL (NM_001699) | GNRHR (NM_000406.2) |
| LEPR (NM_002303) | SEMA3E (NM_012431.3) | KISS1 (NM_002256) |
| PCSK1 (NM_000439) | FGF8 (NM_033163.3) | KISS1R (NM_032551) |
| SOX9 (NM_000346.3) | FGFR1 (NM_001174063) | LHB (NM_000894.2) |
| PNPLA6 (NM_006702) | SEMA7A (NM_003612.4) | TAC3 (NM_013251.3) |
| CHD7 (NM_017780) | HESX1 (NM_003865) | TACR3 (NM_001059) |
| RAB18 (NM_021252.4) | NSMF (NELF) (NM_001130969) | |
| RAB3GAP1 (NM_012233) | PROK2 (NM_001126128.1) | |
| RAB3GAP2 (NM_012414.3) | SPRY4 (NM_030964.3) | |
| SEMA3A (NM_006080) | WDR11(NM_018117.11) | |
| SMCHD1 (NM_015295.2) | PROKR2 (NM_144773) | |

Diagrama de flujo del análisis genético guiado por la presencia o ausencia de anosmia/hiposmia.
HHC: hipogonadismo hipogonadotropo congénito; ISP: insuficiencia suprarrenal primaria; IT: inicio temprano; OB: obesidad; SK: síndrome de Kallmann; UPSIT: University of Pennsylvania Smell Identification Test.
El análisis de variantes se llevó a cabo con la plataforma SOPHiA DDM (SOPHiA Genetics). La presencia de posibles variantes patogénicas se comprobó mediante secuenciación Sanger convencional (analizador 3130, Applied Bio-systems, Foster City, CA, EE. UU.). En casos con muestras disponibles, también se obtuvo el genotipo de los familiares mediante secuenciación Sanger. Las variantes se catalogaron de acuerdo con las recomendaciones del American College of Medical Genetics (ACMG)17.
ReclutamientoEl presente estudio fue revisado y aprobado por el Comité de Ética de la Investigación Hospital Universitario Fundación Jiménez Díaz (código EO 11-2015, fecha de aprobación, 26 de marzo) y se realizó siguiendo los principios de la Declaración de Helsinki. Los participantes firmaron el correspondiente consentimiento informado después de recibir información detallada sobre los procedimientos del estudio. Los participantes no recibieron compensación monetaria.
El resguardo y destino final de las muestras obtenidas fue el biobanco del hospital.
ResultadosEl estudio incluyó a nueve pacientes varones con estudio genético mediante exoma clínico. Cinco (56%) presentaban fenotipo de SK, tres (33%) HHC con olfato normal y uno (11%) sintomatología compatible con síndrome de CHARGE. Los datos clínicos y genéticos de estos pacientes se resumen en la tabla 2.
Descripción del genotipo y fenotipo de los pacientes estudiados
| Paciente | GenLocus | Variante, cigosidad y origen | Clasificación | Volumen testicular (ml) izdo/dcho. Edad a la evaluación(años) | LH (UI/L) pico (P) o basal (B)| Testosterona (ng/mL) |inhibina B (pg/mL) | Puntuación UPSIT | Talla (z) al diagnóstico Rasgos fenotípicos adicionales |
|---|---|---|---|---|---|---|---|
| 1 | SOX1022q13.1 | NM_006941: c.929dupT NP_008872:p.Ser311Glufs*91En heterocigosis, origen sin determinar | Probablemente patogénica | 1/212 | < 0,07(B)| < 0,4|NM | 8/40 | + 0,07.Sordera, micropene |
| 2 | SOX10 22q13.1 | NM_006941: c.473A >C NP_008872:p.Glu158AlaEn heterocigosis, origen sin determinar | Probablemente patogénica | 5/515.5 | < 0,07(B)| < 0,4|NM | 36/40 | -1.Ninguno |
| 3 | - | - | 2/ausente16 | 0,47(P)| < 0,4|NM | 15/40 | -2,12.Criptorquidia | |
| 4 | ANOS1Xp22.31 | NM_000216.4:c.932_941delTCACTATAGT NP_000207:p.Val311Leufs*25En hemicigosis, de novo | Patogénica | 2/215.7 | 0,8(P)|0,39|17 | 7/40 | -1,46.Criptorquidia bilateral, agenesia de bulbos olfatorios |
| 5 | SEMA3A7q21.11 | NM_006080: c.1303G >A NP_006071: p.Val435IleEn heterocigosis, origen sin determinar | VSI | 2/214 | 0,7(P)|0,29|10 | 32/40 | -1,06.Micropene, criptorquidia |
| 6 | - | - | - | 2/319 | 2,1(P)|0,2|12 | 36/40 | + 0,07.Ninguno |
| 7 | FGFR18p11.23 | NM_001174063: c.1196delT NP_001167534.1 p.Met399Argfs*37En heterocigosis, origen sin determinar | Probablemente patogénica | 2/218 | < 0,07(B)|1,77|NM | 14/40 | + 1,23.Rasgos faciales dismórficos, criptorquidia |
| 8 | NSMF9q34.3 | NM_001130969: c.419G >T NP_001124441: p.Arg140LeuEn heterocigosis, origen sin determinar | VSI | 3/322 | 0,4 (B)|0,1|NM | 26/40 | -1,89.Criptorquidia |
| CHD78q12.2 | NM_017780: c.4226T >A NP_060250: p.Val1409GluEn heterocigosis, origen sin determinar | VSI | |||||
| 9 | CHD78q12.2 | NM_017780.3:c.7160C >A NP_060250.2:p.Ser2387En heterocigosis, de novo | patogénica | 2/216 | 0,7 (B)|0,11|NM | ND | -2,7.Displasia del nervio óptico, coloboma del iris, insuficiencia mitral, criptorquidia, sordera, TEA |
LH: hormona luteinizante; ND: no disponible por falta de colaboración (paciente con TEA); NM: no se realizó medición; TEA: trastorno del espectro autista; VSI: variante de significado incierto.
Empleando el panel de 34 genes diseñado para el estudio, se detectaron variantes patogénicas o probablemente patogénicas que explicaban el fenotipo en cinco de los nueve pacientes (55%). En otros dos sujetos, el estudio detectó variables de significado incierto que no explicaban el fenotipo por sí solas, y en los dos restantes no se detectaron cambios genéticos. Todas las variantes relevantes se identificaron con el panel virtual de genes relacionados con el SK. Las variantes se catalogaron según los criterios recomendados por el ACMG17.
A continuación, se presenta información detallada sobre cada una de las variantes detectadas en los pacientes bajo estudio:
SOX10: El paciente 1 era un varón de 12 años que presentaba un volumen testicular inferior a 4 mL, micropene, niveles basales de gonadotropinas indetectables, hiposmia e hipoacusia neurosensorial. El estudio genético detectó una variante patogénica en el gen SOX10, perteneciente a la familia de los factores de transcripción SOX. Estos factores están involucrados en distintos procesos de desarrollo multiorgánicos. SOX10 codifica un factor de transcripción expresado por los precursores de las neuronas productoras de GnRH5. Las variantes de pérdida de función en heterocigosis se asocian a SK con penetrancia incompleta5, y los pacientes afectados también pueden hipoacusia neurosensorial5,7, hipopigmentación del iris y mechones de pelo blanco5. La variante detectada en el paciente 1, c.929dupT; p.Ser311Glufs*91, duplica la timina en la posición 930, cambiando la pauta de lectura con aparición de un codón de parada prematuro que da lugar a una proteína truncada o podría resultar en degradación del ARN mensajero mediada por variantes sin sentido (NMD)18. Esta variante no ha sido descrita previamente (Clinvar19, VarSome20 y GnomAD21). Desafortunadamente, no contamos con muestras de ADN de los progenitores. Aplicando las recomendaciones del ACMG para la interpretación de variantes17, esta fue catalogada como probablemente patogénica.
El paciente 2 era un varón de 17 años con pubertad retrasada, volumen testicular detenido en 5 mL, el mismo volumen medido en la evaluación inicial a los 15,5 años de edad, (tabla 2) y perfil hormonal compatible con hipogonadismo hipogonadotropo, sin evidencia de alteraciones del olfato en el test de UPSIT y antecedentes familiares inciertos de retraso puberal en el padre. El estudio genético detectó una variante de cambio de sentido en heterocigosis en el gen SOX10, c.473A>C. Se trata de una variante no descrita previamente catalogada como deletérea por distintos predictores in silico. La variante se encuentra en el dominio de unión al ADN conocido como HMG-box, en el que otras variantes de cambio de sentido se han catalogado como patogénicas; 38 de las 44 (86%) variantes de cambio de sentido en el gen SOX10 que no son de significado incierto (VSI) son patogénicas, superando el umbral de 51,0%, y 97 de las 152 variantes (63,8%) en el gen SOX10 reportadas en la práctica clínica son patogénicas, superando el umbral del 12,0%19. Esta variante fue catalogada como probablemente patogénica aplicando los criterios del ACMG17. En este caso, tampoco se contó con muestras de ADN de los progenitores.
ANOS1: En el paciente 4, varón de 15,7 años con sintomatología compatible con SK (tabla 2), el estudio detectó la variante c.932_941delTCACTATAGT en el gen ANOS1. Es una variante de cambio de pauta de lectura que ocasiona el cambio de una valina por una leucina en la posición 311 y de la secuencia de aminoácidos posterior, con aparición de un codón de parada prematuro que da lugar a una proteína truncada o podría resultar en NMD18. La variante no se ha descrito anteriormente (Clinvar,19 VarSome20 y GnomAD21). Como otras variantes de cambio de pauta de lectura se han catalogado como patogénicas, se concluyó que esta era patogénica con base en las recomendaciones del ACMG17. El estudio del ADN materno mostró que la variante era de novo. ANOS1 fue el primer gen identificado en relación con el SK22 y presenta un patrón de herencia ligada al cromosoma X.
FGFR1: En el paciente 7, varón de 18 años con volumen testicular inferior a 4 mL y nivel basal de LH indetectable con fenotipo de SK, se identificó una variante con cambio de pauta de lectura en el gen FGFR1, c.1196delT. La variante produce un codón de parada prematuro, dando lugar a una proteína truncada sin el dominio proteína-quinasa o a NMD18. No se ha descrito previamente (Clinvar19, GnomAD21 y VarSome20). El paciente presentaba criptorquidia, HHC y rasgos faciales dismórficos. Aplicando las recomendaciones del ACMG17, esta variable se catalogó como probablemente patogénica. El gen FGFR1 fue el primero identificado como causante de una forma autosómica dominante de SK19–21 por variantes con pérdida de función y penetrancia incompleta1,5,12. Hay que tener presente que las variantes patogénicas de FGFR1 pueden dar lugar al fenotipo SK o el fenotipo de HHC sin alteración del olfato5,7, y que están involucradas en el 10% del total de casos de SK y el 7% de los de HHC sin alteraciones del olfato6. Algunos de estos pacientes presentan patología con anomalías esqueléticas, ectrodactilia, paladar hendido y HHC grave. Cabe destacar que estas manifestaciones se asocian a variantes patogénicas bialélicas del gen FGFR15.
NSMF: El paciente 8 era un varón de 22 años con fenotipo de SK (tabla 2) y una variante de cambio de sentido, c.419G >T, que catalogamos como VSI17. En este mismo paciente también se detectó una variante de cambio de sentido en el gen CHD7, c.4226T >A, que no ha sido descrita previamente ni en bases de datos de población general ni como causante de enfermedad. La variante se encuentra en el dominio helicasa, y los predictores in silico la catalogaron como deletérea. No hubo acceso a muestras de ADN de los progenitores.
A día de hoy, la herencia oligogénica no se ha descrito en el gen CHD723, por lo que no podemos relacionar la presencia de esta variante y de aquella presente en NSMF en el paciente. Al no contar con muestra de los progenitores, no pudimos realizar la segregación de dicha variante.
CHD7: Además de la variante detectada en el paciente 8, en el paciente 9, varón de 16 años con presentación compatible con síndrome de CHARGE (tabla 2), se identificó la variante sin sentido c.7160C >A; p.Ser2387*. Esta resulta en la aparición de un codón prematuro y una proteína truncada o posiblemente NMD18. Ha sido descrita como patogénica por Janssen et al. en pacientes con síndrome de CHARGE24. El análisis de ADN de los progenitores reveló que la variante era de novo.
SEMA3A: En el paciente 5, varón de 14 años que presentó criptorquidia y micropene, se detectó la variante sin sentido c.1303G >A. La base de datos GnomAD reporta una frecuencia poblacional de esta variante de 0,014%21. Se concluyó que era una VSI porque este gen también se asocia a un patrón de herencia oligogénica y no se detectó ninguna otra variante en los genes previamente identificados asociados al HHC oligogénico5,7.
DiscusiónEl rendimiento diagnóstico de nuestro enfoque fue del 55%, en concordancia con la literatura previa1,5,7,12. Además, se identificó a dos pacientes con variantes patogénicas no descritas previamente asociadas a la forma oligogénica de HHC, lo que sugiere que la presencia de una variante patogénica en un gen no es suficiente para causar la enfermedad. Otra hipótesis razonable es que variantes comunes que se han catalogado como benignas en estos genes o en regiones reguladoras o intrónicas podrían ser el factor adicional necesario para causar HHC.
En el presente estudio, todas las variables patogénicas o probablemente patogénicas se identificaron con el panel virtual diseñado para el SK. Esto podría explicarse por una mayor frecuencia de cambios en los genes relacionados con el SK en comparación con los genes asociados al HHC sin alteraciones del olfato12, además del solapamiento fenotípico de los genes involucrados en ambas entidades,1,7 incluso en pacientes sin hiposmia/anosmia. Por lo tanto, en el contexto de la revolución que actualmente se está experimentando en el campo del análisis genómico, en el que muchos laboratorios utilizan el exoma clínico o el exoma completo filtrados por paneles virtuales, tiene sentido diseñar un panel virtual que ofrezca la mayor sensibilidad posible, aunque ciñéndose siempre al concepto de análisis del panel, a pesar de su naturaleza virtual, para minimizar la posibilidad de hallazgos inesperados.
En dos pacientes se detectaron VSI en genes asociados al patrón oligogénico, pero no se identificó la segunda variante requerida para este patrón de herencia, por lo que los resultados no fueron concluyentes. Los genes identificados hasta la fecha en relación con herencia oligogénica de HHC son los siguientes2,5–7,14–16,25–28:
- •
Vía de señalización de FGF8/FGFR1: FGF8, FGFR1, ANOS1, FGF17, KLB, IL17RD, HS6ST1, NSMF, SPRY4, FLRT3 y DUSP6;
- •
Migración neuronal desde la placoda olfatoria hasta el hipotálamo: PROR2, PROKR2, SEMA3A, SEMA3E y SEMA7A; y
- •
Expresión y función de la GnRH: WDR11, GNRHR, KISS1R, TAC3, y TACR3.
Nuestro estudio presenta varias limitaciones. En primer lugar, se utilizó un kit de captura del exoma clínico, el Clinical Exome Solution v. 2 de SOPHiA GENETICS, que incluye 4.493 genes pero no todos relacionados con el HHC. Los 15 genes relevantes que no están incluidos en el kit son CCDC141, DUSP6, FEZF1, FGF17, FLRT3, HS6ST1, IL17RD, KLB, IGSF10, DMXL2, OTUD4, POLR3A, RNF216, STUB1, y TBC1D20. Entre ellos, cabe destacar los genes IL17RD y FGF17, involucrados en el 3% de los casos de HHC15. Además, en la mayoría de los casos no se contó con muestras de ADN de los progenitores. Por último, el tamaño muestral fue pequeño.
En cualquier caso, el rendimiento diagnóstico en nuestro estudio fue similar al reportado por otros investigadores1,4,7,28,29. En nuestra muestra, todos los hallazgos genéticos relevantes se detectaron mediante el panel virtual diseñado para analizar genes asociados al SK. Por lo tanto, en pacientes con HHC sin alteraciones del olfato, el análisis del panel virtual alternativo fue crucial en el diagnóstico. Consideramos que esto tiene que ver con las variaciones en la expresión fenotípica de algunos genes relacionados con el HHC en lo que respecta al olfato2,5,7. Esto nos hizo reflexionar sobre la utilidad del exoma filtrado por dos paneles virtuales distintos con base en la presencia o ausencia de alteraciones del olfato. Es posible que tenga más sentido diseñar un único panel virtual que incluya todos los genes relacionados con el KS y el HHC sin alteraciones del olfato.
A nuestro parecer, el exoma completo filtrado con un panel virtual de genes relacionados con el HHC es una prueba muy sensible y ofrece la posibilidad de hacer una reevaluación en cualquier momento. No obstante, el uso de un panel virtual conlleva las siguientes desventajas: a) desde el punto de vista ético, hay que hablar con los pacientes y/o sus representantes legales sobre la posibilidad de hallazgos inesperados durante la consulta previa a la prueba; y b) la disponibilidad de este tipo de prueba aún es reducida. Por otro lado, los paneles prediseñados no incluyen algunos de los genes identificados más recientemente y no permiten reanalizar datos, aunque están disponibles en la mayoría de los laboratorios.
En conclusión, el estudio genético de sujetos con HHC bien caracterizado con fenotipo de SK o HHC sin alteración del olfato es útil y debería realizarse en todos los pacientes con esta enfermedad. No obstante, la sensibilidad de la prueba variará considerablemente dependiendo de si se utiliza secuenciación de paneles, exoma clínico o exoma completo.
FinanciaciónEstudio apoyado por la Cátedra de Patrocinio de Medicina Genómica de la Universidad Autónoma de Madrid-Fundación Jiménez Díaz (file 081800).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos profundamente a Oliver Shaw por su ayuda en la redacción y organización de la versión en inglés del manuscrito. También damos gracias a los niños que participaron en el estudio, así como a sus familias.
Presentación previa: presentación parcial como comunicación oral bajo el título «Aproximación al diagnóstico molecular del hipogonadismo hipogonadotropo aislado con y sin alteración del olfato», XLI Congreso de la Sociedad Española de Endocrinología Pediátrica; 24-26 de mayo, 2019.

