

Congenital adrenal hyperplasia due to 21-hydroxylase deficiency is an autosomal recessive disorder caused by mutations in the CYP21A2 gene. Cortisol and aldosterone synthesis are impaired in the classic forms (adrenal insufficiency and salt-wasting crisis). Females affected are virilised at birth, and are at risk for genital ambiguity. In this article, we give recommendations for an early as possible diagnosis and an appropriate and individualised treatment. A patient and family genetic study is essential for the diagnosis of the patient, and allows genetic counselling, as well as a prenatal diagnosis and treatment for future pregnancy.
La hiperplasia suprarrenal congénita debida al déficit de 21-hidroxilasa es una enfermedad autosómica recesiva causada por mutaciones en el gen CYP21A2. En las formas clásicas se produce defecto de cortisol y aldosterona (insuficiencia suprarrenal y pérdida salina) y virilizacion de la recién nacida afecta con ambigüedad genital. En este artículo ofrecemos algunas recomendaciones para el diagnóstico, que debe ser lo más precoz posible, y el tratamiento, adecuado e individualizado. El estudio genético del paciente y su familia es clave en el diagnóstico del propio afectado, y también permite establecer el consejo genético, así como el diagnóstico y tratamiento prenatales en futuros embarazos.
Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency is an autosomal recessive disorder caused by impaired corticosteroid synthesis. Between 90% and 95% of affected individuals have 21-hidroxylase deficiency (21OHD, OMIM #201910) with a variable blockade of the synthesis of glucocorticoids and mineralocorticoids and excessive androgen production, leading to a diversity of clinical syndromes that may manifest as early as the neonatal period (classic “salt-wasting” and “simple virilising” forms) or in childhood, adolescence or adulthood (some “simple virilising” and “nonclassic” forms).1,2
The management of classic forms of CAH is complex, individualised and multidisciplinary, and requires the development of a structured treatment and follow-up plan. Thus, these patients should be managed in referral comprehensive care centres (CCCs).
Several scientific societies (European, American, Australian and Japanese paediatric endocrinology societies) and patient associations have published guidelines for the management of this disease.
In this article, the Working Group on Congenital Adrenal Hyperplasia of the Sociedad Española de Endocrinología Pediátrica (Spanish Society of Paediatric Endocrinology) offers general recommendations that may be useful to paediatricians that care for patients with 21OHD based on published guidelines and the group members’ experience in the management of classic forms of disease.
EpidemiologyThe incidence of CAH varies based on the population under study and the method of data collection (whether data were or not collected from early intervention programmes): it is of 1/10000 to 1/20000 for classic forms in newborns,2 while in Spain it ranges between 1/10000 and 1/14000,3 which suggests that 1/50 to 1/60 individuals are carriers of serious mutations.
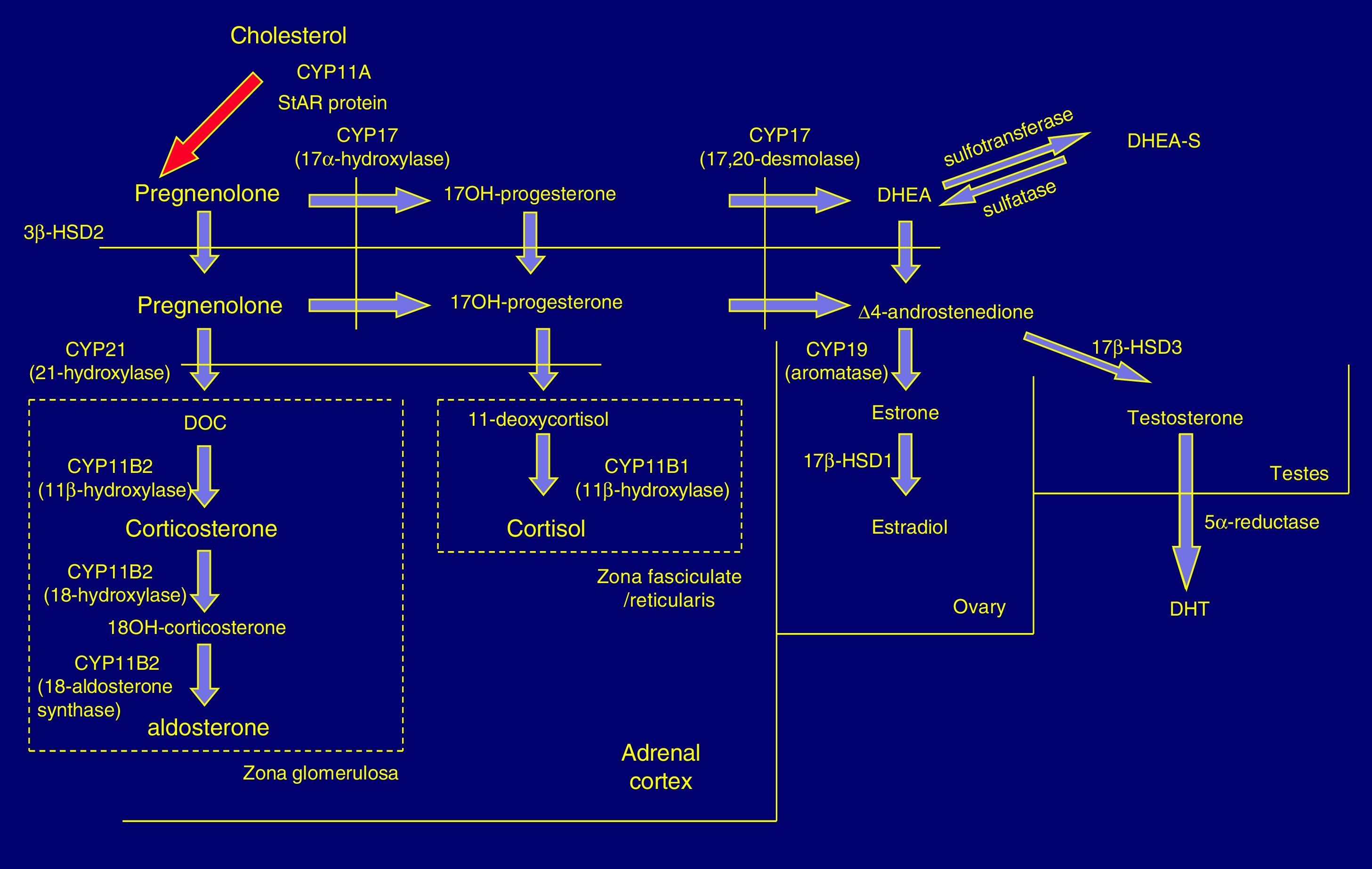
Pathophysiology and forms of diseaseIn the adrenal cortex, hydroxylation of the 21C residue is carried out by the steroid enzyme 21-hydroxylase (also known as cytochrome P450c21), which converts progesterone to deoxycorticosterone and 17-hydroxyprogesterone (17OHP) to 11-deoxycortisol in a process that culminates in the synthesis of aldosterone and cortisol, respectively. Clinical manifestations depend on the severity of the functional impairment of the enzyme (Fig. 1).

In 21OHD, there is a decrease in the synthesis of hormones downstream of the block (glucocorticoids and mineralocorticoids), an increased production of upstream products, such as 17OHP, and increased synthesis in the androgen pathway. Cortisol deficiency leads to a compensatory rise in corticotropin (ACTH) and adrenal hyperplasia.4
In severe forms there are defects in other metabolic pathways: decreased production of adrenalin in the adrenal medulla (tendency to circulatory collapse)5 and activation of dihydrotestosterone (DHT) synthesis by “backdoor” metabolic pathways that result in increased virilisation in female foetuses.6
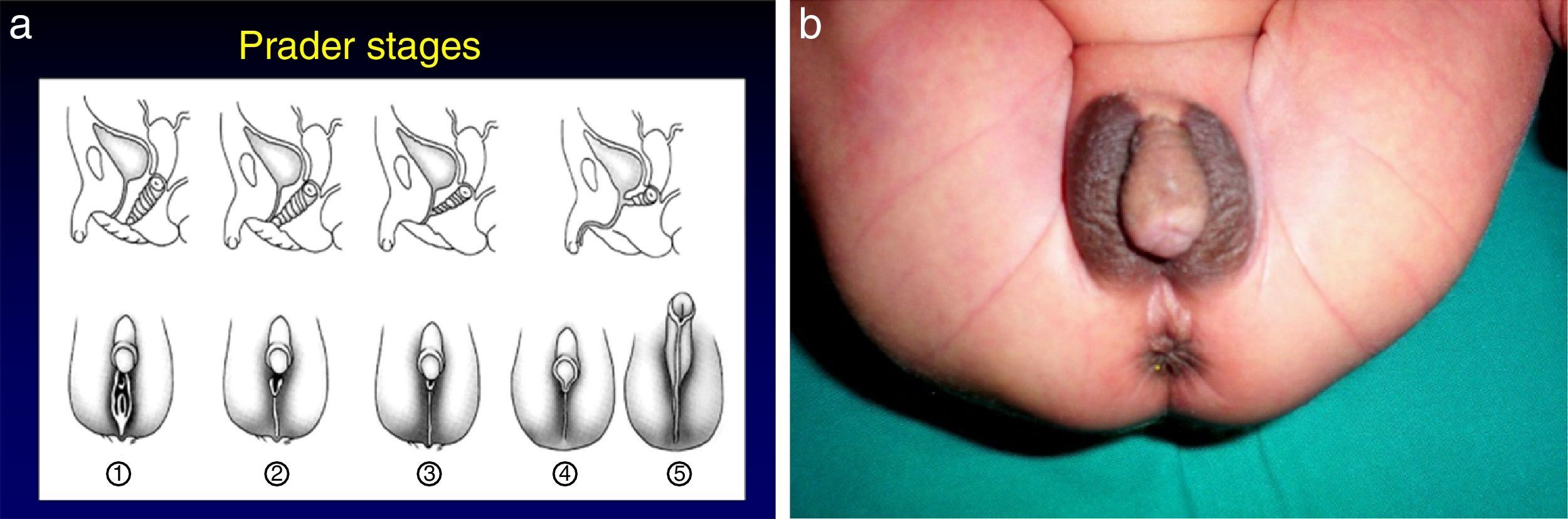
The most severe forms manifest in the neonatal period due to cortisol and aldosterone deficiency with a potentially lethal loss of salt.1 Excess androgens cause virilisation and external genital ambiguity in newborn girls. The Prader scale is very useful to describe the degree of virilisation (Fig. 2). Without adequate treatment, the disease could lead to accelerated bone maturation, premature epiphyseal closure, precocious pseudopuberty and short stature in adulthood.
 Prader stages. Prader 1: female external genitalia; Prader 5: male external genitalia. (b) Female newborn with 21OHD. It would be easy make a male sex assignment in an initial assessment (Prader 5).")
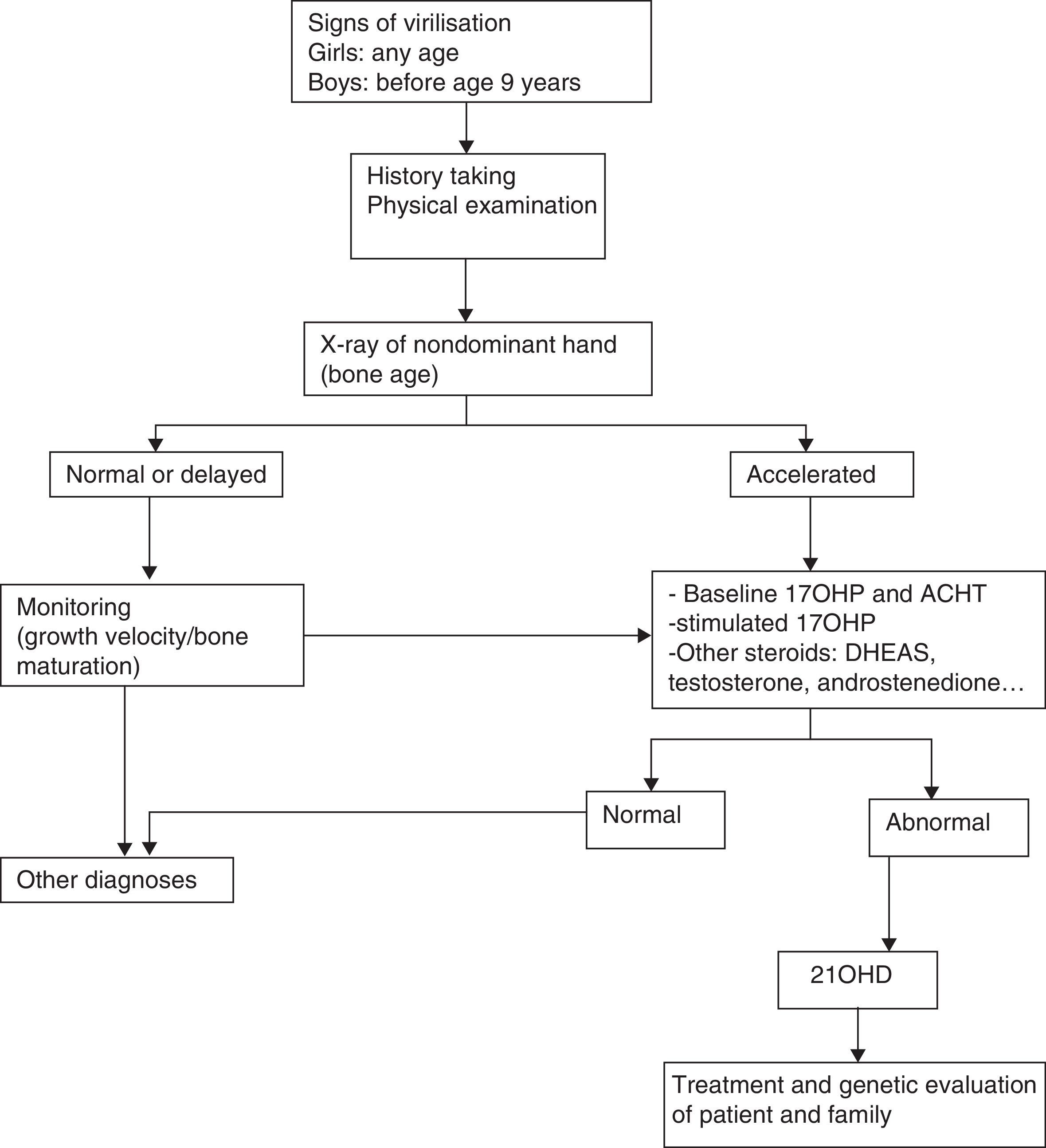
Less-severe forms of disease have onset in late childhood, adolescence or adulthood and are characterised by signs of virilisation: premature pubarche, accelerated growth and bone maturation, hirsutism, severe acne and oligomenorrhea, with no evidence of aldosterone deficiency. They may go undetected, and diagnosis is delayed in a large proportion of cases.
In many cases, 21OHD causes illness of intermediate severity.
Diagnosis of 21-hydroxylase deficiencyThis disease will be suspected in:
- •
Child with symptoms of salt wasting in the early weeks of life.
- •
Girls virilised at birth or with postnatal virilisation, premature pubarche or early puberty.
- •
Boys with virilisation starting in childhood.
The diagnosis is based on testing of the steroid upstream of the block: 17OHP.
Diagnosis in newbornsEarly detection programme: neonatal screeningThe early detection of 21OHD, which is recommended internationally,2 is only performed in a few autonomous communities in Spain. It consists in the measurement of 17OHP levels in capillary blood samples taken on the second day of life for the early-detection screening of various diseases. The purpose of this test is to anticipate salt-wasting crises, prevent incorrect sex assignment and diagnosing simple virilising forms early.7
Each reference laboratory must establish normal ranges for their specific population based on gestational age, birth weight and sex. Although at present 17OHP levels are measured by fluorescence assay (DELFIA), the use of mass spectrometry will alleviate overestimation of 17OHP levels due to cross-reactivity with other adrenal steroids. Genotyping of CYP21A2 is useful as a second-tier test.8
Prenatal administration of glucocorticoids can reduce the levels of 17OHP and give rise to false negatives. Administration of spironolactone can lead to false positives.
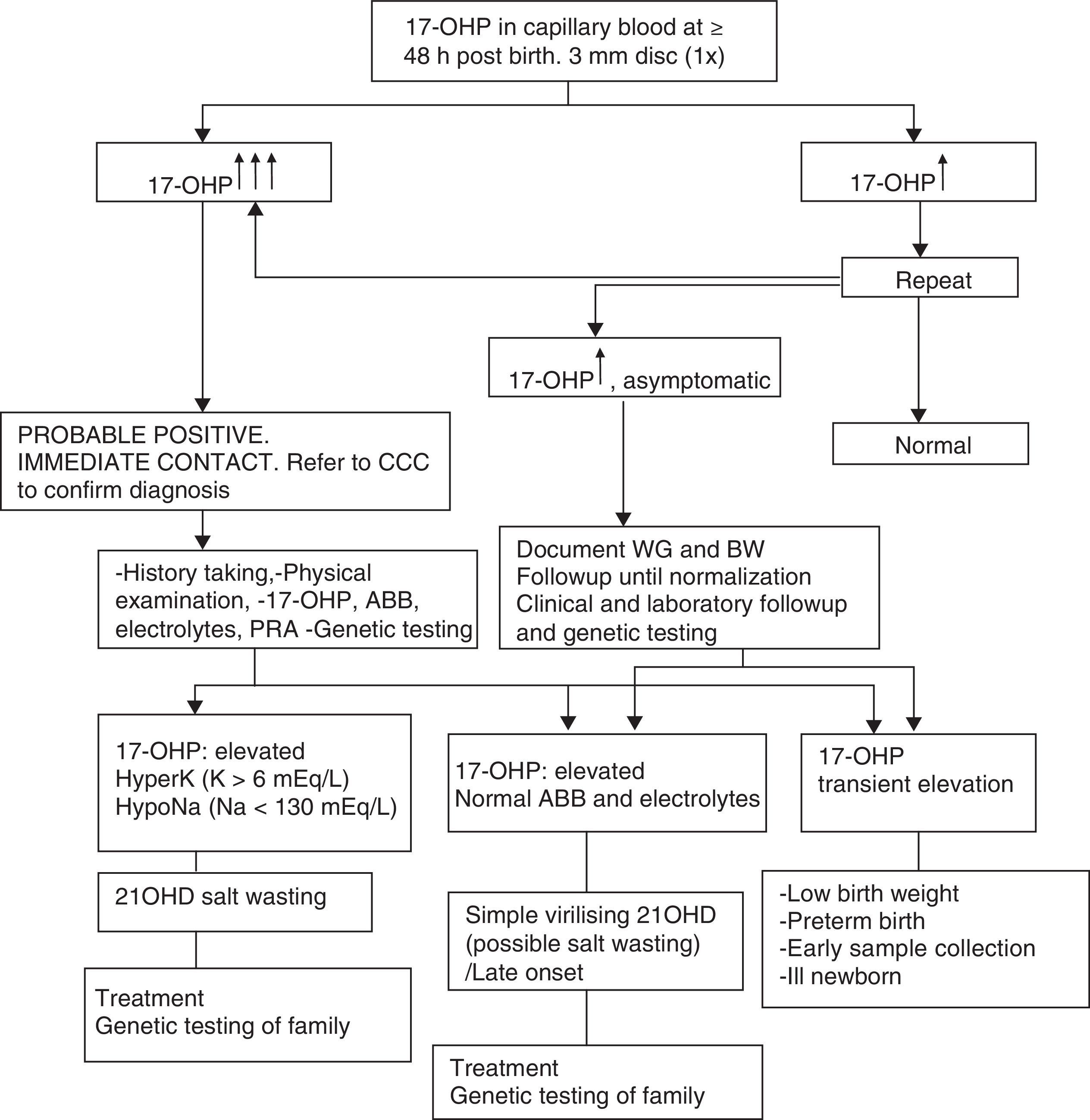
Table 1 presents the diagnostic algorithm for patients with a positive result.
Diagnostic algorithm: newborn with positive detection in neonatal screening (DELFIA) (born to term and with adequate weight for gestational age).
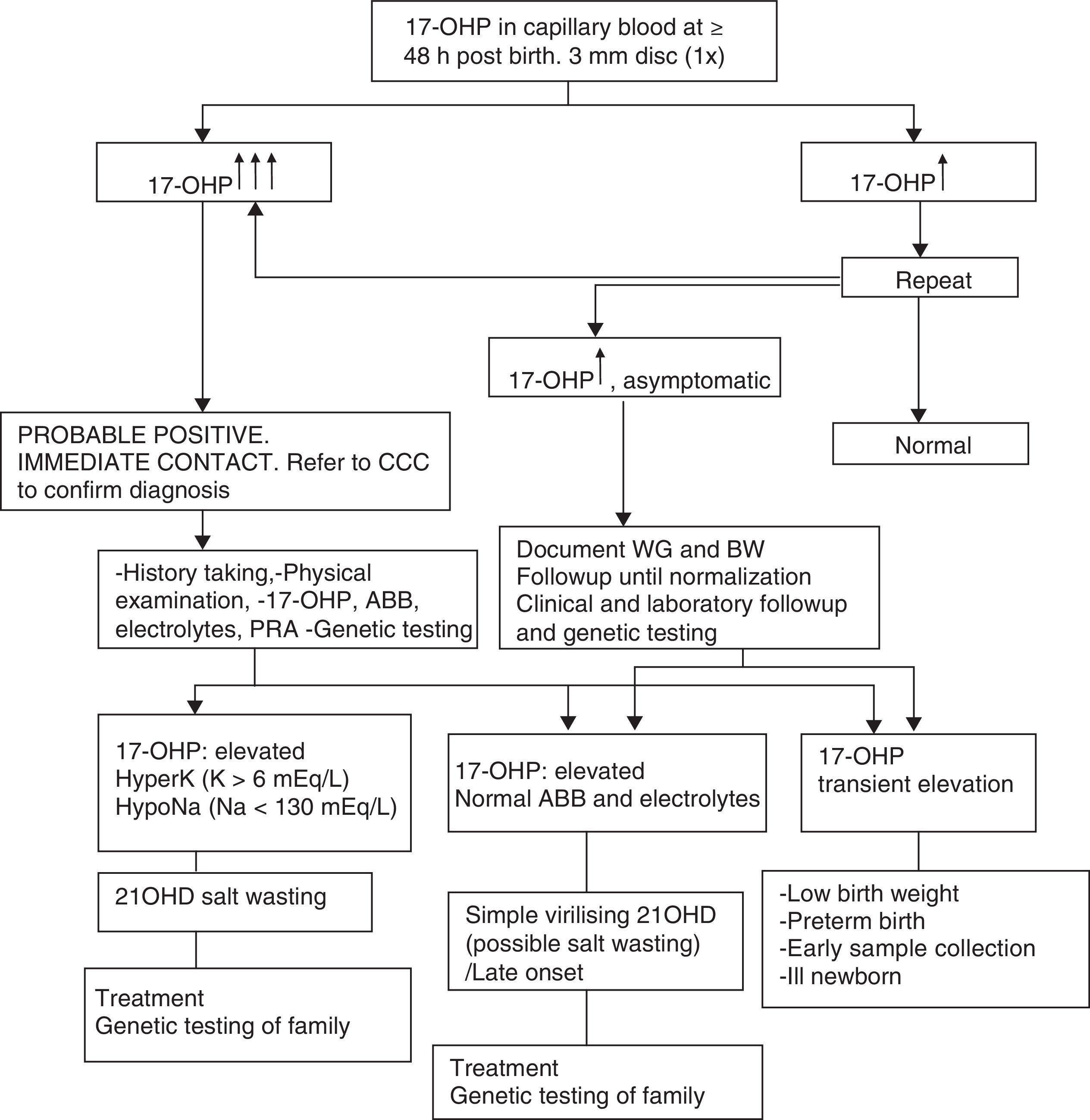
17-OHP, 17-hydroxyprogesterone; 21OHD, 21-hydroxylase deficiency; ABB, acid–base balance; BW, birth weight; CCC, comprehensive care centre; HyperK: Hyperkalaemia; HypoNa, Hyponatraemia; PRA, plasma renin activity; WG, weeks of gestation.
Table 2 presents the approach to the diagnosis of a newborn with suspected 21OHD due to elevation of 17OHP, excluding transient elevations (preterm birth, low weight, severe neonatal illness) that usually normalise within one year.8
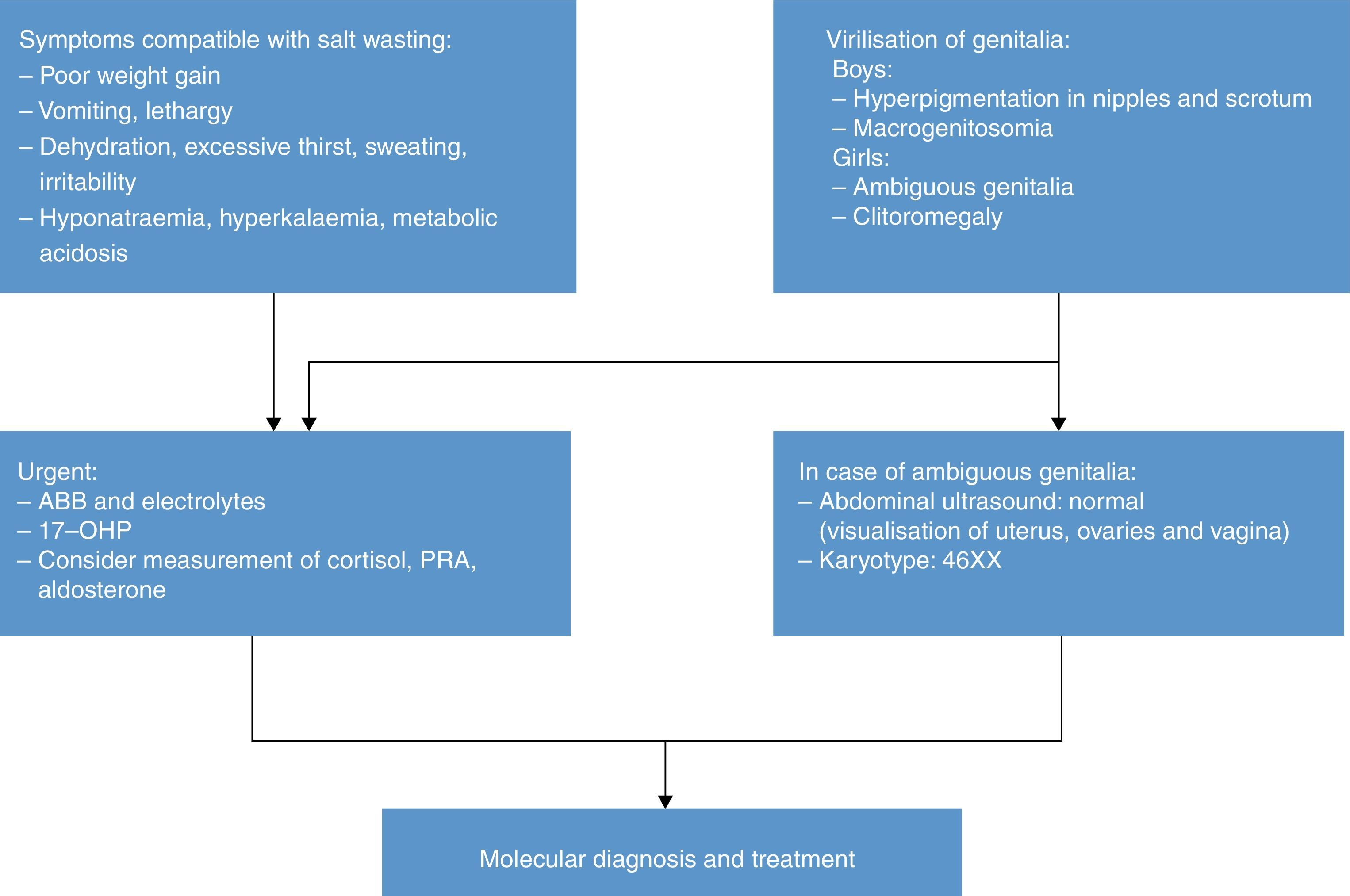
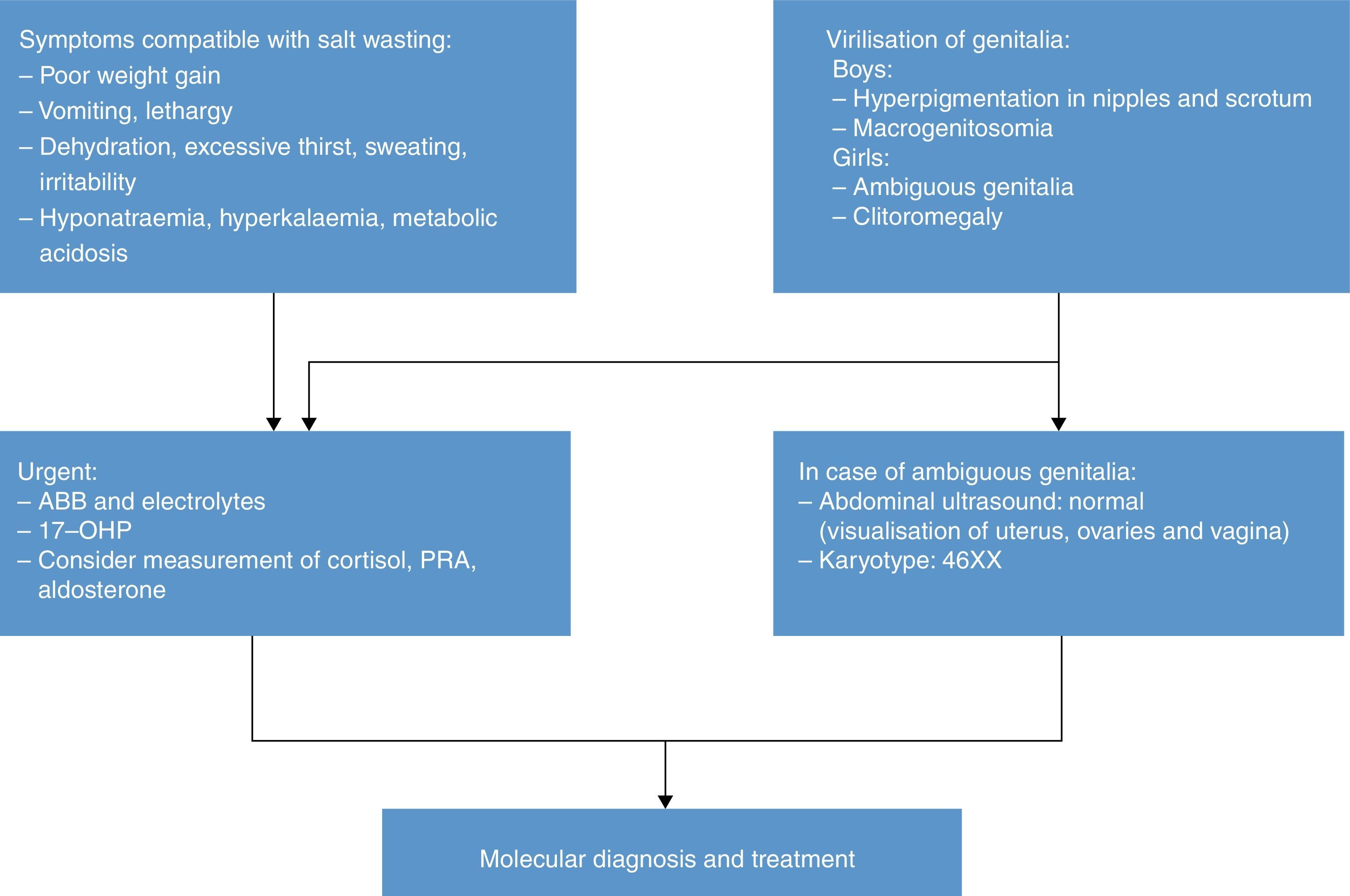
Salt wasting (weight loss, anorexia/excessive thirst, vomiting, lethargy, coma) results in hyponatraemia, hyperkalaemia, increased risk of hypoglycaemia, elevated levels of 17OHP, androstenedione, testosterone, DHEA and ACTH, decreased levels of cortisol and aldosterone, and increased plasma renin activity (PRA).
In the neonatal period, direct immunoassays (the technique used most frequently in Spain) may suffer from cross-reactivity and falsely detect elevations of 17OHP in healthy children or aldosterone levels within “normal” ranges in newborns with salt wasting. Performance of an ACTH stimulation test is not necessary due to the high levels of 17OHP.
Genotyping of the CYP21A2 gene is very useful in confirming the diagnosis.9,10
The karyotype of a female patient with 21OHD and ambiguous genitalia is 46 XX. Ultrasound examination of the abdomen and genitalia can be useful in diagnosis, allowing the visualisation of ovaries, fallopian tubes and uterus.
Diagnosis of older children and adolescentsThe diagnosis of male patients with simple virilising forms may be delayed due to the lack of manifestations in the neonatal period. Some girls may exhibit clitoromegaly and signs of masculinisation during childhood. Table 3 summarises the protocol for the diagnosis of a child or adolescent with suspected 21OHD.
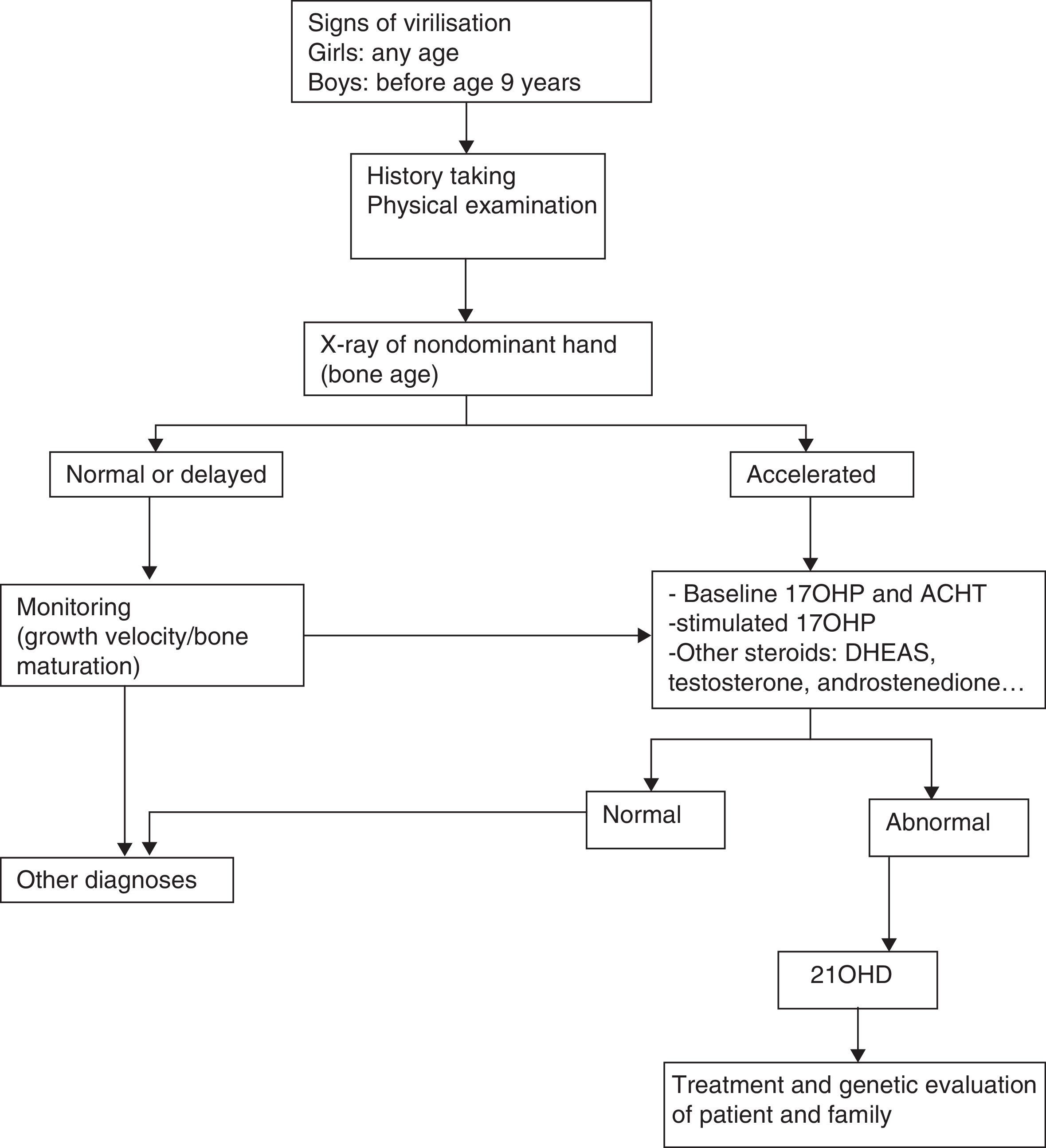
CYP21A2 genotyping is useful to differentiate these patients, as they have severe mutations in both alleles.
Molecular genetic diagnosisThe gene at the root of 21OHD is known as CYP21A2. It is located in the in the HLA class III region of the short arm of chromosome 6p21.3, adjacent to the CYP21A1P pseudogene. Unlike the CYP21A2 gene, CYP21A1P contains a series of mutations that prevent its translation into a functional protein, although both genes are homologous. All symptomatic forms of disease are associated with changes in the CYP21A2 gene. When a patient receives a diagnosis of 21OHD, it is advisable to conduct a family study in order to identify individuals that are carriers or have oligosymptomatic or silent nonclassic forms of disease. Unlike what happens in other diseases with a recessive pattern of inheritance in which the same mutation is frequently found in both alleles, patients with 21OHD frequently are compound heterozygotes with different mutations in the maternal and paternal alleles, or hemizygotes with a point mutation in one allele and a deletion in the other. Homozygous genotypes are only found in patients with highly prevalent mutations or with a history of consanguinity. Carriers and heterozygotes have a mutated gene in only one of the chromosomes and will not develop clinical manifestations, although they may exhibit an increased 17OHP response in the ACTH stimulation test.11,12
Ninety-five percent of the pathological variants of the CYP21A2 gene are recurrent and result from two different mechanisms involving the CYP21A2 and the CYP21A1 pseudogene, namely unequal crossing over and gene conversion. Five percent of the total alleles correspond to mutations of the CYP21A2 gene that cannot be attributed to gene microconversion. De novo mutations amount to 1% of cases (this calls for carrier testing of parents, as it cannot be assumed that either are necessarily carriers).
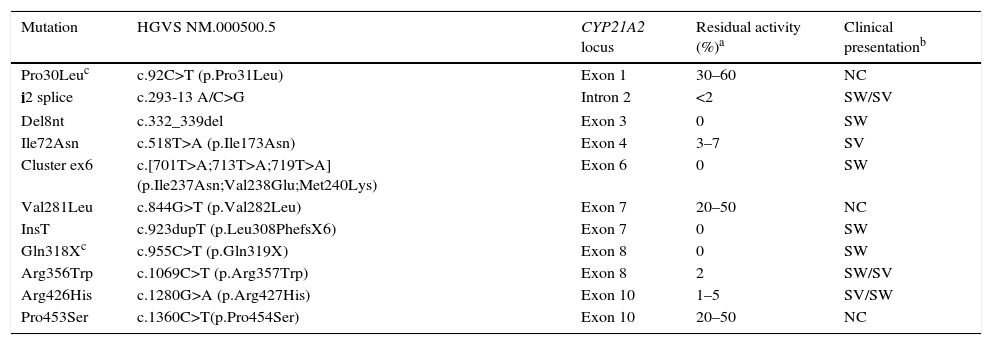
There is a strong genotype/phenotype correlation, as the severity of symptoms depends on the extent of enzymatic disruption, which in turn is determined by the genetic changes found in each allele (Table 4). The genotype-phenotype correlation can only be analysed accurately in patients that undergo whole-exome sequencing; the phenotype correlates to the mildest allele, and severe mutations can be found in patients with severe or with mild phenotypes.12
Residual activity of the most common point mutations in the 21-hydroxylase gene, and genotype-phenotype correlation.
| Mutation | HGVS NM.000500.5 | CYP21A2 locus | Residual activity (%)a | Clinical presentationb |
|---|---|---|---|---|
| Pro30Leuc | c.92C>T (p.Pro31Leu) | Exon 1 | 30–60 | NC |
| i2 splice | c.293-13 A/C>G | Intron 2 | <2 | SW/SV |
| Del8nt | c.332_339del | Exon 3 | 0 | SW |
| Ile72Asn | c.518T>A (p.Ile173Asn) | Exon 4 | 3–7 | SV |
| Cluster ex6 | c.[701T>A;713T>A;719T>A] (p.Ile237Asn;Val238Glu;Met240Lys) | Exon 6 | 0 | SW |
| Val281Leu | c.844G>T (p.Val282Leu) | Exon 7 | 20–50 | NC |
| InsT | c.923dupT (p.Leu308PhefsX6) | Exon 7 | 0 | SW |
| Gln318Xc | c.955C>T (p.Gln319X) | Exon 8 | 0 | SW |
| Arg356Trp | c.1069C>T (p.Arg357Trp) | Exon 8 | 2 | SW/SV |
| Arg426His | c.1280G>A (p.Arg427His) | Exon 10 | 1–5 | SV/SW |
| Pro453Ser | c.1360C>T(p.Pro454Ser) | Exon 10 | 20–50 | NC |
Del, deletion; Ins, insertion; NC, nonclassic; SV, simple virilising; SW, salt-wasting.
Clinical presentation (PS, SV, NC) when the mutation is homozygous, heterozygous or compound heterozygous with another severe mutation (residual activity<7%).
An additional investigation should be performed to assess for a 5′ gene conversion of Pro30 to Leu in association with a 5′ conversion in SV forms, or of SV, or the gene dosage of Gln318X in a gene-duplicated allele that is not associated with deficiency and is not uncommon in the general population.12
There is an alternative nomenclature for CYP21A2 gene mutations that may be featured in some databases, which is particularly confusing for clinicians. The reason for this is that the Human Genome Variation Society (HGVS) chose a sequence for the CYP21A2 gene (NM00500.5) that did not correspond to the sequence that had been used until that moment (M12792.1) and which had been applied to all previous knowledge, publications and databases on this disease. The two sequences differ in a triplet at the beginning of exon 1 in a polymorphic region that includes either 5 or 4 CTG repeats. Thus, amino acids are numbered differently from this position (for example, P.Pro31Leu versus p.Pro30Leu, p.Val282Leu versus p.Val281Leu). Subsequently, the use of the former widespread convention has been promoted (ENST00000448314, which corresponds to M12792.1), although both nomenclatures coexist at the moment.
Table 4 shows the frequency distribution of mutations in patients with classic forms and cases of suspected CAH after neonatal screening in our population. Patients with severe forms of disease have severe mutations in both alleles, while patients with nonclassic and even silent forms have either mild mutations in both alleles, or a mild mutation in one allele and a severe mutation in the other.13
Genetic testing with a single panel including the most common deletions/conversions and point mutations (Table 4) is very informative and can detect 95% of severe alleles. While this may seem like a simple task, this is a complex locus that should be analysed in laboratories with experience in CYP21A2 genotyping.
Genetic counselling should be offered to families of patients with classic forms of CAH, especially due to the need to identify individuals with mild forms of disease that carry severe mutations, the high prevalence of carriers of severe mutations in the general population, and the frequent lack of apparent symptoms in males with virilising forms of disease.
TreatmentThe goals of treatment are replacing the physiological secretion of glucocorticoids and mineralocorticoids to prevent salt wasting, controlling manifestations associated with hyperandrogenism and improving adult outcomes. Surgical reconstruction of the external genitalia is recommended in girls.14
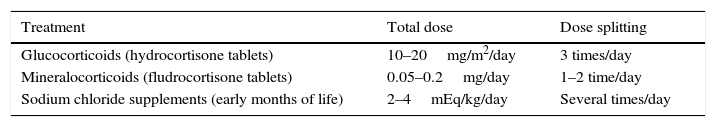
Treatment with glucocorticoidsHydrocortisone is the first-line treatment due to its potency, similar to that of endogenous cortisol, and short half-life. Prednisolone and dexamethasone should not be used in children. Hydrocortisone is administered in tablet form, to be crushed for young children, and not as suspension, since the distribution of the drug in liquid media is uneven and unstable. Administration must be split into at least 3 doses for a total daily dose that is difficult to determine. In newborns with salt wasting, doses are high and administered intravenously (50mg/m2/day) and tapered off gradually to a dose of approximately 20mg/m2/day by age 1 year; children receive 10–15mg/m2/day, and adolescents no more than 15–20mg/m2/day.3,15 The dose is always calculated on a case-to-case basis, as requirements vary based on multiple factors, some known (phenotypic variability, genotype, severity of enzymatic disruption, absorption, metabolism and pharmacokinetics of hydrocortisone) and some unknown.1,14
The goal is to treat patients with the minimum effective dose so they can remain asymptomatic, have a normal growth, weight gain and pubertal development, and achieve an adequate suppression of adrenal androgens. Signs suggestive of undertreatment include accelerated growth, advanced bone age and virilisation. Signs suggestive of overtreatment include suppressed growth, delayed bone age, obesity or high blood pressure. Androstenedione and testosterone are the best markers of adequate glucocorticoid therapy in prepubertal patients. Changes in treatment must be made taking into account the specific clinical manifestations of the patient, and not solely based on laboratory parameters.
In the future, methods for the administration of hydrocortisone that are currently being developed (delayed and extended release, continuous infusion) will better approximate physiological cortisol secretion.16
Treatment with mineralocorticoidsWe use oral 9α-fluorohydrocortisone. Infants need larger doses in the first months of life (0.1–0.2mg/day), while the maintenance dose in older infants and children can be of 0.05–0.1mg/day. Before the introduction of solids, infants will be given oral sodium chloride supplements (2–4mEq/kg/day) split into several doses (NaCl 20%, 1cm3=3.4mEq of sodium) (Table 5).
Patients with simple virilising CAH do not have salt-wasting crises, but may experience increases in PRA (chronic sodium depletion). Maintenance of sodium levels within the normal range reduces the levels of vasopressin and ACTH and allows the use of lower doses of hydrocortisone. In these patients, addition of 9α-fluorohydrocortisone at low doses facilitates normalisation of salt levels.17 The goals of treatment are for the patient to remain asymptomatic and to keep blood pressure, heart rate, electrolyte levels and PRA within normal ranges.
Stress treatmentPatients with severe forms of CAH do not produce enough cortisol in stressful situations, so their maintenance dose of hydrocortisone should be increased by a factor of 2–10 in these situations due to the risk of adrenal crisis (Table 6). Mental stress, mild diseases and physical activity do not require dose increases.
Treatment in stressful situations.
| Supportive care | Physiologic saline 10–20cm3/kg in case of hypovolaemia or shock |
| Treatment of dehydration | |
| Treatment of electrolyte imbalances (hyponatraemia, hyperkalaemia, metabolic acidosis, hypoglycaemia) | |
| Aetiologic treatment | Mild stress: double the dose of oral hydrocortisone for 3–4 days |
| Severe stress: parenteral hydrocortisone | |
| Initial dose based on chronological age: | |
| - Up to 6 months: 10–20mg | |
| - <2 years: 25mg | |
| - 2–12 years: 50mg | |
| - >12 years: 100mg | |
| Successive doses: 100mg/m2/day (or 3–4 times the maintenance dose) every 6h. Decrease to usual dose as soon as clinical situation permits. | |
These children must be vaccinated according to the official schedule.
Surgical treatmentUnlike other diseases that lead to changes in sexual differentiation, there is widespread consensus that girls with this condition should undergo surgery for reconstruction of female genitals. These patients have a female 46 XX karyotype and normal ovaries, fallopian tubes and uterus, and their pubertal development and fertility can be normal with appropriate treatment. The paediatric surgeon will determine the sequence of surgical interventions to be performed in facilities with demonstrated experience, at an early stage and before age 24 months so the girl can develop an appropriate body schema. In the absence of complications, surgery is not recommended between ages 2 years and adolescence. Surgical outcomes should be re-evaluated in adolescence and adulthood, managing potential complications (urethrovaginal fistula, vaginal stenosis) and assessing whether the morphology of the genitalia is consistent with normal female anatomy. The patient should undergo as few genital examinations as possible, and only for the purpose of improving the care plan. If performed, it should be in the presence of the fewest possible people and/or under sedation so as to not traumatise the girl.18,19
Short statureOverproduction of adrenal androgens and high-dose glucocorticoid therapy are the factors that lead to short stature in adulthood. Patients may also experience peripheral precocious puberty, which requires treatment with gonadotropin-releasing hormone analogues.
Aromatase inhibitors (anastrazole) and growth hormone therapy should only be used in patients with a very short predicted final stature or in clinical trials.15
Adrenal tumoursRises in ACTH can cause hyperplasia of sensitive tissues such as the adrenal glands and regions with ectopic adrenal rest tissue (testes and ovaries). The gold standard for evaluation is periodic ultrasound examination. Adrenal tumours require intensification of glucocorticoid therapy and, in some cases, surgery.20
FertilityFemale patients with CAH may have reduced fertility. Androgen excess causes anovulatory cycles and alterations in the hypothalamic–pituitary–ovarian axis. They may experience painful intercourse due to stenosis of the vaginal introitus or anatomical abnormalities resulting from virilisation or surgery.
During pregnancy, glucocorticoid and mineralocorticoid therapy should continue at the previous doses, and mothers should be given parenteral hydrocortisone during labour, as it constitutes a highly stressful situation for the foetus.
Genetic counselling should be offered before conception.
Male patients with classic forms may also experience reduced fertility due to the associated hypogonadism and the presence of ectopic adrenal rest tissue in the testes. Inadequate treatment allows an increased production of androgens of adrenal origin that suppress FSH, leading to decreased sperm production.2
Metabolic changesChronic steroid therapy is a risk factor for osteoporosis that calls for prophylaxis with physical activity and adequate calcium and vitamin D intake.2
Obesity, hypertension, insulin resistance and carbohydrate intolerance are side effects that need to be prevented and treated.
Prenatal diagnosis and treatmentPrenatal diagnosis should be considered in foetuses with a 1 in 4 probability of having severe disease (two parents carriers of severe mutations, one parent with disease, or history of CAH diagnosis in the family). Families with a known case of disease should undergo a full evaluation before planning a pregnancy.
The goal of prenatal treatment is to prevent the virilisation of genitalia in affected female foetuses, for whom there is a 1/8 probability in each pregnancy. It consists in the administration of dexamethasone to the mother in the early weeks of gestation (before week 7–8). Dexamethasone can cross the placenta and thus curb excess androgen production by the foetal adrenal gland, preventing the virilisation of female foetuses. When diagnostic test results become available, treatment is discontinued if the foetus is male or an unaffected female. The method that can be performed earliest to obtain an exclusively foetal sample for testing, usually around 12–14 weeks’ gestation, is chorionic villus sampling.
This treatment has been used since the 1990s and is effective in preventing virilisation of girls with 21OHD, the need for surgical interventions and the impact that the birth of a girl with virilised genitals has on the family.21,22
However, this approach entails the unnecessary exposure to dexamethasone of 7 out of 8 foetuses (all male foetuses and 3 unaffected female foetuses out of every 4). There is evidence that dexamethasone has teratogenic effects in mice, and its long-term effects are not well understood. It may also cause side effects in pregnant mothers.23
Currently, we recommend dexamethasone treatment as long as it is performed in facilities with an experienced staff and after obtaining informed consent.24
Analysis of foetal DNA in maternal blood can determine the sex of the foetus from 6 weeks of gestation, although its sensitivity does not reach 100% until week 10. It cannot identify pregnancies that require treatment so it cannot prevent treatment in female foetuses that are unaffected or carriers. Massive DNA sequencing of free foetal DNA will probably allow very early diagnosis (week 6) in foetuses with 21OHD, although this technique has yet to be introduced in the clinical setting. Due to the mixed presence of maternal and foetal DNA, testing requires the analysis of polymorphic markers, as the complexity of the CYP21A2 locus precludes a direct analysis.25
Preimplantation genetic diagnosis is not yet widely available. It must be done in experienced facilities, as it is performed on a single cell and requires in vitro amplification due to the particular characteristics of the locus of interest.
Referral comprehensive care centresPatients with severe forms should be managed in CCCs with proven experience in CAH to avoid the fragmentation, delays and inadequacies in care that may otherwise occur despite the efforts of paediatric endocrinologists. Care must be based in tertiary-level hospitals with sufficient human and technical resources.26
Transition to adulthoodAdolescents are likely to skip checkups and fail to adhere to treatment. Adult gonadal function depends on adequate adherence to treatment; ovarian dysfunction in women and testicular masses in men worsen when adherence is poor. In female patients, it is important to address issues related to genital anatomy and past surgical history. The plan for transitioning to adult care must be developed collaboratively and with the agreement of the patient, the paediatric endocrinologist and the endocrinologist.27
Conflicts of interestThe authors have no conflicts of interest to declare.
M. Alonso (Hospital Ramón y Cajal, Madrid, Spain).
S. Berrade (Complejo Hospitalario de Navarra, Pamplona, Spain).
M. Bonet (Hospital del Mar, Barcelona, Spain).
M. Gallego (Hospital Universitario 12 de Octubre, Madrid, Spain).
M. Gussinyé (Hospital Vall d’Hebron, Barcelona, Spain).
A. Gutiérrez (Hospital del Mar, Barcelona, Spain).
F. Hermoso (Hospital Clínico Universitario de Valladolid, Spain).
J.L. Lechuga (Hospital Universitario Puerta del Mar, Cádiz, Spain).
C. Luzuriaga (Hospital Marqués de Valdecilla, Santander, Spain).
A. Oliver (Hospital Universitario La Paz, Madrid, Spain).
S. Quintero (Hospital Materno Insular, Las Palmas, Spain).
B. Roldan (Hospital Ramón y Cajal, Madrid, Spain).
Appendix A lists the members of the Working Group on Congenital Adrenal Hyperplasia of the Sociedad Española de Endocrinología Pediátrica.
Please cite this article as: Rodríguez A, Ezquieta B, Labarta JI, Clemente M, Espino R, Rodriguez A, et al. Recomendaciones para el diagnóstico y tratamiento de pacientes con formas clásicas de hiperplasia suprarrenal congénita por déficit de 21-hidroxilasa. An Pediatr (Barc). 2017;87:116.e1–116.e10.
- Documento de manejo clínico del paciente pediátrico con infección por SARS-CoV-2.
(Actualizado el 26 de Noviembre de 2020) - Test de diagnóstico rápido en las consultas de Pediatría de Atención Primaria y Urgencias Pediátricas en la era COVID-19: más que una recomendación
(Actualizado el 21 de septiembre de 2020) - Consenso nacional sobre diagnóstico, estabilización y tratamiento del Síndrome Inflamatorio Multisistémico Pediátrico vinculado a SARS-CoV-2 (SIM-PedS).
(Actualizado el 27 de Julio de 2020) - Recomendaciones para el manejo del recién nacido en relación con la infección por SARS-CoV-2.
(Actualizado el 27 de Mayo de 2020) - Manejo del paciente pediátrico ante sospecha de infección por el nuevo coronavirus SARS-CoV-2 en atención primaria (COVID-19). AEPap-SEIP/AEP-SEPEAP.
(Actualizado el 27 de Abril de 2020) - Propuesta de adaptación de las recomendaciones de reanimación cardiopulmonar pediátrica avanzada a la infección por coronavirus.
(Actualizado el 11 de Abril de 2020)


Anales de Pediatría sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas