


The term brachydactyly encompasses a group of bone dysplasias involving the phalanges and/or metacarpal/metatarsal bones of the hands and feet. There are 5 types (A–E) and several subtypes (A1–A4; E1–E3). It has an autosomal dominant pattern of inheritance with variable penetrance. Cases with recessive inheritance are extremely rare.
Brachydactyly type C (BDC) is characterised by a shortening of the middle phalanges of the second, third and fifth finger and the first metacarpal, and may also present with ulnar deviation of the index finger, polydactyly, or a distinctive hypersegmentation of the proximal or middle phalanges of the second and third fingers. The fourth digit is least affected and usually longest.1–3 “Angel-shaped” phalanges (Fig. 1A), while characteristic of BDC, are not pathognomonic, as they may also occur in the disorder known as angel-shaped phalango-epiphyseal dysplasia. It is possible that this disorder and BDC are part of the same clinical spectrum.1 In BDC, this feature normalises when physeal closure of the hand bones is completed, ending as simple brachydactyly.1 Other anomalies associated with BDC are short stature with delayed bone age, Madelung deformity, hip dysplasia, talipes valgus or equinovarus or absence of middle phalanges in the toes.2,3
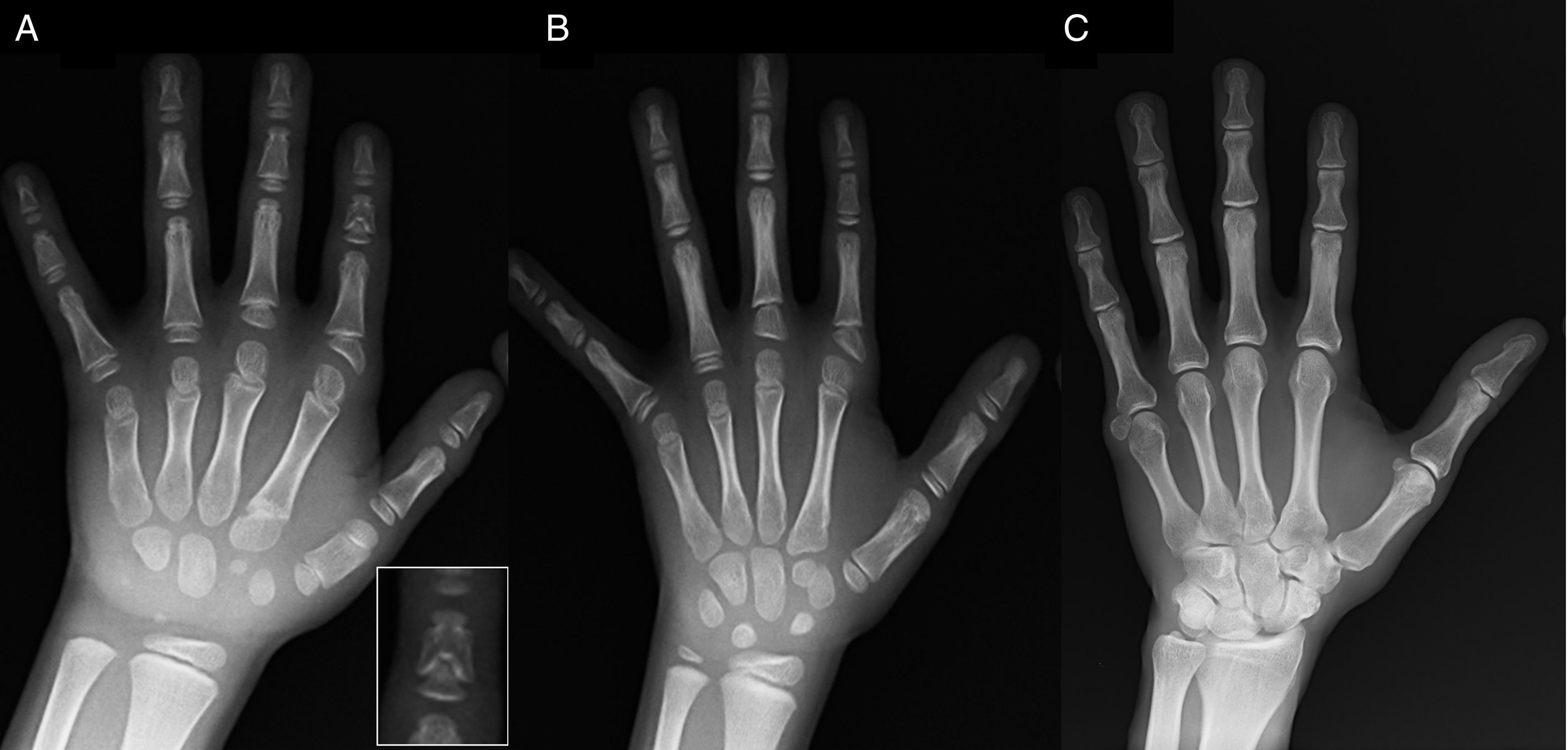
(A) Radiograph of the left hand of the proband (chronological age, 7 years; bone age, 5 years), with shortening of an anomalous first metacarpal (double proximal and distal epiphyseal plates) and the middle phalanges of the second, third and fifth fingers. The second finger exhibits ulnar deviation, and the fourth finger is the least affected and longest in the left hand. The proximal epiphyses of the second and third finger are dysplastic, with a conspicuous angel-shaped middle phalanx in the second finger (inset in A). (B). Radiograph of the left hand of the proband's sister (aged 5½ years with no delay in bone age), with shortening of the middle phalanges of the second, third and fifth fingers and a normal fourth finger. In this case, the first metacarpal was normal. The most salient feature is the triangular shape of the proximal epiphysis of the second finger's proximal phalanx, similar to the brother's, and the trapezoidal shape of the phalange in the middle finder. Like her brother, her second finger exhibits ulnar deviation. (C) Radiograph of the left hand of the father of the proband, which reveals only a bony remnant of a post-axial hexadactyly that was surgically corrected in childhood.
We present the case of a boy aged 7 years with radiological features compatible with BDC referred for evaluation of short stature (z-score, −1.8). His father had unilateral post-axial polydactyly, which was also present bilaterally in a paternal uncle. Radiographic examination of the left hand and wrist revealed a bone age that was 2 years younger than chronological age and anomalies that prompted performance of a skeletal survey. The survey detected anomalies in the hands (Fig. 1A) and subtle abnormalities in the feet (mild epiphyseal dysplasia in the proximal phalanges of some toes). The radiological evaluation of a sister aged 6 years revealed lesions in the hand similar to those of the proband, although less pronounced (Fig. 1B). A radiograph of the father's hand (Fig. 1C) only revealed a bone remnant of the post-axial hexadactyly that had been surgically corrected in childhood.
Sequencing of the GDF5 gene in the proband revealed a heterozygous point substitution in exon 2 (c.1462A>T) resulting in a premature stop codon (p.Lys488*, nonsense mutation) and a truncated protein 14 amino acids shorter than the wild protein (Fig. 2A). This mutation was also found in the father and sister of the patient, but not in the healthy mother (Fig. 2B). Fig. 2C shows the family's pedigree. This is a novel variant that is probably pathogenic and with an autosomal dominant pattern of inheritance.
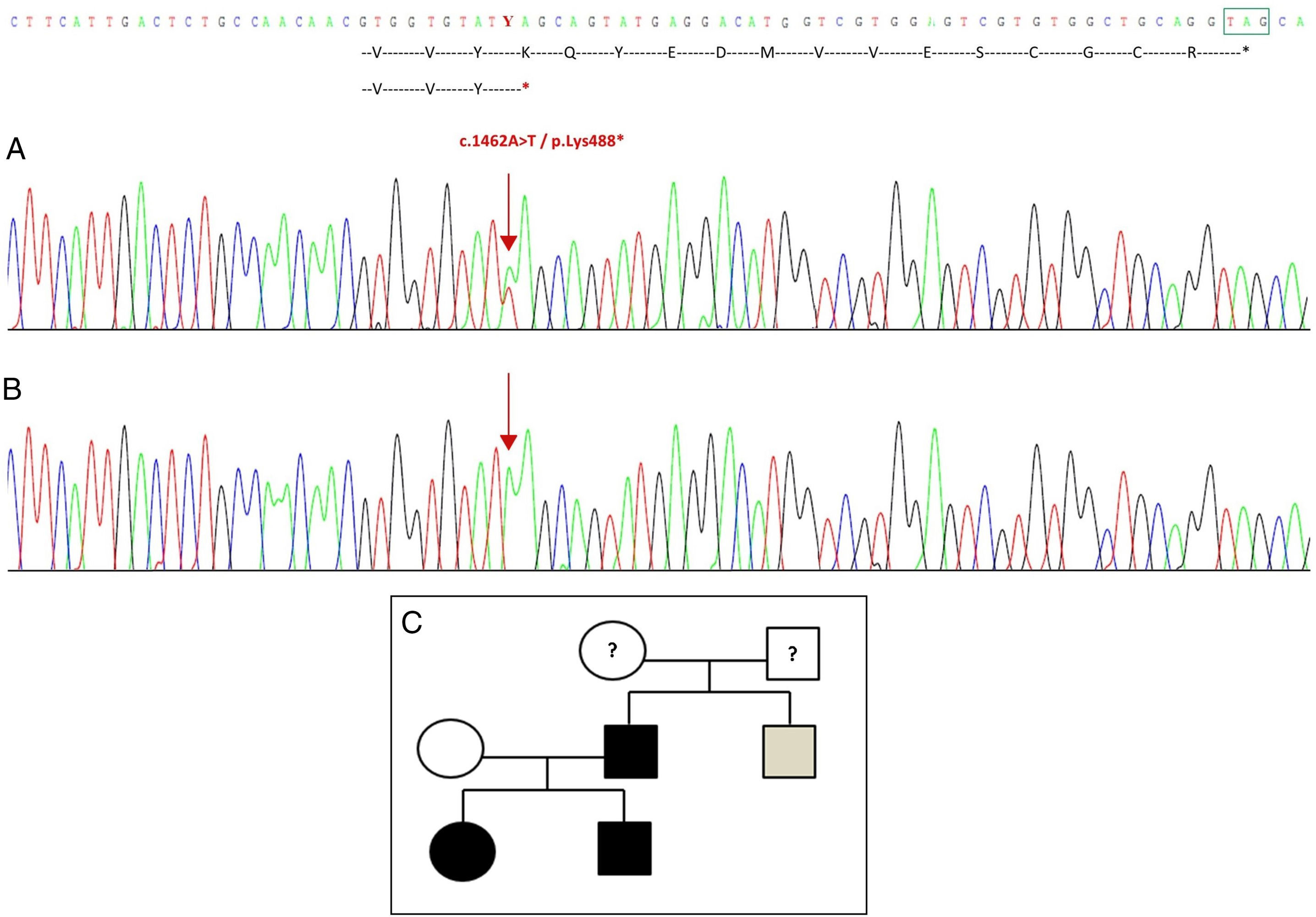
Novel mutation detected in the GDF5 gene. (A) Gene sequence of exon 2 in the proband, showing a c.1462A>T mutation that results in a premature stop codon and a truncated protein (p.Lys488*) (we have indicated the normal position of the stop codon with a green square). (B) Normal sequence of the same region in the healthy mother. (C) Pedigree of the family: the father and both children, in black, have a confirmed mutation in GDF5. The healthy mother appears in white. The grey square represents a paternal uncle with bilateral post-axial polydactyly who probably has the mutation.
Growth differentiation factor 5 (GDF5) is closely associated with bone morphogenetic proteins and belongs to the transforming growth factor β superfamily, with is involved in embryonic skeletal and joint development.4 The GDF5 gene is a mutational hotspot for disorders associated with skeletal malformations.5 Most homozygous or compound heterozygous mutations are associated with severe diseases: Grebe type chondrodysplasia (OMIM 200700), Hunter–Thomson type acromesomelic dysplasia (OMIM 201250) or Du Pan syndrome (OMIM 228900). On the other hand, heterozygous mutations associated to milder skeletal dysplasias: proximal symphalangism 1 B (OMIM 615298) and multiple synostosis syndrome type 2 (OMIM 610017), both associated with missense mutations with gain of function, and brachydactyly type A1 and A2, also associated to missense mutations, but with loss of function.5 Brachydactyly type C is associated with heterozygous mutations with loss of function, although 3 cases with recessive inheritance have also been reported.6 Most mutations associated with BDC are frameshift mutations in the prodomain part of the gene, while most mutations in the mature domain are missense mutations, with a highly variable phenotypic expression.4
The family that we present here has a nonsense mutation in the region that codes for the active domain of the protein, resulting in the elimination of its last 14 amino acids. This is the second nonsense mutation affecting the active mature domain described in the literature.3 The first one is a similar mutation in the amino acid immediately preceding the one mutated in the family that we describe here (p.Tyr487*/c.1461T>G), which suggests that both give rise to mutant monomers and functional haploinsufficiency of GDF5, thus causing BDC.
FundingThis study was partially funded by projects PI13/00467 and PI13/01295, integrated in the 2013–2016 R&D&I plan of the Spanish Government and co-funded by the Deputy General-Directorate of Research Evaluation and Promotion of the Instituto de Salud Carlos III (ISCIII), the European Regional Development Fund (ERDF) and the Centro de Investigación Biomédica en Red sobre Fisiopatología de la Obesidad y la Nutrición (Biomedical Research Networking Centre on the Physiopathology of Obesity and Nutrition [CIBERobn]), ISCIII, Madrid.
Please cite this article as: Travieso-Suárez L, Pereda A, Pozo-Román J, Pérez de Nanclares G, Argente J. Braquidactilia tipo C debida a mutación de parada en el gen GDF5. An Pediatr (Barc). 2018;88:107–109.
