



Advances in next-generation sequencing (NGS) technologies have made the detection of the molecular causes of paediatric diseases increasingly affordable, accessible and rapid. While exome sequencing and genome sequencing were until recently only available for research, they are now used in health care practice. The clinical application of NGS has raised many challenges in genetic counselling for families in terms of the interpretation of test results and incidental findings, as well as technical limitations in the event of inconclusive results. Given the impact of genetic results in clinical decision-making, specialized knowledge is required of the techniques and methods used in genetic studies, their advantages and limitations, and their potential psychosocial, legal and ethical impact on patients, relatives and health care professionals. The ethical implications of parents giving consent to genetic testing in their offspring and the potential disclosure of genetic diseases for which there are limited therapeutic options are still under debate. In this review, we provide an overview of all these aspects, including the advantages and limitations of current NGS techniques, and discuss the possibilities of upcoming solutions.
Los avances en las tecnologías de secuenciación de nueva generación (NGS) han hecho que la detección de las causas moleculares de las enfermedades pediátricas sea cada vez más asequible, accesible y rápida. Si bien hasta hace no mucho la secuenciación del exoma y la secuenciación del genoma sólo estaban disponibles para la investigación, ahora se utilizan en la práctica asistencial. La aplicación clínica de la NGS ha planteado muchos retos a la hora de asesorar a las familias sobre la interpretación de los resultados diagnósticos y los hallazgos incidentales, así como las limitaciones técnicas ante resultados genéticos no concluyentes. Dadas las implicaciones de los resultados genéticos en la toma de decisiones médicas se requiere de un conocimiento especializado de los procedimientos técnicos de los estudios genéticos, sus ventajas y limitaciones, así como del posible impacto (psico)social, legal y ético de los mismos en pacientes, familiares y profesionales sanitarios. Las implicaciones éticas que derivan del hecho de que sean los progenitores quienes otorguen el consentimiento para el estudio de su descendiente y la posible revelación de enfermedades genéticas con opciones terapéuticas limitadas siguen siendo objeto de reflexión. En esta revisión planteamos una pincelada sobre todos estos aspectos, incluyendo las ventajas y limitaciones de las técnicas actuales de NGS, y mostramos las posibilidades de las soluciones que están por venir.
Approximately 400 million people worldwide directly suffer from a rare disease, in 70% of cases with onset in childhood. At present, the median time from disease onset to diagnosis is 4.8 years.1 Reducing this delay is important in order to improve the quality of life of these patients and, in some cases, may provide a window for therapeutic intervention.
Advances in DNA sequencing technologies and the associated decrease in cost have led to the use of whole exome sequencing (WES) and/or whole genome sequencing (WGS) in the clinical setting. Although the application of these technologies has greatly facilitated the identification of genetic variants associated with diseases (which in turn has involved the development of consensus recommendations and standards for the classification of genetic changes),2 the diagnostic rate for rare diseases overall continues to be lower than 50%.3 How can it be that, with all this technology at our disposal, we are unable to give an answer to so many of the families that come to our clinics? The aim of this article is to describe the molecular diagnosis process in paediatrics, from the initial visit of the patient until the evaluation is complete, with or without a conclusive result, highlighting what we consider to be the main limitations and problems that need to be addressed and briefly discussing possible solutions.
Basic foundationsWe often compare the human genome to an encyclopedia of 23 or 46 volumes in which individual letters in the text would correspond to the four nucleotides of DNA, the phrases or words to genes, and the volumes to chromosomes.
Thus, when we explain possible abnormalities that may cause a disease, we can talk to patients about letter changes that alter a word to describe point variants in the DNA sequence, which would, in turn, result in an incorrect instruction; or about the loss of book chapters, one copy of the book or a series of books (trilogy, pentalogy or “polylogy”) when we discuss exon, gene or chromosome deletions, respectively.
But, as professionals, it is important to be aware that changes in genetic material constitute a continuum ranging from changes at the chromosomal level to changes in a single nucleotide. And that the most efficient techniques for diagnosis may vary depending on the expected type of abnormality (Fig. 1).4
 or based on the affected cell (somatic or germline variants). Also, based on their effects and according to the scheme proposed by the American College of Medical Genetics (ACMG), they can be classified as benign, likely benign, of uncertain significance, likely pathogenic and pathogenic.2")
Classification of genetic variants. Variants can be classified based on their size (chromosome or gene variants) or based on the affected cell (somatic or germline variants). Also, based on their effects and according to the scheme proposed by the American College of Medical Genetics (ACMG), they can be classified as benign, likely benign, of uncertain significance, likely pathogenic and pathogenic.2
In Spain, Law 14/2007 on Biomedical Research stipulates that any person undergoing genetic testing must be provided adequate genetic counseling by qualified professionals before and after testing.
Genetic counseling is a communication process in which a specialized professional provides complex medical information to the patient and/or family members in clear and simple terms regarding the genetic disorder, its transmission, the risk of recurrence and the options open to them5 (Fig. 2). This process requires establishing rapport to be able to find out the motivation and expectations of the patients (or parents) in seeking consultation while also establishing what they already know. This atmosphere of trust allows the counselor to clarify the purpose, possible misconceptions and goals of genetic counselling and of potential genetic testing.
 form has been signed, the pertinent genetic tests are performed. The results must be interpreted by qualified staff, and may include: (likely) pathogenic variants, (likely) benign variants and variants of uncertain significance (VUS). In addition, depending on what was specified in the signed informed consent, incidental findings may also be reported.")
Stages of genetic counseling in paediatrics. Genetic counseling is a patient- and family-centered process. After the clinical diagnosis, the patient is referred to genetic counseling, where the counselor explains the characteristics of the disorder, associated risks, etc. Then, after the informed consent (IC) form has been signed, the pertinent genetic tests are performed. The results must be interpreted by qualified staff, and may include: (likely) pathogenic variants, (likely) benign variants and variants of uncertain significance (VUS). In addition, depending on what was specified in the signed informed consent, incidental findings may also be reported.
The aim of professionals involved in the genetic counseling process is to help the patient and/or family members6:
- •
Comprehend medical facts including the diagnosis, probable course and causes of the disorder and the available management
- •
Understand the way heredity contributes to the disorder and the risk of recurrence or present occurrence in the family
- •
Understand the alternatives for reducing the risk of occurrence, recurrence or transmission
- •
Choose the most appropriate course of action in view of their risk, their available options and their ethical and religious standards
- •
If genetic testing is available, understand the implications, advantages and limitations of the test and the results that it may provide as well as its consequences.
- •
To provide guidance and emotional support to help them make informed decisions that are the most appropriate for the genetic disorder or risk.
- •
The ultimate objective of genetic counseling is to facilitate decision-making taking into account the values and beliefs of the family and to act in accordance with the resulting choices
- •
Genetic counseling differs from other types of clinical consultations in that it does not focus solely on the individuals that attend the visit, but also has implications for their family.
The initial visit includes an assessment of the personal and family history through the information provided by the patient or family, which is visually represented in a family tree (known as pedigree) using the international standardized nomenclature7,8 (Fig. 3), with the aim of estimating the probability of an inherited predisposition to a disease. The pedigree should include a minimum of 3 generations and allows visualization of the members of the family, indicating which are affected or unaffected. The information includes current age, health status, age at time of death and cause of death and medical diagnoses with any associated environmental exposures. Usually, information about the personal relationships between the different members of the family can be obtained indirectly while working on the pedigree.9
 Family with a history of miscarriage in several members. The patient wanted to know her risk of having another affected child. Based on the pedigree, the patient was informed that she was most likely carrying a balanced translocation and that a new pregnancy could result in another miscarriage, a child with congenital disease due to inheritance of the unbalanced translocation, a healthy child carrying the same balanced translocation or a child without chromosomal abnormalities. The patient was offered the option of karyotyping. B) In the context of a family history of achondroplasia, the risk of recurrence in the current pregnancy was discussed. Since this disease has an autosomal dominant pattern of inheritance and neither parent was affected, the couple was informed about the possibility of germline mosaicism and the risks and benefits of performing an amniocentesis to screen for the familial variant. C) Based on the pedigree, the most likely scenario was that both parents carried the variant responsible for the hearing loss. Therefore, the a priori risk of the new offspring to present the disease is 25%. The couple was offered the option of genetic testing of both partners.")
Examples of family pedigrees. The symbols indicate the gender of the individual: square for male, circle for female and diamond for unknown or nonbinary gender; uncrossed triangles represent spontaneous miscarriage and crossed triangles voluntary termination of pregnancy; crossed symbols represent deceased individuals and the letter “P” a current pregnancy. The arrow points at the individual who sought consultation or is the reason for consultation, and an arrow next to a letter “P” indicates the proband. Roman numerals symbolize generations and Arabic numerals symbolize individuals. AMAB, assigned male at birth.
A) Family with a history of miscarriage in several members. The patient wanted to know her risk of having another affected child. Based on the pedigree, the patient was informed that she was most likely carrying a balanced translocation and that a new pregnancy could result in another miscarriage, a child with congenital disease due to inheritance of the unbalanced translocation, a healthy child carrying the same balanced translocation or a child without chromosomal abnormalities. The patient was offered the option of karyotyping.
B) In the context of a family history of achondroplasia, the risk of recurrence in the current pregnancy was discussed. Since this disease has an autosomal dominant pattern of inheritance and neither parent was affected, the couple was informed about the possibility of germline mosaicism and the risks and benefits of performing an amniocentesis to screen for the familial variant.
C) Based on the pedigree, the most likely scenario was that both parents carried the variant responsible for the hearing loss. Therefore, the a priori risk of the new offspring to present the disease is 25%. The couple was offered the option of genetic testing of both partners.
The evaluation of the family pedigree allows differentiating genetic (hereditary) factors from environmental factors (lifestyle, exposure to toxic substances, etc) and is very useful as an educational tool for health promotion and disease prevention. It is important for clinicians to remain aware that in many instances this information may be affected by recall bias, as the individuals that attend the visit may provide inaccurate information about ages at diagnosis or at death or even medical diagnoses. When such information is relevant for the purposes of counseling, it is advisable to ask the patient or family to verify this information (or to obtain the consent of the family members in question so the provider can check their health records) and make an additional visit.
With this information, the provider can establish the possible pattern of inheritance and inform the patient and family about the risk of recurrence (probability that the parents of the affected patient will have another child with the same disease) and of transmission (the probability that the affected patient will pass the disease to future offspring).9
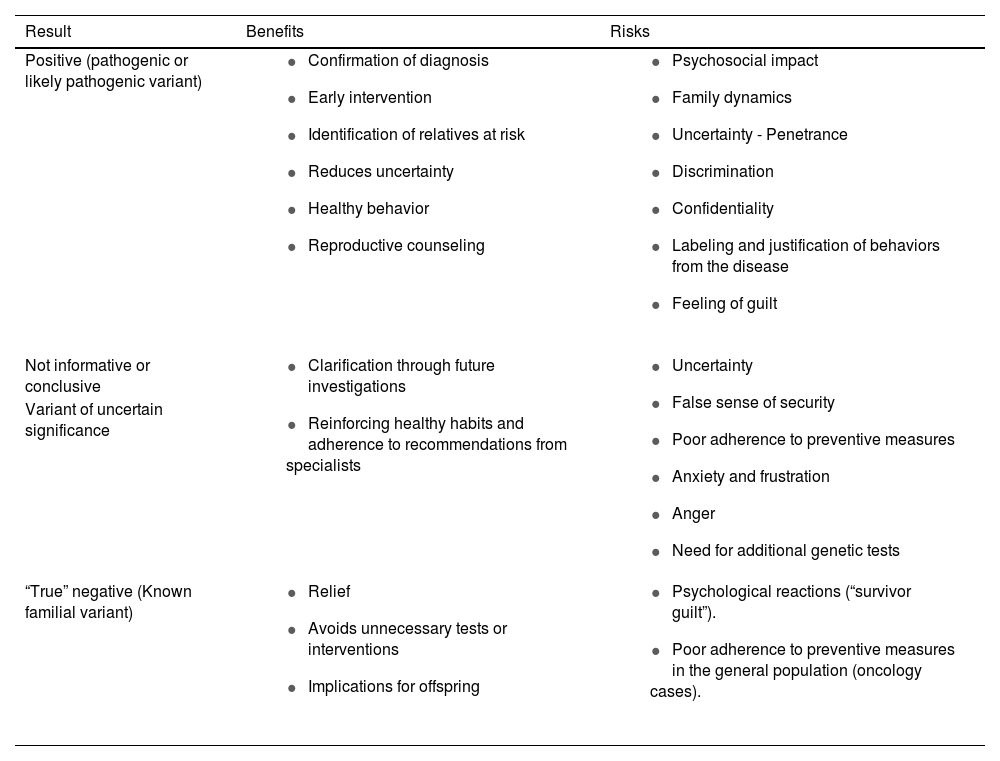
In addition, the provider gives information about the genetic or molecular tests that may be available for the disease, the probability of a hereditary predisposition, the possible results (Table 1), the probability of transmission and of recurrence, the medical implications of the disease, measures of prevention and early diagnosis, reproductive care options, etc. This should include a description of the procedure and its potential benefits and limitations.
Possible results that can be obtained after genetic testing for evaluation of the presenting disorder, including some the benefits and risks. The result is considered positive when genetic testing identifies a genetic variant that is likely to be the cause of the disease (pathogenic). Genetic testing is considered non-informative or inconclusive when it does not identify pathogenic or likely pathogenic variant(s) that explain the disease. The result of genetic testing is considered to be (a true) negative when the proband does not carry the familial variant.
| Result | Benefits | Risks |
|---|---|---|
| Positive (pathogenic or likely pathogenic variant) |
|
|
| Not informative or conclusive |
|
|
| Variant of uncertain significance | ||
| “True” negative (Known familial variant) |
|
|
There is a widespread belief that a genetic counseling visit involves collection of a sample for genetic testing, when in fact the goal is to gather information and provide education on testing risks, the potential implications of results and prevention and management measures with a non-directive approach. Since genetic counseling does not always lead to recommendation of genetic testing, it is important that the referring clinician informs the patient that the purpose of the referral is to determine whether genetic testing would be appropriate, rather than undergoing testing. Patients may feel frustrated if they come to the visit with the sole expectation of being tested and this does not happen.
On the other hand, if available, genetic testing is optional and voluntary; the autonomy of patients and legal guardians is safeguarded, and they can choose to accept or refuse genetic testing once they have the necessary information to make the decision. It is not infrequent for families who arrive to genetic counseling convinced of the need of testing to choose to defer it after the visit, either to share the obtained information with other family members (which is very frequent when only one of the parents attends the visit) or the referring clinician, or to think carefully about the potential repercussions of testing and the opportunities it may offer. In the case of massive sequencing techniques, making a decision is particularly challenging for parents once they understand the implications of testing in terms of both health10 and ethics.11
If the patient or family express an interest in undergoing testing, the provider should inform them of who in the family is the best candidate for testing, the one that would be most “informative”, which is not always the referred patient. It is important to explain why one individual should be tested earlier than another and their relevance in potential results. When the decision is made for the candidate to undergo testing, the provider should obtain the informed consent, which should be signed by both parents or legal guardians in the case of candidates aged less than 12 years, the minor (if capable of consent) and one parent or legal guardian in candidates aged more than 12 years, or, in the case of an adult patient, the candidate (article 5.3 of Royal Decree 1090/2015; article 20.1 of Law 14/2007). Recommendations have been proposed regarding the crucial elements that must be included in the informed consent for massive sequencing tests.12
Genetic tests are usually performed on blood specimens collected for the purpose following the informed consent process. We once again underscore that the decision whether or not to perform genetic testing is a personal one and should always be taken by the patient or legal guardians after being appropriately informed.
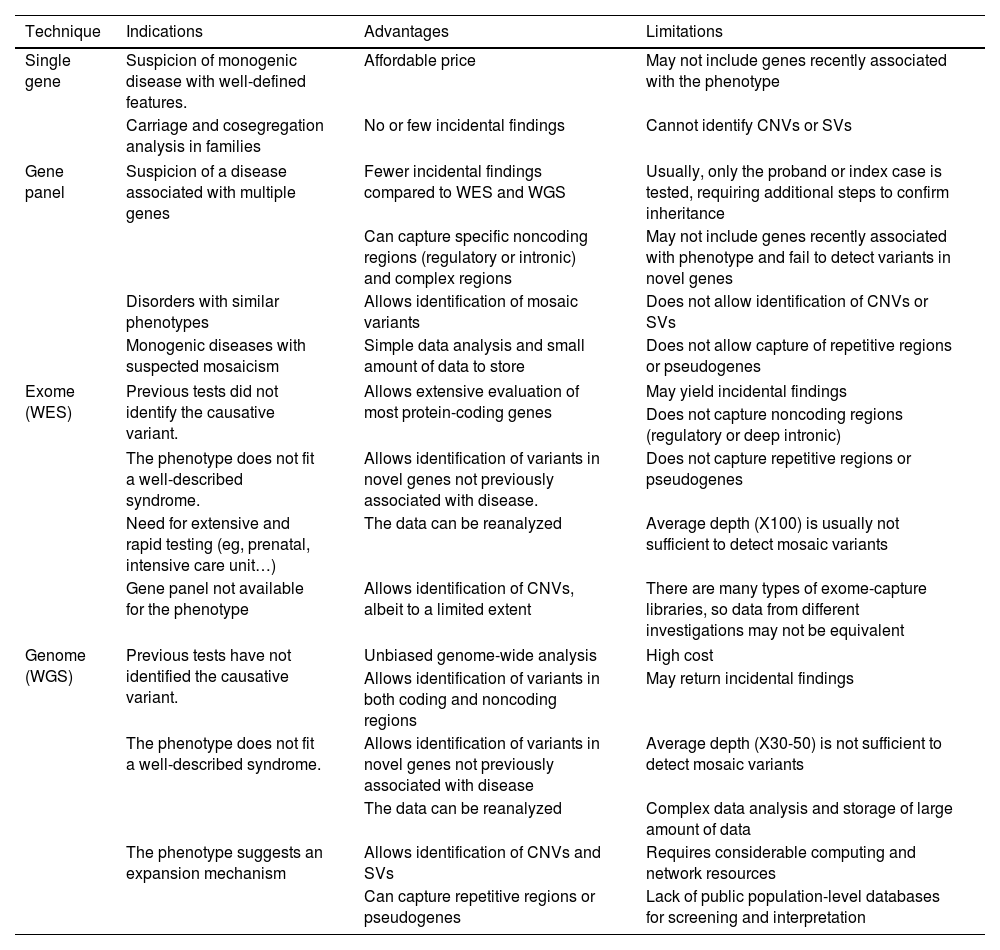
Genetic testing and molecular diagnosisThanks to next-generation sequencing (NGS) technology, there are different options for genetic testing ranging from the sequencing of a single gene to the entire genome. Thus, NGS can be used to sequence specific regions of the genome or groups of genes (what is commonly known as “gene panels”), all coding regions or exons in the genome (whole exome sequencing [WES]) or all coding, noncoding and intergenic regions (whole genome sequencing [WGS]). Each of these strategies has advantages and limitations that must be considered in selecting the most appropriate test for the patient (Table 2).
Advantages and limitations of each of the NGS techniques, and the indications for which each technique is best suited.
| Technique | Indications | Advantages | Limitations |
|---|---|---|---|
| Single gene | Suspicion of monogenic disease with well-defined features. | Affordable price | May not include genes recently associated with the phenotype |
| Carriage and cosegregation analysis in families | No or few incidental findings | Cannot identify CNVs or SVs | |
| Gene panel | Suspicion of a disease associated with multiple genes | Fewer incidental findings compared to WES and WGS | Usually, only the proband or index case is tested, requiring additional steps to confirm inheritance |
| Can capture specific noncoding regions (regulatory or intronic) and complex regions | May not include genes recently associated with phenotype and fail to detect variants in novel genes | ||
| Disorders with similar phenotypes | Allows identification of mosaic variants | Does not allow identification of CNVs or SVs | |
| Monogenic diseases with suspected mosaicism | Simple data analysis and small amount of data to store | Does not allow capture of repetitive regions or pseudogenes | |
| Exome (WES) | Previous tests did not identify the causative variant. | Allows extensive evaluation of most protein-coding genes | May yield incidental findings |
| Does not capture noncoding regions (regulatory or deep intronic) | |||
| The phenotype does not fit a well-described syndrome. | Allows identification of variants in novel genes not previously associated with disease. | Does not capture repetitive regions or pseudogenes | |
| Need for extensive and rapid testing (eg, prenatal, intensive care unit…) | The data can be reanalyzed | Average depth (X100) is usually not sufficient to detect mosaic variants | |
| Gene panel not available for the phenotype | Allows identification of CNVs, albeit to a limited extent | There are many types of exome-capture libraries, so data from different investigations may not be equivalent | |
| Genome (WGS) | Previous tests have not identified the causative variant. | Unbiased genome-wide analysis | High cost |
| Allows identification of variants in both coding and noncoding regions | May return incidental findings | ||
| The phenotype does not fit a well-described syndrome. | Allows identification of variants in novel genes not previously associated with disease | Average depth (X30-50) is not sufficient to detect mosaic variants | |
| The data can be reanalyzed | Complex data analysis and storage of large amount of data | ||
| The phenotype suggests an expansion mechanism | Allows identification of CNVs and SVs | Requires considerable computing and network resources | |
| Can capture repetitive regions or pseudogenes | Lack of public population-level databases for screening and interpretation | ||
CNV, copy number variant; SV, structural variant; WES, whole exome sequencing; WGS, whole genome sequencing.
Sequencing of a single specific gene may be a suitable approach for disorders in which changes in a particular gene are the main cause of the disease. This would be the case, for instance, of patients with achondroplasia, of who 99% carry the heterozygous pathogenic variant c.1138G>A in gene FGFR3 and 1% the c.1138G>C variant,13 or with cystic fibrosis, who carry biallelic variants in the CFTR gene.14
However, due to the genetic and phenotypic heterogeneity of most inherited pediatric diseases, single gene sequencing is often not the most efficient or effective approach.
Another application of single gene sequencing or targeted sequencing is to screen family members for the specific variant previously identified as causing disease in the family.
Single gene tests can also be used for genetic carriage screening in the reproductive partner of a patient with a known autosomal recessive disorder or who is a known carrier for such a disorder. Determining the carrier status of both partners allows a more accurate assessment of the risk to their offspring.15
These tests can be carried out with conventional Sanger sequencing or NGS.
Gene panelsMany paediatric diseases are genotypically and phenotypically heterogeneous, involving more than one disease-causing gene, or with a series of phenotypes resulting from different variants of the same gene. For example, hundreds of disease-causing genes have been reported as being involved in hereditary hearing loss,16 epilepsy17 or skeletal dysplasias.18 In such cases, it is useful to sequence all of them with a gene panel, which is based on the use of NGS technology to select or capture exons and sequence the selected regions or genes. After sequencing, a bioinformatic analysis is necessary to identify potential disease-causing variants.
An important advantage of this approach is the capacity of simultaneously sequencing many genes rather than having to sequence one gene after another. In addition, panels generally have a high sequencing depth (the same nucleotide is sequenced multiple times), which allows the detection of mosaic variants.
The disadvantages of panels include a greater cost compared to targeted or single gene sequencing, the identification of variants of uncertain significance and the potential for incidental findings (see below).
Whole exome sequencing (WES) and whole genome sequencing (WGS)As sequencing becomes more affordable, both WES and WGS are increasingly used for diagnostic purposes.19,20 Both methods offer exhaustive identification of genetic variants in coding regions. Whole genome sequencing can also identify genetic variants in noncoding regions (promoters, intergenic regions, regulatory regions…). These methods have the advantage of discovering new disease-causing genes and expanding the knowledge of the phenotypic spectrum of known disease-causing genes. Lastly, it is also important to remember that, depending on the bioinformatics pipeline, in addition to detecting sequence changes, it is possible to analyse large-scale chromosomal abnormalities, like deletions or duplications.21
There are some limitations to WES. It usually does not detect intronic variants (unless immediately flanking a targeted exon), also does not detect trinucleotide repeat expansions or methylation abnormalities (the latter are also not detectable by WGS), and may offer only limited detection of copy-number variants.19
Like gene panels, WES and WGS require sophisticated bioinformatics analysis (more extensive in their case, as they identify a larger number of variants) to determine which of the DNA variants detected through sequencing may be pathogenic.
For the purpose of discovering new genes, it may be useful to extend sequencing to other members of the family (trio genome/exome),19 and in fact results from different families may be needed to obtain robust evidence that a new gene is disease-causing. The disadvantages of WES and WGS include their greater cost, increased data storage requirements, longer turnaround times, need of highly qualified staff for the bioinformatics analyses and, last of all, the potential for incidental findings (see below).
Tests for detection of structural variantsGenomic structural variation is defined as changes greater than 1 kilobase (kb) that may be unbalanced (deletions, insertions or duplications, also known as CNVs) or balanced (translocations and inversions).22
While, as mentioned above, some bioinformatics methods allow the detection of some structural variants in the analysis of NGS data, in some cases specific methods or technologies are required, such as fluorescence in situ hybridization (FISH) or multiplex ligation-dependent probe amplification (MLPA), when the region that may be affected is known; or comparative genomic hybridization (CGH), single nucleotide polymorphism (SNP) chips, when the whole genome needs to be analysed.
Structural variants may explain a significant part of the “missing heritability” in paediatric disorders when sequencing tests could not identify (or did not look for) these abnormalities.23 It is important to remember that comprehensive genetic testing in patients with hereditary diseases should include both sequencing and analysis of CNVs.
Interpreting the results of genetic testingNext generation sequencing generates thousands of sequence variants potentially associated with diseases. As the number of sequenced genes increases, so does the number of variants that need to be interpreted. Bioinformatic analyses are an essential step in identifying variants that actually cause disease. In addition, providing a precise description of the clinical phenotype using HPO terms (https://hpo.jax.org/app/) can be useful when submitting patient samples for genetic testing. This information can guide variant prioritization.24
The American College of Medical Genetics (ACMG) has published guidelines for the interpretation of sequence variants using population data, computational data (in silico), functional data, clinical data and segregation data to classify variants as pathogenic, likely pathogenic, of uncertain significance, likely benign or benign2. ClinGen (Clinical Genome Resource, https://clinicalgenome.org/), GenCC (Gene Curator Coalition, https://search.thegencc.org/), Franklin (https://franklin.genoox.com/clinical-db/home), ClinVar (www.ncbi.nlm.nih.gov/clinvar) and LOVD (www.lovd.nl) provide additional resources for variant interpretation.
Variants of uncertain significanceVariants of uncertain significance (VUS) may have some characteristics of disease-causing variants, but there is insufficient evidence to support either a pathogenic classification or a benign classification. An example of a VUS is a variant in a known disease-causing gene that is rarely found in the population, but with no functional data to support its pathogenicity. Other evidence that would support pathogenicity might include a functional evaluation or analysis of co-segregation with disease in the family or in another family carrying the same variant and affected by the same disease.
Variants of uncertain significance can be reclassified over time,25 so it is important for individuals in whom genetic testing yields such results to keep in touch with the clinician who ordered or performed testing to receive updates should new information on the variant become available.
Incidental findingsMassive sequencing tests may identify variants that cause diseases other than the one for which testing was originally indicated. Such unsolicited results can be divided into secondary findings and incidental findings. While both terms refer to (likely) pathogenic variants (that is, associated with a disorder) unrelated to the primary indication for testing, secondary findings refer to variants located in genes actively screened by the medical laboratory and analysed as part of the minimum gene list proposed by the ACMG,26 which includes genes selected on account of their association with diseases for which there is a reliable genetic test and effective management or treatment. On the other hand, incidental findings are (likely) pathogenic variants unrelated to the primary indication for testing that are identified by chance during genetic testing.27
There is no consensus regarding whether or not incidental findings should be systematically reported to patients.28,29 This is an even greater dilemma when genetic testing is performed in children.11 We must not forget that in the case of trio exome sequencing, these unsolicited results, whereby a variant is found in the patient, could be included in the report with additional information regarding its inheritance, and thus could lead to diagnosis in a parent at the same time as the offspring. Even if sequencing is not originally performed in other family members, the incidental detection of a variant in a child can have implications for the entire family, as cascade testing may be recommended to unaffected members. This potential consequence for the parents of genetic testing results in children could affect the decisions they make about whether or not to test their offspring.11
Post-test counselingGiven the broad range of possible results (from pathogenic variants to VUS or incidental findings), this visit ideally includes the clinician who referred the patient for testing and who will be in charge of the followup.
In this visit, the patient and/or parents are notified that the results are available and asked one more time whether they are interested in knowing them. If they expressed an interest in learning about incidental findings during the pretest visit, they are asked to confirm that they are still interested. Any question or concern that they may have had while they awaited the results is addressed at this time.
The counselor then informs the patient and parents of the results, explaining them in the context of the presentation, the medical implications, the pattern of inheritance, the potential risk of other relatives and reproductive planning options. Doubts and concerns that may arise are clarified and addressed. The counselor also informs of the recommended referrals to specialists depending on the identified genetic disorder to ensure followup appropriate for the level of risk, and the patient (and family members) are then referred to the corresponding specialists.
In addition, it may be necessary to report and explain incidental findings and their implications, both in the proband and in other family members.
Usually, this session involves more than the delivery of medical information and tends to focus on providing support to families to cope with the emotional, psychological, medical, social and economic results of the test. Specifically, the counselor addresses psychological issues such as denial, anxiety, anger, suffering or guilt, and, if needed, provides a referral for in-depth psychosocial assessment.
In this visit, the patient or family is given a written report summarizing the reason for consultation, genetic tests performed and test results,30 as well as recommendations and educational material to share with families or information on community resources and support groups or patient associations (if there are any). Whenever possible, contact information should also be provided to establish an open-door relationship in case there are any questions or further consultation is needed.
If testing returns positive results, clinicians may consider testing of other relatives of the proband and consequently offer referring other members of the family for risk assessment.
Limitations and next stepsBetween 50% and 60% of individuals with a presumed Mendelian disorder remain undiagnosed after performance of extensive genetic testing (although the rate of diagnosis varies based on the disorder).
Several factors contribute to the failure to reach a confirmed genetic diagnosis:
- •
The genetic basis of many Mendelian disorders remains unknown.
- •
In the case of diseases in which the causative gene or genes are known, it is possible that the ordered test does not target the appropriate gene or genes (for instance, single gene test or gene panel including several genes), the type or types of variants (for instance, triplet repeat expansion) or epigenetic changes (eg, methylation status).
- •
Technical limitations can hinder the detection of a pathogenic variant (for instance, detection of a CNV using WES).
- •
There may not be sufficient evidence to interpret the pathogenicity of a variant. This is exacerbated by the fact that the interpretation by diagnostic laboratories can vary significantly due to differences in their approach to the pathogenicity classification of a variant,31 although the standardization of variant classification and efforts data sharing should mitigate this effect.32,33
- •
Incomplete penetrance and the challenges involved in determining whether a phenotype results from large-effect alleles or a complex pattern of inheritance (for instance, digenic or oligogenic) may hinder the identification of the molecular etiology of the phenotype.
- •
Historically, the diagnostic yield has depended on the thoroughness of the phenotypic evidence available for variant classification.34 Although there are no established guidelines for the systematic use of phenotype evidence, the use of human phenotype ontology terms and phenopackets could set a standard for the exchange of phenotype data between laboratories, clinicians and researchers.24
- •
It is also important to remain aware that non-genetic factors, such as infection or toxic exposure, can be the cause of some of these diseases.
To address some of these limitations, different laboratories may opt for reanalysis of NGS data and/or reinterpretation of identified variables, on the request of the referrer or even the patient, if additional affected family members are identified or new clinical manifestations emerge.35 Some authors even propose carrying out such revisions routinely at intervals ranging from one36 to three years.37 In the case of systematic reanalysis, it would be important to consider the ethical, economic, legal and (psycho)social repercussions that it may have, both for patients/families and for the health care professionals involved.38
Lastly, we cannot forget promising emerging technologies, such as long-read sequencing39 or optical genome mapping,40 which will help provide an answer to some patients still awaiting a diagnosis.
FundingThis research did not receive any external funding.
Conflicts of interestThe authors have no conflicts of interest to declare.

