


Newborn screening (NBS) for cystic fibrosis (CF) is well-established in many countries and provides the opportunity for an early diagnosis and treatment before the development of irreversible structural lung damage.
In 1999, Catalonia, Castilla-León, and the Balearic Islands started the NBS programme for CF. In the last 10 years its implementation rapidly spread and all the autonomies offer the NBS programme for CF since 2015. There are many different strategies across Spain. It is believed that it is very opportune to have an updated and consensual guide for the diagnosis, follow-up, and treatment of patients diagnosed by neonatal screening.
El diagnóstico de fibrosis quística (FQ) a través del cribado neonatal (CN) está bien establecido en muchos países y brinda la oportunidad de un diagnóstico y tratamiento temprano antes del desarrollo de daño estructural pulmonar irreversible.
En 1999, Cataluña, Castilla-León y las Islas Baleares iniciaron el programa CN para FQ. En los últimos 10 años su implementación se extendió rápidamente y todas las autonomías ofrecen el programa CN para FQ desde 2015. Hay varias estrategias diferentes en toda España. Creemos que es muy oportuno contar con una guía actualizada y consensuada para el diagnóstico, el seguimiento y el tratamiento de los pacientes diagnosticados de FQ mediante CN.
Cystic fibrosis (CF) is the most frequent severe genetic disorder with an autosomal recessive inheritance pattern in the Caucasian population, with an incidence of 1 per 1800 to 25000 live births depending on the geographical region or ethnicity. In 2016, the Canadian Cystic Fibrosis Foundation reported a median survival of 53.3 years.1 The substantial increase in survival in these patients is due to a series of factors, among which the implementation of neonatal screening programmes for early diagnosis have played a significant role.
Cystic fibrosis meets the criteria warranting early detection and NS for the purpose of determining the actual incidence of the disease, early genetic counselling and immediate initiation of treatment with the purpose of preventing or minimising lung damage, as molecules that can target and correct the defect in the abnormal cystic fibrosis transmembrane conductance regulator (CFTR) protein are currently available.
Since 2015, neonatal screening is performed in every autonomous community in Spain.
We think it would be very convenient to have an updated consensus-based guideline for the diagnosis of CF and the follow-up and treatment of patients that meet the diagnostic criteria. This document summarises all the multidisciplinary aspects involved in the management of these patients.
Benefits and risks of neonatal screening for cystic fibrosisAt present, most professionals devoted to the care of patients with CF agree that a well-designed neonatal screening programme involves a positive risk-benefit ratio where the benefits (nutritional, respiratory, of early eradication of microorganisms, genetic counselling and participation in early intervention clinical trials) outweigh the disadvantages (especially in the psychosocial dimension) and are cost-effective in the long term.2–5 To guarantee the beneficial effect of neonatal screening, patients with CF should be managed appropriately (following standard care protocols) immediately after diagnosis in specialised CF units.6,7 Most of the risks of neonatal screening are minimised by taking the following measures6: (a) establishing a neonatal screening programme adapted to the characteristics of the population, (b) communicating effectively with parents and giving them useful information throughout the diagnostic process, both in case of affected patients and of patients who are carriers or that received a false positive result, trying to keep the time elapsed to establishment of the definitive diagnosis to a minimum; (c) offering medical follow-up to affected children.
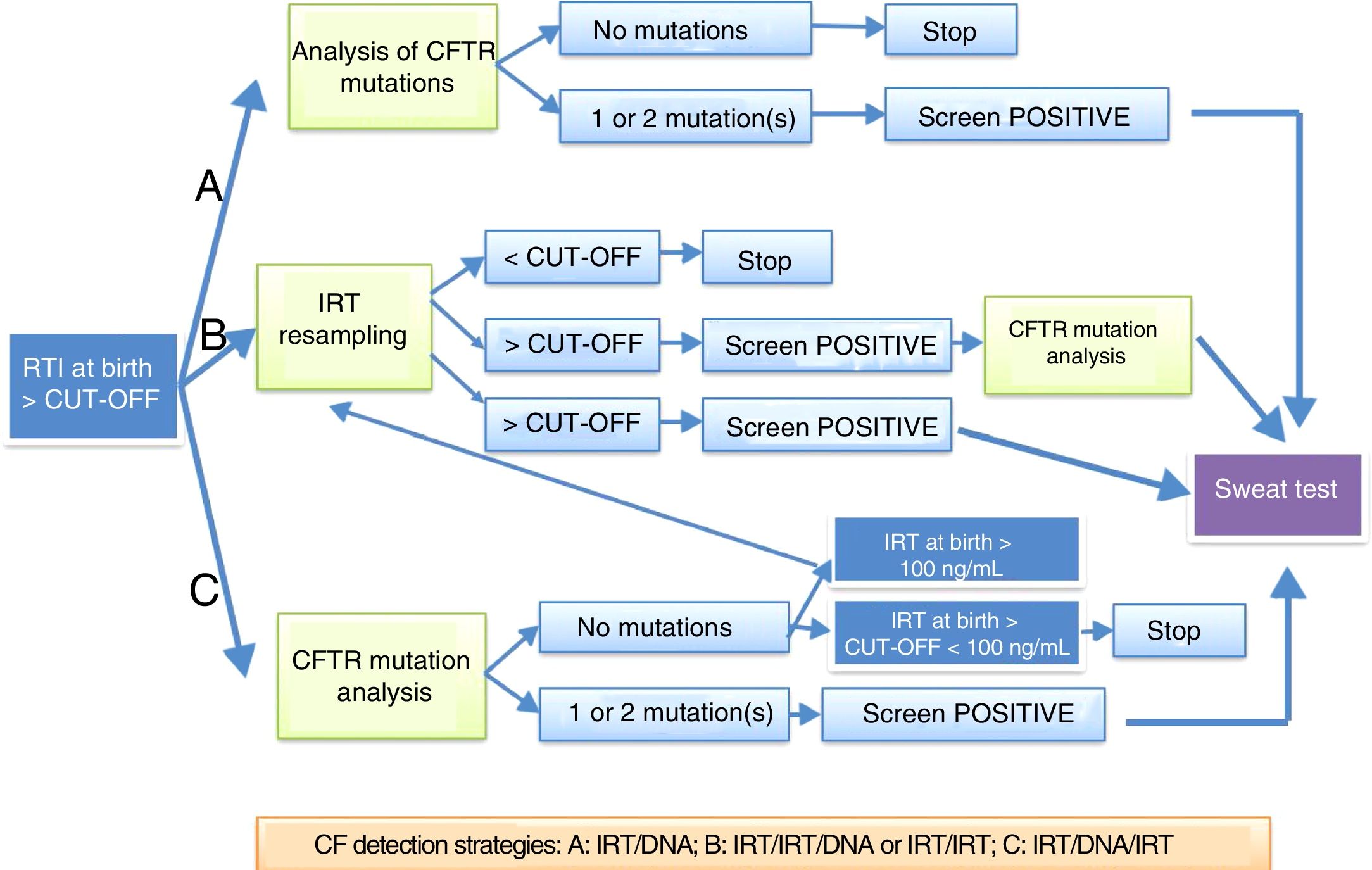
Neonatal screening protocols in SpainImmunoreactive trypsinogenSerum levels of immunoreactive trypsinogen (IRT) are higher in newborns with CF and remain elevated longer compared to newborns who are not affected by the disease.8,9
An elevation of IRT at birth is not specific for CF, as there are healthy newborns that experience a transient elevation of this enzyme. The specificity of elevation of IRT in a single sample is low, so protocols have been developed that use a DNA test in the first neonatal sample (IRT+DNA), the protocol most frequently used in Spain, or that request a second sample for repeat measurement of IRT followed by a DNA test (IRT+IRT+DNA), or a sweat test (Fig. 1). It is important to remember that up to 30% of newborns with meconium ileus have normal IRT levels. The IRT+IRT strategy is applied in only a few autonomous communities.

Protocols for diagnosis of cystic fibrosis through neonatal screening proposed by the European Best Practice Guidelines for Cystic Fibrosis Neonatal Screening7 applied in Spain.
In Spain, there are 3 different strategies currently in use: IRT+IRT, IRT+IRT+DNA and IRT+DNA+IRT.
Genetic testing for cystic fibrosisGenetic testing is part of most protocols for neonatal screening for CF and is performed in samples with IRT levels exceeding the established threshold. In each population, the applied protocol should be adapted to its mutation spectrum.
There are 4 possible outcomes of genetic testing in a newborn with elevated IRT:
- a)
If the test detects 2 CF-causing mutations, the newborn should undergo a sweat test and DNA segregation analysis should be performed to confirm that each mutation comes from a different parent.
- b)
In case of a single mutation, a sweat test should be performed. If the results of the sweat test are inconclusive, genetic testing should be expanded to attempt to identify a second mutation. If the sweat test is negative, the newborn is considered a carrier.
- c)
Cases where no mutation is found and the sweat test is normal are considered false positives of screening.
There is another group comprising those newborns in whom a conclusive diagnosis cannot be made, even with use of ancillary tests, for which the Working Group on CF of the European Cystic Fibrosis Society has proposed the label cystic fibrosis screen positive, inconclusive diagnosis (CFSPID).10 This group includes:
- a)
Newborns with a CF mutation in only one allele and intermediate sweat chloride values (30–59mmol/L).
- b)
Newborns with a CFTR mutation in each allele, of which only 1 is classified as causing CF, and normal sweat chloride values (<30mmol/L).
This group of patients requires ongoing follow-up, as in some cases CF is diagnosed at a later age due to onset of symptoms with elevation of sweat chloride levels.
Sweat chloride testThe sweat test is the cornerstone of diagnosis of CF. Newborns with a positive neonatal screen test for CF must be referred for a sweat test exclusively to specialised CF units accredited for diagnosis.
The sweat test is divided in 3 stages: (1) Stimulation by pilocarpine iontophoresis. (2) Collection of a sample over 30min by either of the following methods: Macroduct collection system or the original Gibson and Cooke method (collection in pre-weighed filter paper or gauze). The sample should have a volume of at least 15μL (Macroduct) or weigh at least 75mg (gauze). (3) Chloride determination by coulometric titration with a chloridometer.11
The diagnosis cannot be confirmed based exclusively on the sweat conductivity measurements obtained by the nanoduct or sweat check systems (Macroduct). Samples must be analysed as soon as they are collected.
The test result is classified as “normal” if the chloride concentration is less than 30mmol/L; “intermediate” if it is between 30 and 59mmol/L and “positive” if it is of 60mmol/L or higher.12
Patients with persistent intermediate values should undergo a diagnostic evaluation, including extended DNA testing and repetition of the sweat test at 6 months.
Appointment for reporting the results of neonatal screeningNeonatal screening elicits a varying degree of anxiety in parents. Thus, it is advisable to report the results as soon as possible, preferably on the same day that testing is performed.
If the newborn has CF, parents will be given information on all the tests performed to diagnose the disease and of the encouraging outcomes of new treatments and the extensive research that is underway, thanks to which it is now possible to have an optimistic perspective on the disease.
At the end of the visit, if the newborn is not affected, parents will receive a report stating that the child does not have CF or that the child is a carrier and offering genetic counselling to the parents.13
Initial appointment with a newborn with cystic fibrosisThe diagnosis of CF through a neonatal screening test comes as a surprise to parents, since in most instances the newborn does not have any symptoms. For this reason, the psychosocial impact on the family of the new diagnosis must be managed with care.14
The most important aspects to address are the following:
- •
Provide an overview of the disease.
- •
Introduce the concept of the “care team”.
- •
Manage the expectations of the family.
- •
Offer psychological support.
One of the key goals of the visit is to provide basic information in a positive light with a sensitive and empathic attitude and using simple language.15
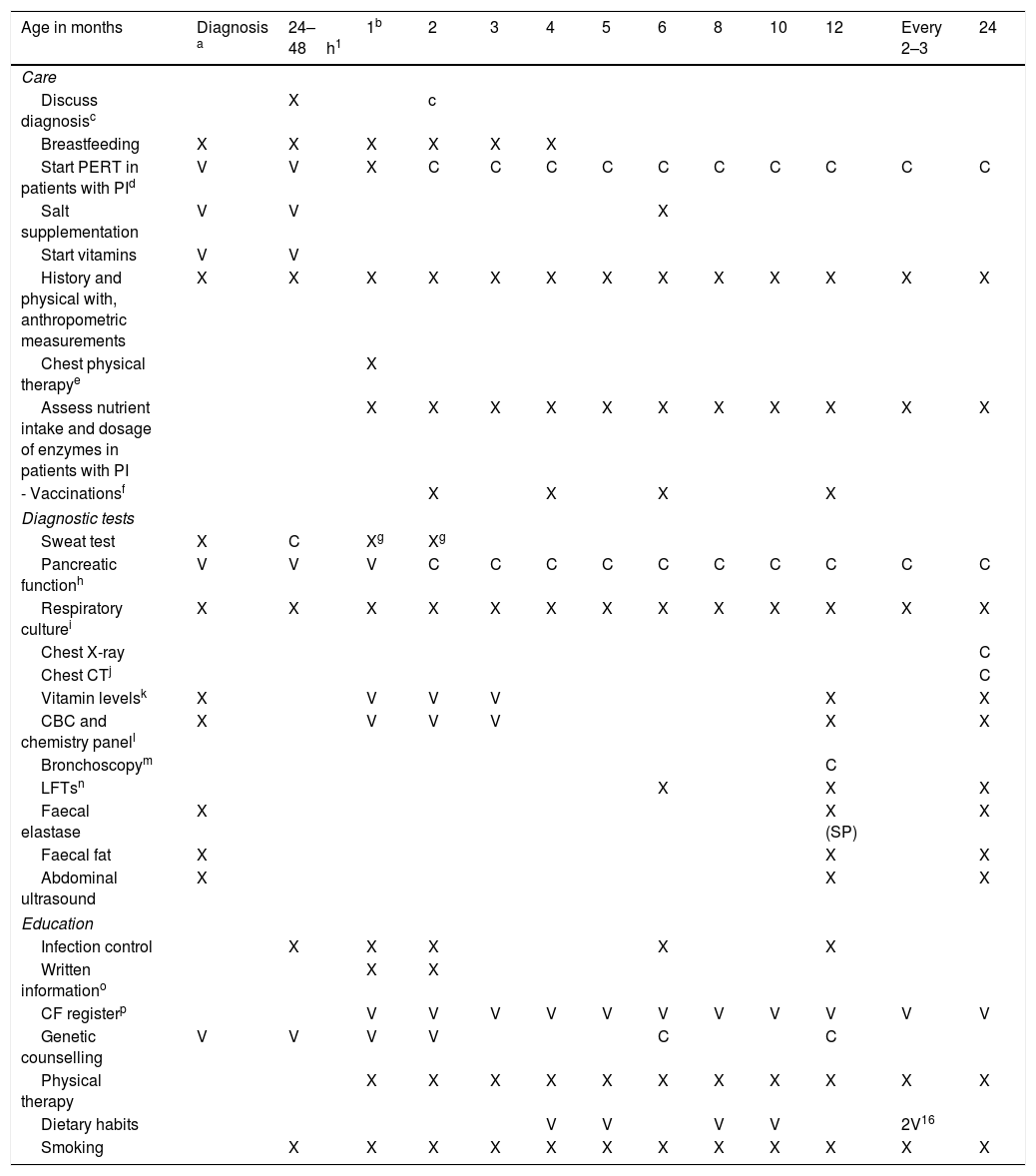
Table 1 specifies the actions to be taken during the first visit.
Routine visit schedule.
| Age in months | Diagnosis a | 24–48h1 | 1b | 2 | 3 | 4 | 5 | 6 | 8 | 10 | 12 | Every 2–3 | 24 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Care | |||||||||||||
| Discuss diagnosisc | X | c | |||||||||||
| Breastfeeding | X | X | X | X | X | X | |||||||
| Start PERT in patients with PId | V | V | X | C | C | C | C | C | C | C | C | C | C |
| Salt supplementation | V | V | X | ||||||||||
| Start vitamins | V | V | |||||||||||
| History and physical with, anthropometric measurements | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Chest physical therapye | X | ||||||||||||
| Assess nutrient intake and dosage of enzymes in patients with PI | X | X | X | X | X | X | X | X | X | X | X | ||
| - Vaccinationsf | X | X | X | X | |||||||||
| Diagnostic tests | |||||||||||||
| Sweat test | X | C | Xg | Xg | |||||||||
| Pancreatic functionh | V | V | V | C | C | C | C | C | C | C | C | C | C |
| Respiratory culturei | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Chest X-ray | C | ||||||||||||
| Chest CTj | C | ||||||||||||
| Vitamin levelsk | X | V | V | V | X | X | |||||||
| CBC and chemistry panell | X | V | V | V | X | X | |||||||
| Bronchoscopym | C | ||||||||||||
| LFTsn | X | X | X | ||||||||||
| Faecal elastase | X | X (SP) | X | ||||||||||
| Faecal fat | X | X | X | ||||||||||
| Abdominal ultrasound | X | X | X | ||||||||||
| Education | |||||||||||||
| Infection control | X | X | X | X | X | ||||||||
| Written informationo | X | X | |||||||||||
| CF registerp | V | V | V | V | V | V | V | V | V | V | V | ||
| Genetic counselling | V | V | V | V | C | C | |||||||
| Physical therapy | X | X | X | X | X | X | X | X | X | X | X | ||
| Dietary habits | V | V | V | V | 2V16 | ||||||||
| Smoking | X | X | X | X | X | X | X | X | X | X | X | X | |
C, consider performing in this visit; LFT, lung function test; PERT, pancreatic enzyme replacement therapy; V, perform in any of these visits; X, perform in this visit.
Explain how it was determined that the child has CF and provide details regarding the diagnosis, genetics and impact on other siblings and family members.
Initiate PERT if patient has symptoms of malabsorption or a faecal elastase concentration <200mcg/g or 2 CFTR mutations associated with PI.
Train on techniques that facilitate mucociliary clearance: percussion and postural drainage. There is controversy as to which of the 2 is most appropriate in asymptomatic children.
Coordination with routine primary care visits. Include vaccination against varicella, S. pneumoniae and rotavirus. Vaccinate children aged more than 6 months against influenza and administer palivizumab to infants aged less than 1 year.
If PI is not present at the time of diagnosis, repeat the faecal elastase test at least twice in the first year of life and annually thereafter, as PI may develop at a later time.
Routine performance recommended from age 2 years in asymptomatic children, or before if the patient has symptoms. Expiratory CT scan with low-dose radiation and expiratory views to assess peripheral airway disease.
Repeat at 1–2 months from initiation of supplementation. Measure more frequently in case of abnormal results.
Complete blood count, electrolytes, urea, creatinine, albumin, aspartate aminotransferase, alanine aminotransferase, gamma-glutamyl transferase, bilirubin, alkaline phosphatase.
Some hospitals perform a bronchoalveolar lavage at approximately 3 months and 1 year of age for early detection of infection, inflammation markers or aspiration/reflux, and a bronchial biopsy for clinical investigation.
Follow-up by a paediatric gastroenterologist is recommended, with visits every 1–2 weeks immediately after diagnosis to ensure adequate nutrition and thereon every month for the first 3 months. If the patient is in good condition, subsequent visits may be held at 2- to 3-month intervals.
NutritionThe main goal of nutritional interventions in CF is to promote normal growth and development for age (Table 2).
Guide for prescription of nutritional interventions18
| <2 years | 2–18 years | |
|---|---|---|
| Optimal nutritional status: preventive nutritional counselling | Weight for height ≥P50 | BMI≥P50 |
| Suboptimal status: dietary modification and oral nutrient supplementation | Weight for height P10−P50 Weight loss Faltering growth | BMI=P10−P50 Weight loss in 2–4 months or absence of weight gain in 2 months |
| Persistent malnutrition: enteral nutrition (nasogastric or gastrostomy tube) | Weight for height a<P10 Persistent weight loss or faltering growth despite oral supplementation | BMI<P10 Drop of 2 percentile points despite oral supplementation |
BMI, body mass index calculated as weight (kg)/height square (m2); P10, 10th percentile; P50, 50th percentile.
We recommend encouraging breastfeeding on demand. Whatever the feeding modality, patients with pancreatic insufficiency (PI) will receive pancreatic enzymes.16,17
The introduction of complementary foods should conform to the guidelines for the general population, starting at age 6 months in exclusively breastfed infants and at age 4 to 6 months in formula-fed infants.18,19
Pancreatic and liver functionAs early as the neonatal period, children with CF may exhibit hypovitaminosis and PI.
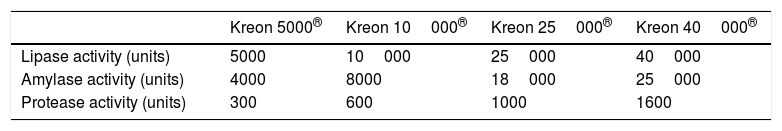
In case of clinical manifestations suggestive of PI or the presence of 2 mutations associated with PI, the patient should receive pancreatic enzyme replacement therapy and vitamin supplementation (Table 3), and while treatment should not be delayed unduly, it is preferable and recommended to have the results of a faecal elastase test before starting treatment.
Recommendations for pancreatic enzyme replacement therapy in children with pancreatic insufficiency.
| Infants 0–12 months | 2000–4000IU lipase/120mL or per breastfeeding episode |
|---|---|
| Children 12 months to 4 years | 1000IU lipase/kg per feeding,a maximum 2500IU/kg/day or 10000IU/kg/day or 4000 UI/g fat in diet |
| Children >4 years through adulthood | 500IU lipase/kg per feeding,a maximum 2.500IU/kg/day or 10000IU/kg/day or 4000IU/g fat in diet |
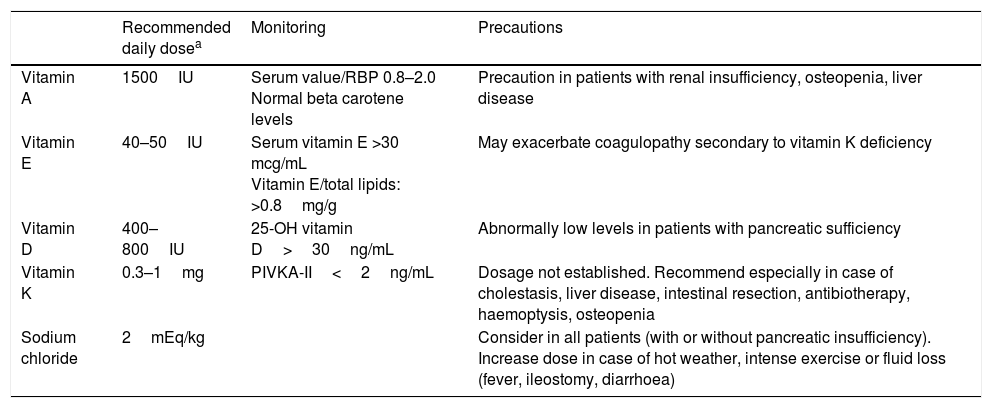
Different enzyme formulations are currently available (Table 4). Pancreatic enzymes should be taken with every meal. Administration with water or acidic foods such as apple juice or sauce is recommended to prevent the microbeads from opening prematurely.20 Routine supplementation with fat-soluble vitamins (A, D, E and, to a lesser degree, K) is indicated in all infants with PI (Table 5).16
Recommended doses of fat-soluble vitamins and minerals in infants aged less than 1 year with pancreatic insufficiency.
| Recommended daily dosea | Monitoring | Precautions | |
|---|---|---|---|
| Vitamin A | 1500IU | Serum value/RBP 0.8–2.0 Normal beta carotene levels | Precaution in patients with renal insufficiency, osteopenia, liver disease |
| Vitamin E | 40–50IU | Serum vitamin E >30 mcg/mL Vitamin E/total lipids: >0.8mg/g | May exacerbate coagulopathy secondary to vitamin K deficiency |
| Vitamin D | 400–800IU | 25-OH vitamin D>30ng/mL | Abnormally low levels in patients with pancreatic sufficiency |
| Vitamin K | 0.3–1mg | PIVKA-II<2ng/mL | Dosage not established. Recommend especially in case of cholestasis, liver disease, intestinal resection, antibiotherapy, haemoptysis, osteopenia |
| Sodium chloride | 2mEq/kg | Consider in all patients (with or without pancreatic insufficiency). Increase dose in case of hot weather, intense exercise or fluid loss (fever, ileostomy, diarrhoea) |
Based on the recommendations of the European Cystic Fibrosis Society.16
Serum retinol (mg/dL)/retinol binding protein (RBP) (mg/dL)×0.0734.
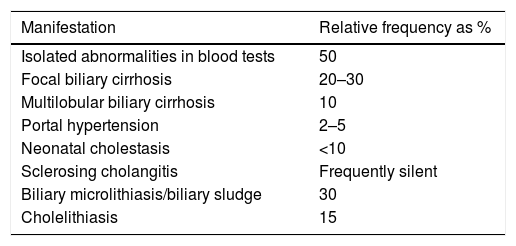
These patients can experience liver impairment of a heterogeneous nature (Table 6).
Liver involvement in cystic fibrosis.
| Manifestation | Relative frequency as % |
|---|---|
| Isolated abnormalities in blood tests | 50 |
| Focal biliary cirrhosis | 20–30 |
| Multilobular biliary cirrhosis | 10 |
| Portal hypertension | 2–5 |
| Neonatal cholestasis | <10 |
| Sclerosing cholangitis | Frequently silent |
| Biliary microlithiasis/biliary sludge | 30 |
| Cholelithiasis | 15 |
It is important to remember sodium chloride supplementation in hot weather.
Respiratory managementTechniques to improve mucociliary clearanceThe dehydrated secretions of these patients hinder mucociliary clearance, giving rise to a vicious cycle of inflammation and chronic lung infection, which calls for early initiation of chest physical therapy.21 There are several chest physical therapy techniques that can be applied in the paediatric age group, such as percussion, postural drainage and high-frequency chest compression. The evidence currently available is insufficient to establish which method is best.
Treatment with recombinant human DNase reduces the viscosity of the mucus and facilitates mucociliary clearance. In children aged more than 6 years, administer 2.5mL/day in nebulised form (following administration of a bronchodilator); in younger children, this treatment will only be considered in select cases.
Hypertonic saline solution has an osmotic effect that restores fluid to the airway surfaces. Nebulisation of 4mL of 7% hypertonic saline (following administration of a bronchodilator) in patients aged more than 6 years was found efficacious in improving lung function and pulmonary exacerbations.22,23
Treatment of inflammationAzithromycin modulates cytokine production and plays an indirect role in biofilm formation. In cases where it is indicated (for instance, with persistent detection of Pseudomonas in culture), it should be given 3 times a week at the usual dose while monitoring liver function, taking into account that co-administration with inhaled tobramycin could impair the response to the latter.24
Respiratory infectionsPrevention of infectionNeonatal screening for CF provides an opportunity to educate the family and the child from a young age on the importance of hand hygiene in the household.25
In the clinic setting, it is recommended that waiting times are kept to a minimum and that the child wears a mask and plays with their own toys.26 At present, there is absolute consensus that these patients should always be segregated. Vaccination according to the routine schedule is recommended, including vaccination against influenza (in children aged more than 6 months), while attendance to a child care centre is recommended against.
Treatment of primary bacterial infectionsThe presence of viscous secretions in the airways characteristic of these patients facilitates recurrent infections by different microorganisms. The most frequently involved pathogens are the following:
Staphylococcus aureus(A) Methicillin-sensitive S. aureus
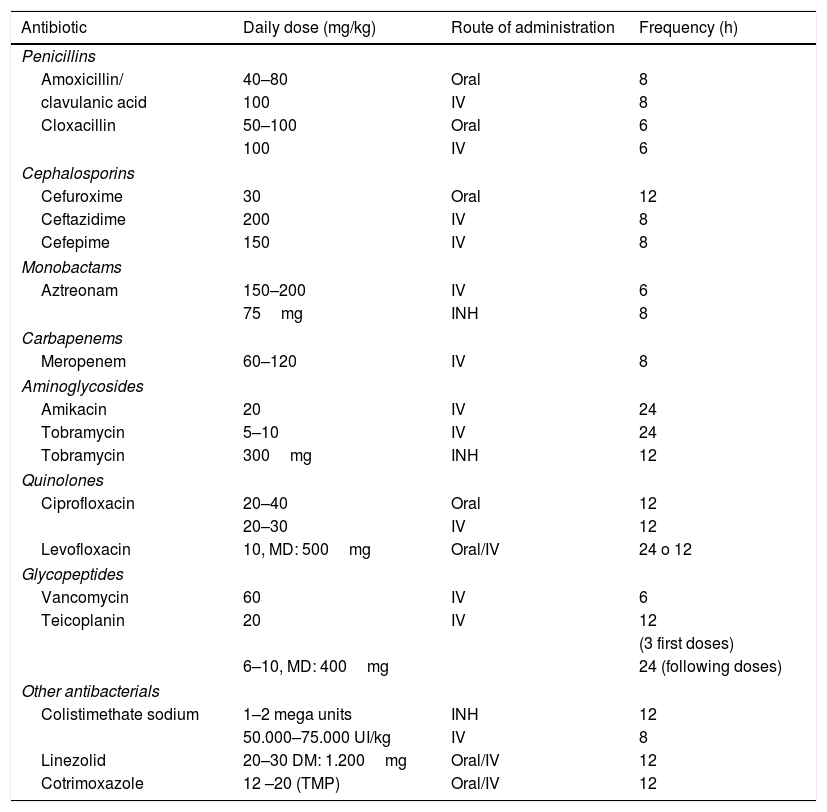
The current evidence is insufficient to recommend or discourage eradication of S. aureus or antibiotic prophylaxis.27 The drugs used most frequently are amoxicillin-clavulanic acid, cefadroxil y trimethoprim-sulfamethoxazole (TMP-SMX) given orally for 14–21 days. Performance of a respiratory culture 1 week after completion of treatment is recommended (Table 7).
Antibiotic agents used in the management of respiratory disease in cystic fibrosis.
| Antibiotic | Daily dose (mg/kg) | Route of administration | Frequency (h) |
|---|---|---|---|
| Penicillins | |||
| Amoxicillin/ | 40–80 | Oral | 8 |
| clavulanic acid | 100 | IV | 8 |
| Cloxacillin | 50–100 | Oral | 6 |
| 100 | IV | 6 | |
| Cephalosporins | |||
| Cefuroxime | 30 | Oral | 12 |
| Ceftazidime | 200 | IV | 8 |
| Cefepime | 150 | IV | 8 |
| Monobactams | |||
| Aztreonam | 150–200 | IV | 6 |
| 75mg | INH | 8 | |
| Carbapenems | |||
| Meropenem | 60–120 | IV | 8 |
| Aminoglycosides | |||
| Amikacin | 20 | IV | 24 |
| Tobramycin | 5–10 | IV | 24 |
| Tobramycin | 300mg | INH | 12 |
| Quinolones | |||
| Ciprofloxacin | 20–40 | Oral | 12 |
| 20–30 | IV | 12 | |
| Levofloxacin | 10, MD: 500mg | Oral/IV | 24 o 12 |
| Glycopeptides | |||
| Vancomycin | 60 | IV | 6 |
| Teicoplanin | 20 | IV | 12 |
| (3 first doses) | |||
| 6–10, MD: 400mg | 24 (following doses) | ||
| Other antibacterials | |||
| Colistimethate sodium | 1–2 mega units | INH | 12 |
| 50.000–75.000 UI/kg | IV | 8 | |
| Linezolid | 20–30 DM: 1.200mg | Oral/IV | 12 |
| Cotrimoxazole | 12 –20 (TMP) | Oral/IV | 12 |
INH, inhaled; IV, intravenous; MD, maximum dose; TMP, trimethoprim.
(B) Methicillin-resistant S. aureus
In patients with no symptoms or mild symptoms: TMP-SMX, linezolid, 2–4 weeks.28 In patients with severe symptoms, the recommended treatment is intravenous vancomycin or teicoplanin for 2–3 weeks. In case of chronic infection, one possible option is inhaled vancomycin for 2–3 weeks.
Haemophilus influenzaeIf the patients have symptoms, the most frequently used drugs are amoxicillin-clavulanic acid and TMP-SMX (2–4 weeks).
Pseudomonas aeruginosaTreatment for eradication must be initiated as soon as P. aeruginosa is detected for the first time. None of the eradication protocols published to date has proven superior to the others. Based on the Spanish consensus guidelines, the management of a first episode with isolation of P. aeruginosa29 consist in:
(1) Asymptomatic patients: oral ciprofloxacin for 2–4 weeks+nebulised antibiotics—colistin given daily (maximum 3–6 months) or 1–3 on-off cycles of tobramycin or aztreonam (maximum 6 months).
(2) Symptomatic patients: systemic+inhaled antibiotherapy.
- Mild: oral ciprofloxacin for 2–4 weeks.
- Moderate–severe: intravenous ceftazidime+tobramycin (2–3 weeks).
Performance of a respiratory culture is recommended 1 week after completion of treatment. If the culture is negative, we recommend maintenance of daily colistin (for a maximum of 3–6 months) or 1–3 on–off cycles of tobramycin or aztreonam (maximum 6 months).
Treatment of pulmonary exacerbationsA consensus definition of pulmonary exacerbation in infants and preschool-aged children with CF is necessary. Some groups have defined it as any change in the usual respiratory symptoms.
The greater the number of exacerbations in the first 2 years of life, the more the disease progresses.30 If case of respiratory infection, a respiratory culture must be ordered immediately along with initiation of antibiotherapy based on the age of the patient, the clinical presentation and previous microbiological results15 for a duration of 2–4 weeks.
If there is no improvement despite adequate treatment, it is possible that the infection is caused by microorganisms resistant to the prescribed antibiotics (in this case, assess the need for flexible bronchoscopy and bronchoalveolar lavage), or that other diseases are at play, such as gastro-oesophageal reflux, swallowing problems, etc.14,15,30 Chest physical therapy and adequate nutrition and hydration should never be neglected.
Treatment of the underlying CFTR protein defectTreatments are currently available that target the underlying defect that causes the disease. These new therapies aim to improve CFTR function using compounds that act on the protein, which may be correctors (class II) or potentiators (classes III and IV). Ivacaftor (Kalydeco®) is a potentiator that has proven to be efficacious (improvements in lung function and body mass index, and reductions in the chloride concentration in sweat and of the number of exacerbations by up to 52%) and safe in patients with class III mutations.31 The dosage consists of one 150mg tablet every 12h, to be ingested with fatty foods. In Spain, its used is approved starting at age 6 years, and in the United States starting at 12 months.
The combination of a CFTR corrector and a CFTR potentiator, lumacaftor+ivacaftor (Orkambi®), is a new strategy for treatment of patients homozygous for the F508del mutation (class II) that has been shown to improve lung function, the frequency of exacerbations and quality of life.32 The dosage consists of 2 tablets every 12h. It is authorised for use starting at age 2 years in the United States, but is not yet available in Spain.
Follow-up of the newborn with a diagnosis of cystic fibrosis through neonatal screeningProgrammed visits and annual evaluationTable 5 shows the schedule of routine visits recommended in patients who are asymptomatic or clinically stable, although in practice it should be individualised based on the symptoms and the needs of the family.14
If something happens or questions arise, families should be able to contact the care team and be acquainted with the care protocol. Should there be any changes in symptoms suggestive of a pulmonary exacerbation, the care team should facilitate performance of a respiratory culture and determine whether empirical antibiotherapy is indicated.33
Ancillary diagnostic testsA chest radiograph is currently recommended in case a complication is suspected or of moderate to severe pulmonary exacerbation.
Chest computed tomography (CT) is the gold standard among imaging tests for the early detection of initial abnormalities (air trapping, bronchiectasis) and their progression, as it is more sensitive than chest radiography.
Bronchiectasis can be found in up to 50% of CF patients by age 5 years.34,35 The CT protocol for assessment of CF should include inspiratory and expiratory views with low-dose radiation. Most hospitals perform a routine CT scan starting at age 2 years (without anaesthesia) and every 2 years thereafter at the time of the annual evaluation, while others do the initial scan at age 4 to 5 years with less frequent follow-up scans.
Lung function in early childhoodSpirometry is the most commonly used technique to assess lung function. The forced expiratory volume in 1 second (FEV1) is useful to define the presence of an exacerbation and the response to treatment. However, there is evidence that even in the presence of structural abnormalities, lung function may be normal in preschool-aged children.
Lung clearance indexThe lung clearance index is a new and very sensitive test that detects abnormalities earlier than spirometry and can be used to assess ventilation inhomogeneity.36 At present it is used in clinical trials.37,38
ConclusionsNeonatal screening for CF offers a unique opportunity for early diagnosis that has been proven to improve nutritional status, lung function and survival and to reduce hospital admissions, all of which may reduce health care costs. It is widely implemented worldwide and in Spain. Screening can only have beneficial effects if it is followed by strict adherence to the established standards of care, which is why we thought it was essential to publish these guidelines for the diagnosis and follow-up of patients with CF identified through neonatal screening.
Conflicts of interestThe authors have no conflicts of interest to declare.
María José Alonso: Instituto de Biología y Genética Molecular (IBGM), Universidad de Valladolid, Valladolid.
Marina Álvarez: Paediatric Gastroenterology, Hepatology and Nutritional Support Unit, Hospital Universitari Vall d’Hebron, Barcelona.
Anselmo Andrés Martín: Department of Paediatric Pulmonology, Department and Clinical Management Unit of Paediatrics, Hospital Universitario Virgen Macarena, Seville.
María Isabel Barrio Gómez de Agüero: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Universitario La Paz, Madrid
María Jesús Cabero Pérez: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Marqués de Valdecilla, Santander.
Pilar Caro Aguilera: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Materno-Infantil Universitario de Málaga, Malaga.
María Cols Roig: Paediatric Pulmonology and Cystic Fibrosis Unit, Department of Paediatrics, Hospital Sant Joan de Déu, Barcelona.
Isidoro Cortell Aznar-Pérez: Department of Paediatric Allergy and Pulmonology, Hospital Universitario La Fe, Valencia.
Jordi Costa Colomer: Paediatric Pulmonology and Cystic Fibrosis Unit, Department of Paediatrics, Hospital Sant Joan de Déu, Barcelona.
Isabel Delgado Pecellín: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Universitario Virgen del Rocío, Seville.
Amparo Escribano Montaner: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Clínico Universitario de Valencia, Universitat de València, Valencia.
Joan Figuerola Mulet: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Universitario Son Espases, Palma de Mallorca.
Gloria García Hernández: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Universitario 12 de Octubre, Madrid.
Pilar Guayarte: Paediatric Gastroenterology Unit, Hospital Universitario Parc Taulí, Sabadell.
David Gil Ortega: Paediatric Gastroenterology, Hepatology and Nutrition Unit, Hospital Universitario Virgen de la Arrixaca, Murcia.
David Gómez Pastrana: Paediatric Pulmonology Unit, Hospital de Jerez, Jerez de la Frontera.
Adelaida Lamas Ferreiro: Cystic Fibrosis Unit, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Madrid.
José Luis Marín Soria: Neonatal Screening Laboratory of Catalonia, Hospital Clinic, Barcelona.
Carlos Martín de Vicente: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Universitario Miguel Servet, Zaragoza.
Martín Navarro Merino: Department of Paediatric Pulmonology, Department and Clinical Management Unit of Paediatrics, Hospital Universitario Virgen Macarena, Seville.
Concepción Oliva Hernández: Hospital Universitario Nuestra Señora de la Candelaria, Santa Cruz de Tenerife.
Javier Pérez Frías: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Materno-Infantil Universitario de Málaga, Malaga.
Estela Pérez Ruiz: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Materno-Infantil Universitario de Málaga, Malaga.
Sandra Rovira Amigo: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Vall d’Hebron, Barcelona.
Antonio Salcedo Posadas: Cystic Fibrosis Unit, Hospital Universitario del Niño Jesús-Hospital Universitario Gregorio Marañón, Madrid.
Manuel Sánchez-Solís: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Universitario Virgen de la Arrixaca, Murcia.
Josep Sirvent Gómez: Paediatric Pulmonology Unit, Hospital Materno Infantil, Complexo Hospitalario Universitario A Coruña, A Coruña.
Carlos Vázquez Cordero: Paediatric Pulmonology and Cystic Fibrosis Unit, Hospital Universitario Cruces, Bilbao.
José Ramón Villa Asensi: Pulmonology and Cystic Fibrosis Unit, Hospital Universitario del Niño Jesús, Madrid.
Please cite this article as: Gartner S, Mondéjar-López P, Asensio de la Cruz Ó, Grupo de Trabajo de Fibrosis Quística de la Sociedad Española de Neumología Pediátrica. Protocolo de seguimiento de pacientes con fibrosis quística diagnosticados por cribado neonatal. An Pediatr (Barc). 2019;152:251.
The names of the authors of the Cystic Fibrosis Working Group of the Spanish Society of Pediatric Pulmonology are listed in Appendix A.
- Documento de manejo clínico del paciente pediátrico con infección por SARS-CoV-2.
(Actualizado el 26 de Noviembre de 2020) - Test de diagnóstico rápido en las consultas de Pediatría de Atención Primaria y Urgencias Pediátricas en la era COVID-19: más que una recomendación
(Actualizado el 21 de septiembre de 2020) - Consenso nacional sobre diagnóstico, estabilización y tratamiento del Síndrome Inflamatorio Multisistémico Pediátrico vinculado a SARS-CoV-2 (SIM-PedS).
(Actualizado el 27 de Julio de 2020) - Recomendaciones para el manejo del recién nacido en relación con la infección por SARS-CoV-2.
(Actualizado el 27 de Mayo de 2020) - Manejo del paciente pediátrico ante sospecha de infección por el nuevo coronavirus SARS-CoV-2 en atención primaria (COVID-19). AEPap-SEIP/AEP-SEPEAP.
(Actualizado el 27 de Abril de 2020) - Propuesta de adaptación de las recomendaciones de reanimación cardiopulmonar pediátrica avanzada a la infección por coronavirus.
(Actualizado el 11 de Abril de 2020)


Anales de Pediatría (English Edition) follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals