
La acondroplasia requiere un seguimiento multidisciplinario, con el objetivo de prevenir y manejar las complicaciones, mejorar la calidad de vida y favorecer su independencia e inclusión social. Esta revisión se justifica por las múltiples publicaciones generadas en los últimos años que han llevado a cabo un cambio en su gestión. Se han desarrollado diferentes guías y recomendaciones, entre las que destacan la realizada por la Academia Americana de Pediatría en 2005 recientemente actualizada (2020), la guía japonesa (2020), el primer Consenso Europeo (2021) y el Consenso Internacional sobre el diagnóstico, abordaje, enfoque multidisciplinario y manejo de individuos con acondroplasia a lo largo de la vida (2021). Sin embargo, y a pesar de estas recomendaciones, actualmente existe una gran variabilidad a nivel mundial en el manejo de las personas con acondroplasia, con consecuencias médicas, funcionales y psicosociales en los pacientes y sus familias. Por ello, es fundamental integrar estas recomendaciones en la práctica clínica diaria, teniendo en cuenta la situación particular de cada sistema sanitario.
Achondroplasia requieres a multidisciplinary follow-up, with the aim of preventing and managing complications, improving the quality of life and favoring their independence and social inclusion. This review is justified by the multiple publications generated in recent years that have carried out a change in its management. Different guidelines and recommendations have been developed, among which the one made by the American Academy of Pediatrics in 2005 recently updated (2020), the Japanese guide (2020), the first European Consensus (2021) and the International Consensus on the diagnosis, approach multidisciplinary approach and management of individuals with achondroplasia throughout life (2021). However, and despite these recommendations, there is currently a great worldwide variability in the management of people with achondroplasia, with medical, functional and psychosocial consequences in patients and their families. Therefore, it is essential to integrate these recommendations into daily clinical practice, taking into account the particular situation of each health system.
La acondroplasia es la displasia esquelética más frecuente que asocia talla baja desproporcionada. Se estima una incidencia de 1/10.000-1/30.000 recién nacidos vivos, sin diferencias por sexo o raza. Un total de 360.000 personas en el mundo tienen esta enfermedad rara1. La prevalencia mundial es 4,73/100.000, siendo en Europa 3,62/100.0002. Dicha displasia se produce por una variante patogénica en heterocigosis en el gen FGFR3, situado en el cromosoma 4p16.3, que codifica el receptor del factor de crecimiento fibroblástico tipo 3, que se traduce en una ganancia de función por parte del receptor mediante la activación de la vía de señalización MAPK, inhibiendo la proliferación y diferenciación de los condrocitos en la placa crecimiento3,4. Otras vías de señalización como STAT, Wnt/β-catenina, PI3K/AKT y PLCϒ están también implicadas5.
Esta alteración genera un conjunto de comorbilidades esqueléticas derivadas de la afectación de la osificación endocondral (huesos largos y esqueleto axial). Esto se traduce en un fenotipo característico con talla baja disarmónica, debida al acortamiento rizomélico de miembros y una altura en sedestación cercana a la talla estándar6. Los individuos afectos presentan macrocefalia, frente abombada, hipoplasia medio facial y mano en tridente. La talla final media adulta esperada es de 130cm (con rango de 120-145cm) en varones y 125cm (con rango de 115-137cm) en mujeres, de acuerdo a las tablas de crecimiento específicas7,8. Dicha talla implica una altura media entre –6 y –7 desviaciones estándard (DE) por debajo de la media respecto a la talla de población no afecta de acondroplasia6.
Como consecuencia de las manifestaciones esqueléticas se derivan otras alteraciones neurológicas (estenosis del foramen magnum, radiculopatía lumbar), otorrinolaringológicas y/o psicológicas, entre otras.
Algunas investigaciones indican que el FGFR3 es expresado en tejidos diferentes al esquelético, lo que sugiere que la actividad del FGFR3 podría contribuir a las manifestaciones extraesqueléticas características de población acondroplásica.
Es por ello que resulta necesario realizar un seguimiento multidisciplinar, con el objetivo de prevenir y manejar las complicaciones, mejorar su calidad de vida y favorecer su independencia e inclusión social9,10.
Con objeto de optimizar dicho seguimiento se han desarrollado diferentes guías y recomendaciones, entre las que destaca la realizada por la Academia Americana de Pediatría en 2005 recientemente actualizada9, la guía japonesa11, el primer Consenso Europeo10 y el Consenso Internacional sobre el diagnóstico, abordaje multidisciplinar y manejo de los individuos con acondroplasia a lo largo de la vida1. Sin embargo, y a pesar de dichas recomendaciones, actualmente existe una gran variabilidad mundial en el manejo de las personas con acondroplasia, con consecuencias médicas, funcionales y psicosociales en los pacientes y sus familias1.
Por tanto, resulta imprescindible integrar dichas recomendaciones en la práctica clínica diaria teniendo en cuenta la situación particular de cada sistema sanitario.
DiagnósticoDiagnóstico prenatalCuando no existen antecedentes familiares, se puede sospechar a partir de la semana 22 de edad gestacional, aunque es difícil de detectar antes de la semana 26. Este hecho implica un diagnóstico ecográfico prenatal tardío en el tercer trimestre, después del screening de la semana 20, por una ecografía realizada por otro motivo.
Los hallazgos ecográficos sugestivos de acondroplasia son el acortamiento rizomélico, incurvación del fémur y desaceleración del crecimiento del fémur a partir de la semana 26, proporción longitud fémur/longitud pie <1, braquidactilia con mano en tridente, tórax estrecho sin hipoplasia pulmonar, perfil facial (frente prominente, hipoplasia mediofacial y puente nasal aplanado), crestas iliacas redondeadas y planas y polihidramnios.
El diagnóstico genético es posible por biopsia de vellosidad corial a partir de la semana 12 o amniocentesis a partir de la semana 16, en los casos de alta sospecha durante el primer trimestre, que acontece en hijos de parejas con acondroplasia o en hijos de parejas sanas con antecedente de hijo previo con acondroplasia. Se están utilizando formas no invasivas para el estudio de la variante patogénica en FGFR3 en ADN fetal extraído de sangre materna (en caso de madre no afecta), como alternativa a la amniocentesis, que, acompañado de la ecografía, puede ser una forma segura y costo-efectiva de diagnóstico no invasivo de acondroplasia12.
Es recomendable y necesaria una visita prenatal con un genetista clínico y un pediatra experto, tras la sospecha diagnóstica intraútero, para proporcionar la información correcta. Los profesionales responsables de dar la primera noticia y el asesoramiento genético deben estar cualificados para hacerlo. Este proceso de comunicación debe llevarse a cabo en presencia de ambos progenitores, en un lugar adecuado y con el tiempo necesario. Han de tener conocimientos actualizados en torno a los recursos disponibles y la formación para llevar a cabo el afrontamiento. La participación de representantes de asociaciones de pacientes en el proceso puede ser beneficioso (http://gat-atenciontemprana.org/wp- content/uploads/2019/05/primera-noticia-web.pdf) (http://assets.comitedebioetica.es/files/documentacion/consejo-genetico-prenatal.pdf).
El parto se planificará en el lugar y momento adecuado para manejar las complicaciones potenciales que pueda tener el recién nacido a corto plazo. Se hará de forma coordinada con un equipo que incluya: obstetra con experiencia en esta patología, genetista, neonatólogo y pediatra. En cuanto al parto generalmente se recomienda cesárea electiva, debido a la desproporción pélvico-fetal por la macrocefalia y el riesgo de compresión espinal cervical en la fase de expulsivo debido a un foramen magnum restrictivo9.
Diagnóstico clínico-radiológicoLa sospecha clínica de acondroplasia en el neonato se establece con los siguientes hallazgos: macrocefalia, frente plana ancha, raíz nasal plana, mano en tridente, talla baja disarmónica con un patrón de acortamiento rizomélico en extremidades (húmero y fémur, segmentos proximales desproporcionadamente más cortos que los segmentos distales), aumento de pliegues en extremidades, tronco largo y estrecho e hipotonía neonatal13.
Ante la sospecha clínica, no se recomienda de rutina un mapa óseo completo, sino radiografía de cráneo anteroposterior y lateral, columna vertebral a nivel toracolumbar lateral, tórax y pelvis anteroposterior, extremidad superior e inferior anteroposterior y mano izquierda dorsopalmar1. Radiológicamente es característico de esta patología una forma cuadrada de la pelvis con acetábulo horizontal y escotadura sacrociática pequeña, pedículos vertebrales cortos con estrechamiento interpedicular desde la zona torácica baja a la lumbar, acortamiento rizomélico (proximal) de los huesos largos, radiolucencia femoral proximal, forma de galón de la epífisis femoral distal, hipercifosis toracolumbar y falanges proximales y medias cortas9.
Diagnóstico genéticoEl diagnóstico de acondroplasia se establece por las características clínicas y radiológicas, si bien, el estudio genético es necesario para la confirmación molecular y el asesoramiento reproductivo1.
El 99% de los pacientes presentan la variante patogénica c.1138G>A en heterocigosis en el gen FGFR3 y un 1% la variante c.1138G>C. La penetrancia es del 100% y el patrón de herencia autosómico dominante. En el 80% de los casos es una variante de novo; no heredada de los progenitores, y asociada a una edad paterna avanzada (generalmente mayor de 35 años). La acondroplasia forma parte de alteraciones minoritarias denominadas trastornos RAMP —trastornos recurrentes, autosómicos dominantes, masculinos, de efecto de edad paterna— los cuales podrían surgir debido a su efecto de selección positiva en la espermatogonia. No obstante, el riesgo de recurrencia de la enfermedad en la descendencia de una pareja no afecta de acondroplasia con un hijo afecto se estima en un 1%, por la posibilidad de mosaicismo germinal9. Si un progenitor está afecto tendrán un 50% de posibilidades de que su hijo lo esté; si ambos están afectos tienen un 25% de posibilidades de tener un hijo sano, 50% de un hijo con acondroplasia y en un 25% de los casos una forma homocigota letal.
Variaciones patogénicas en el mismo gen son las causantes de enfermedades incluidas dentro del diagnóstico diferencial de esta enfermedad, la displasia tipo SADDAN (acondroplasia grave con retraso en el desarrollo y acantosis nigricans) y la hipocondroplasia (entidad similar a la acondroplasia, pero con un fenotipo más leve)3.
El diagnóstico genético, tras la sospecha clínica, se hará por estudio dirigido de las dos variantes de FGFR3 relacionadas con la acondroplasia. Si bien, en casos de presentación atípica, el diagnóstico puede derivar del uso de técnicas de secuenciación masiva como paneles de genes relacionados con displasias óseas, exoma clínico o exoma completo1.
SeguimientoEs necesario un seguimiento multidisciplinar desde el diagnóstico, ya que asocia una tasa incrementada de morbilidad y complicaciones que pueden llegar a ser mortales, beneficiándose de un seguimiento programado. Esta atención multidisciplinar debe llevarse a cabo en un centro de referencia de displasias esqueléticas o en el que haya profesionales expertos en acondroplasia. En el Primer Consenso Europeo de acondroplasia y en el Consenso Internacional, ambos publicados en 2021, enfatizan en este punto, especialmente en los dos primeros años de vida1,10. El principal objetivo es permitir la anticipación, identificación y tratamiento de los posibles problemas, favoreciendo la autonomía, el bienestar y la independencia. Este seguimiento es recomendable también en la adolescencia, incluyendo transición a la edad adulta, asesoramiento genético, bienestar psicosexual y manejo del embarazo10.
La Asociación Americana de Pediatría establece un cronograma con el seguimiento específico en cada etapa de la vida (tabla 1), requiriendo en cada visita: antropometría, exploración física, neurológica y revisión por Otorrinolaringología. No se debe olvidar advertir signos de complicaciones graves para anticiparnos a ellas9. La vacunación se realizará según el programa de inmunización específico de cada país1.
Cronograma adaptado de la Asociación Americana de Pediatría (2020) para el seguimiento de pacientes con acondroplasia17
| Nacimiento-2 años | 2-13 años | Adolescentes | Adultos | |
|---|---|---|---|---|
| Antropometría (talla, peso, perímetro craneal) | X | X | X | X |
| Examen físico | X | X | X | X |
| Exploración neurológica | X | X | X | X |
| Evaluación del desarrollo | X | X | ||
| Neuroimagen | X | X | X | X |
| (entre 6 meses y 1 año o ante clínica sugestiva) | (si indicación) | (si indicación) | (si indicación) | |
| Polisomnografía | X | X | X | X |
| (antes del año, a ser posible antes del mes de vida) | (si indicación) | (si indicación) | (si indicación) | |
| Evaluación otorrinolaringológica | X | X | X | X |
| Radiografía para cifosis, genu varo o arqueamiento de extremidades | X | X | X | X |
| (si indicación) | (si indicación) | (si indicación) | ||
| Advertir signos de posibles complicaciones | X | X | X | X |
| Prevención y/o abordaje de obesidad | X | X | X | |
| Informar sobre grupos de apoyo y soporte psicosocial | X | X | X | X |
| Asesoramiento genético | X | X |
En esta etapa de la vida es primordial el seguimiento exhaustivo, si es posible, cada 2-4 meses1.
Antropometría y proporcionalidad corporalLa exploración antropométrica debe incluir parámetros básicos como el peso, la talla, la talla en sedestación, el perímetro cefálico y la braza. De la relación entre estas variables se deben registrar el IMC y variables relacionadas con la proporcionalidad corporal entre las que destacan la relación entre el tronco y el segmento inferior, la altura y el perímetro cefálico, la altura y la braza. Dichas variables deben ser recogidas por personal entrenado, y analizadas según las curvas de crecimiento de cada paciente y comparadas con las tablas específicas para población con acondroplasia en cohorte europea, siendo las últimas publicadas y recomendadas para el seguimiento, las tablas de Neumeyer et al.8, aunque previamente se han publicado otras tablas ajustadas a población con acondroplasia7,14,15,16.
Neurología/NeurocirugíaLas personas con acondroplasia tienen más riesgo de complicaciones a nivel neurológico debido a que la base del cráneo es de osificación endocondral y, por lo tanto, se afecta la unión craneocervical. Pueden presentar estenosis del foramen magno y del canal vertebral cervical superior, así como una posición y forma anormal del odontoides e hiperlaxitud ligamentosa en la médula cervical17.
Se ha observado un aumento de la incidencia de muerte súbita, con respecto a la población general, del 7,5% en el primer año de vida, que parece deberse a la compresión del troncoencéfalo y la médula a nivel de foramen magno17. También tienen un riesgo aumentado de apnea del sueño central, aunque es más frecuente la obstructiva18,19. Por este motivo es importante realizar una polisomnografía el primer año de vida para descartar apneas no percibidas por los padres o en cualquier momento ante la presencia de síntomas1. Algunos autores sugieren la evaluación del riesgo neurológico a través de polisomnografía, recomendando su realización lo antes posible, a ser posible al mes de vida9.
Es fundamental realizar una exploración neurológica minuciosa con el objetivo de anticipar las posibles complicaciones que pueden tener, al menos hasta los 2 años de vida, siendo menos frecuentes estos problemas en edades posteriores1.
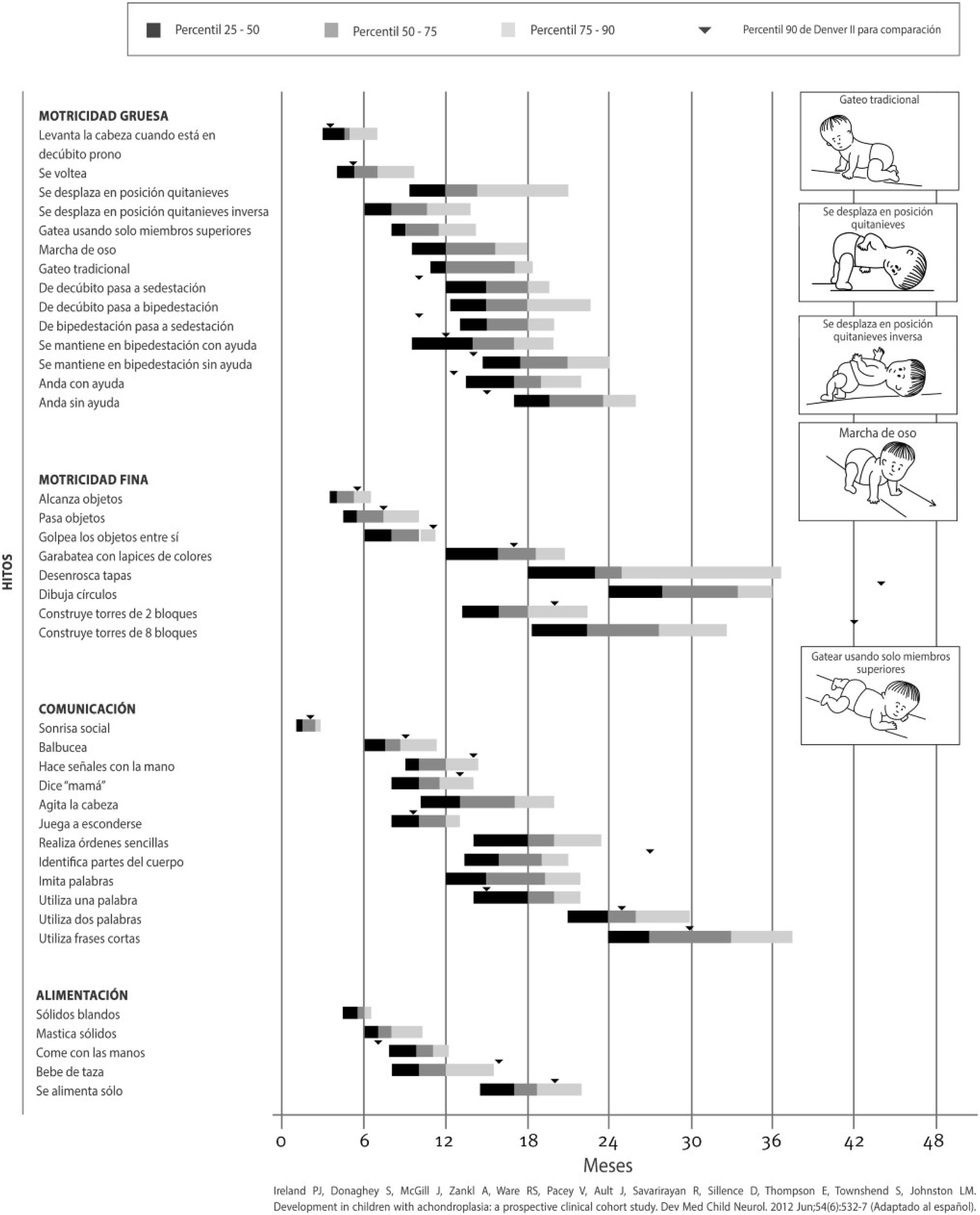
Los niños con acondroplasia presentan un retraso en los hitos motores, debido a las particularidades de su anatomía, pero sin la afectación en otras áreas de neurodesarrollo, por lo que si observamos un retraso de un hito no motor puro hay que sospechar una posible complicación. Existen gráficas adaptadas a la adquisición de los hitos motores, destacando la deambulación con una media de 20 meses9 (fig. 1).
 traducida al español y reproducida con permiso de la autora40.")
Herramienta de evaluación del desarrollo en niños con acondroplasia creada por Ireland et al. (2012) traducida al español y reproducida con permiso de la autora40.
La macrocefalia con líquido extraaxial excesivo y la ventriculomegalia asintomática son una variante de la normalidad en esta entidad que no requiere abordaje quirúrgico. La hidrocefalia puede ser una complicación, pero es poco frecuente11.
La monitorización del perímetro craneal se recomienda cada 2-4 meses durante el primer año. Un crecimiento excesivo del perímetro craneal junto a otros síntomas o signos neurológicos deben hacer sospechar compresión craneocervical o hidrocefalia1. Es importante explorar la fontanela junto con los siguientes signos y síntomas de alarma: dificultad para la deglución, vómitos, hipertensión, bradicardia, apnea prolongada durante el sueño, cianosis, asimetría de movimientos de miembros inferiores, disminución de tono axial y periférico, pérdida de habilidades adquiridas, incremento de reflejos, respuesta plantar extensora o preferencia por un miembro superior1,9,11. Es necesario evitar los traumatismos craneoencefálicos y cervicales, siendo recomendable el uso de asientos adecuados con soporte cervical9. Se debe informar de la importancia de evitar situaciones como la realización de volteretas, uso de trampolines y deportes de impacto donde puede colisionar la cabeza.
Con respecto a la prueba de imagen, la tomografía computarizada (TAC) posibilita comparar el tamaño de foramen magno con valores de referencia, no precisa sedación, pero no permite analizar el troncoencéfalo y la médula superior con tanta definición como la resonancia. La TAC está siendo sustituida por la resonancia magnética (RM), con la desventaja de requerir anestesia20. Visualiza mejor tronco encéfalo y médula superior y ayuda a evidenciar si existe compresión dinámica del cordón y alteración del flujo de líquido cefalorraquídeo, que es el mejor indicador de la necesidad de intervención9. En 2021 se publicó la primera clasificación basada en resonancia magnética para screening de estenosis del foramen magno, siendo el estadio 0 normal y el 4 el más grave21.
Aunque la revisión de las guías no permite una recomendación clara, puede deducirse que para la evaluación del riesgo neurológico sería recomendable la realización de polisomnografía lo antes posible, incluso en el primer mes de vida. La realización de prueba de imagen (TAC versus RM, según disponibilidad, pero siendo más recomendable la RM) se aconseja entre los 6 meses y el año de vida, así como ante la presencia de síntomas sugestivos.
Se recomienda la derivación a Neurocirugía para valoración de una descompresión del foramen magno ante clínica sugestiva, una exploración neurológica anormal, un estancamiento ponderal no justificado, una polisomnografía patológica o una prueba de imagen que muestre foramen magno pequeño, deformación de la médula superior o pérdida de líquido cefalorraquídeo alrededor de la médula espinal9. Si la cirugía es necesaria, deberá realizarla un centro de referencia con un equipo de Neurocirugía con experiencia previa en este procedimiento en niños con acondroplasia1, así como con un equipo de anestesia especializado (ver más adelante en el apartado de anestesia).
OtorrinolaringologíaPresentan un riesgo aumentado de apneas obstructivas debido a la hipertrofia amigdalar, glosoptosis, laxitud y redundancia de la pared faringolaríngea18,19. Debido a la anatomía del conducto auditivo tienen más riesgo de padecer otitis durante la infancia, por eso es importante un seguimiento otorrinolaringológico anual, ya que pueden asociar pérdida de audición por este motivo. Hay que confirmar que se ha realizado el cribado auditivo al nacimiento y completar estudio en aquellos que el resultado esté alterado13. Ante una infección de vías altas o irritabilidad hay que descartar siempre una infección de oído medio y no demorar su tratamiento. Pueden asociar retraso del lenguaje secundario a la pérdida de audición9 y esto será criterio de derivación a terapia de refuerzo del lenguaje o logopedia1.
TraumatologíaA nivel del raquis uno de los problemas más frecuentes en los primeros años es la cifosis toracolumbar (90-95%). Los padres deberán ser instruidos para proporcionar un adecuado soporte de espalda hasta que el tronco adquiera fuerza muscular, evitando asientos donde este soporte no sea adecuado. Para ello puede ser necesaria la ayuda de un terapeuta ocupacional o un rehabilitador de manera temprana. Durante la alimentación, espalda y cabeza deben estar alineadas mediante el uso de cojines firmes. Si la cifosis toracolumbar es leve mejora cuando comienza a andar, y si es moderada (20-40 grados) un año después de la deambulación requiere revisión por Traumatología, siendo excepcional que precise cirugía. En aquellos que persiste (10%) presentarán evolutivamente más riesgo de neuropatía/estenosis lumbar en etapas posteriores1,9,13.
En las extremidades destaca la hipotonía y la hiperlaxitud articular, lo cual favorece los tropiezos y las caídas al iniciar la deambulación, información de la que deben disponer los padres para extremar precauciones, aunque animándolos a que permanezcan activos1. Destaca también la rotación externa de caderas, que suele desaparecer de forma espontánea cuando inician la deambulación9. Pueden presentar gateos peculiares que no son patológicos.
AnestesiaEs de especial importancia que el anestesista tenga experiencia en esta patología, ya que precisan de control cervical muy cuidadoso (vía área difícil y riesgo de compresión craneocervical). Además, la medicación debe ser dosificada por peso, no por edad, y puede ser dificultoso el acceso venoso por la imposibilidad de extender el codo por completo. Por último, es recomendable evitar la anestesia epidural o intradural a no ser que se haya realizado una prueba de neuroimagen en la que se observe un adecuado espacio y no haya signos de compromiso neurológico.
Seguimiento de 2 a 13 añosEn esta etapa las revisiones serían recomendables cada 6-12 meses, individualizando cada caso 1.
AntropometríaRecomendable en cada visita.
Neurología/NeurocirugíaSe recomienda continuar vigilando los signos y síntomas de compresión cervicomedular.
Hay que recordar que el cierre fontanelar es tardío y se produce de manera habitual hacia los 5- 6 años de edad.
TraumatologíaA nivel del raquis, se recomienda continuar monitorizando la cifosis toracolumbar y la hiperlordosis lumbar, completando el seguimiento con la exploración neurológica.
A nivel de miembro superior debe monitorizarse el rango articular de codos debido al flexo característico.
A nivel de miembro inferior, es frecuente la desviación en varo de la tibia afectando al 40-70% de los pacientes22,23. Dicha deformidad en varo puede desencadenar síntomas, entre los que se incluye el dolor mecánico y la limitación en la marcha y otras actividades físicas. Por ello, se debe realizar de manera protocolaria una evaluación clínica seriada del miembro inferior mediante exploración en decúbito prono, supino, bipedestación y durante la marcha con el objetivo de analizar la deformidad de manera integral valorando la deformidad en varo y recurvatum, la torsión tibial interna y la inestabilidad mediolateral característica1. Si dicha deformidad es sintomática y/o progresiva la exploración se completará mediante estudio radiológico con telerradiología anteroposterior y lateral en carga de miembros inferiores. No se recomienda el uso de ortesis para tratar dicha comorbilidad1.
Otorrinolaringología/NeumologíaSe requiere mantener la monitorización de la audición, las infecciones de oído y el desarrollo del lenguaje. Si precisan la realización de una timpanostomía, el otorrino debe buscar signos de bulbo yugular alto, ya que tienen más riesgo de disección24. En esta época hay que seguir vigilando las apneas, y si son obstructivas por hipertrofia amigdalar valorar la amigdalectomía, aunque no es el único factor (presentan una morfología craneofacial peculiar), por lo que a veces no se resuelven tras la cirugía19. Ante un retraso del lenguaje a una edad superior a los 2 años se recomienda excluir problemas de audición.
Si se sospecha apnea obstructiva requerirá también valoración por Neumología. La enfermedad pulmonar restrictiva es rara, siendo la obstructiva más común. Pueden asociar enfermedad por reflujo gastroesofágico, en especial los que presentan problemas neurorrespiratorios9.
ObesidadEs un problema común ya previo a la epidemia mundial de obesidad, por lo que se postula que factores de la propia patología pueden favorecerla. Es muy precoz y de predominio abdominal. Estos pacientes tienen limitación para la actividad física, cierta compulsión por la comida, un metabolismo basal disminuido comparado con personas de su sexo y edad y además parece que la propia variante en FGFR3 favorece la diferenciación de la célula mesenquimal común hacia adipocitos25. Es muy importante la prevención y vigilancia, en cada consulta, del peso y el IMC en tablas específicas26, puesto que empeora los problemas neurológicos y traumatológicos, las apneas y el riesgo de enfermedades cardiovasculares a una edad más precoz, principal causa de mortalidad en la acondroplasia. El tratamiento es dietético junto a ejercicio físico adecuado a cada paciente1 y apoyo psicológico si precisan25.
Atención tempranaSe entiende por atención temprana el conjunto de intervenciones dirigidas a la población infantil de 0 a 6 años, a la familia y al medio, que tienen por objeto atender lo más precozmente posible las necesidades transitorias o permanentes que presentan o que tienen el riesgo de presentar. La coordinación interdisciplinar es una exigencia necesaria para el desarrollo de la intervención en atención temprana. Para ello debe haber una relación técnica efectiva, una comunicación fluida y una participación activa por parte de todos los profesionales que trabajan de forma directa o indirecta. Es necesaria la coordinación interdisciplinar entre sistema sanitario, educativo, social y de atención temprana, así como de asociaciones de pacientes.
En atención temprana de niños con acondroplasia se deben trabajar precozmente todas las áreas del desarrollo. Es importante la valoración inicial y evolutiva en las áreas más destacadas que podrían estar afectadas, como área motora gruesa y fina, área del lenguaje y comunicación, área cognitiva y área de autonomía personal y relación social (https://www.infad.eu/wp-content/uploads/ConOtraMirada_guia_acondroplasia.pdf).
- 1.
Área motora gruesa: La intervención en esta área se llevará a cabo a través de fisioterapia con 2 objetivos primordiales, que son: igualar el desarrollo motor grueso a su edad cronológica y prevenir las complicaciones que puedan derivarse. Es importante tener algunas consideraciones específicas, como la de evitar el gateo. Una vez lograda la deambulación, por el riesgo de hiperlordosis lumbar, deben evitarse posturas de decúbito prono con elevación de la cabeza. Se han de inculcar maneras correctas para pasar del suelo a la bipedestación, evitando la posición intermedia con hiperextensión de las rodillas.
- 2.
Área de lenguaje y comunicación: Los niños con acondroplasia no suelen tener serios problemas de lenguaje, pero sí se ha de tener especial vigilancia de su adquisición, cuya aparición no debe retrasarse más allá de los 2 años, ya que un retraso puede poner de manifiesto el infradiagnóstico de hipoacusia. La intervención del lenguaje se llevará a cabo a través de logopedia con los objetivos generales de educar la fonoarticulación y conseguir una adecuada expresión y comunicación. Los objetivos específicos pretenden abordar las posibles dificultades respiratorias ocasionadas por el estrechamiento de vías nasales y la motricidad fina en los órganos bucofonatorios para conseguir una pronunciación correcta de determinados fonemas, así como conseguir una masticación y deglución normal, teniendo en cuenta algunas consideraciones específicas como el prognatismo mandibular, el paladar ojival y la macroglosia que conllevan mala oclusión de los maxilares, dificultad para la movilidad de la lengua y por tanto dificultad para la articulación y pronunciación de determinados fonemas. Con el fin de establecer un habla y audición adecuadas se trabajan actividades relacionadas con la respiración, soplo, discriminación auditiva, praxias bucofonatorias, articulación, impostación vocal, prosodia, ritmo y deglución.
- 3.
Área motora fina: Los objetivos generales en esta área son mejorar las habilidades manipulativas y el desarrollo de la percepción óculo-manual. Los objetivos específicos son igualar el desarrollo motor fino a su edad cronológica, favoreciendo el desarrollo de la pinza digital y el desarrollo de la grafomotricidad. En esta área se tendrá en cuenta la necesidad de adaptaciones como el uso de un lápiz más corto, adaptador, tijeras más pequeñas, papel apaisado…
- 4.
Área cognitiva: El área cognitiva no tiene por qué estar afectada, pero es importante tener en cuenta que el desfase en otras áreas de desarrollo puede alterar el progreso de esta. Pueden ser determinantes: el retraso en el desarrollo motor grueso en los primeros años, las dificultades en la motricidad fina, el retraso en el habla, los problemas de autoimagen y las dificultades en autonomía personal. El trabajo en el área cognitiva se puede realizar de forma transversal con el resto de áreas.
- 5.
Área de autonomía personal y social: Como objetivos generales se tienen en cuenta la adquisición progresiva de autoconfianza y una imagen ajustada y positiva, siendo objetivos específicos la consecución de la confianza en las propias posibilidades para adaptarse al medio, adquiriendo coordinación y control dinámico en el juego y en actividades de la vida cotidiana, utilizando elementos adaptativos adecuados para una mejor autonomía.
Es necesario un seguimiento por un odontólogo desde edades tempranas y con frecuencia, el uso de ortodoncia24.
Adolescentes-adultos jóvenesEn esta etapa las revisiones serán individualizadas en cada caso1.
Es necesario mantener una monitorización endocrinológica (obesidad), otorrinolaringológica (apneas, audición), odontológica y traumatológica. A nivel neurológico pueden tener signos de compresión nerviosa debida a estenosis espinal, por lo que es importante explorar los reflejos, el tono, la sensibilidad y la incontinencia. Se debe investigar sobre la presencia de dolor crónico, que es frecuente en estos pacientes1,9.
Hay que facilitarles el asesoramiento genético, proporcionarles información sobre anticoncepción y a las mujeres información sobre test prenatales, el embarazo y sus posibles complicaciones (restricción pulmonar, morbilidad cardiovascular, riesgo anestésico) y la recomendable finalización en cesárea9.
AdultosSe sabe que pueden tener una esperanza de vida reducida, con 10 años de vida perdidos en relación con el aumento de mortalidad cardiovascular1,25. La funcionalidad y la salud se pueden deteriorar de forma notable en la cuarta década de la vida. La calidad de vida puede verse disminuida debido a la discapacidad funcional y a los desafíos psicosociales, presentando casi un 50-60% problemas de autoestima-ansiedad10.
Apoyo psicológicoDesde el momento del diagnóstico, tanto prenatal como posnatal, hay que ofrecer apoyo psicológico, información sobre el manejo del niño, incluyendo expectativas futuras, y dar a conocer las asociaciones y grupos de apoyo, que son una ayuda importante para la familia1,10. Este proceso también debe ser llevado a cabo con los hermanos de estos pacientes, resolviendo sus preguntas, preocupaciones o conflictos que pueden surgir en la familia1. Durante la infancia y adolescencia se realizará de forma periódica una valoración funcional para identificar limitaciones en la deambulación, el autocuidado y el funcionamiento diario, proporcionando los medios necesarios para conseguir su independencia. Conforme van creciendo precisarán adaptar el domicilio, el colegio, el baño y el asiento del coche para fomentar la autonomía. Algunos adolescentes pueden evitar usar equipos adaptados en la escuela por la preocupación de que sus compañeros se burlen de ellos o les hagan preguntas incómodas, por lo que se debe ofrecer ayuda para resolver estos obstáculos, siendo los grupos de apoyo entre iguales una ayuda esencial a esta edad1. Los fisioterapeutas y terapeutas ocupacionales son un pilar muy importante en esta patología1,9. Tanto los pacientes como sus padres presentan una calidad de vida disminuida comparada con niños de su edad, por lo que hay que optimizar los recursos para mejorarla27. Se afrontará, como en todos los adolescentes, el tema de drogas, alcohol y tabaco, relaciones sexuales, reproducción, expectativas de futuro y la necesidad de vehículos adaptados, siendo esencial el apoyo psicológico9.
TratamientoCirugíaLa mayor parte de las cirugías se enmarcan en el tratamiento de las complicaciones. Entre ellas, destacan en los primeros años de vida, los procedimientos neuroquirúrgicos (estenosis del foramen magno y derivaciones ventriculoperitoneales), procedimientos otorrinolaringológicos (drenajes timpánicos, amigdalectomía y adenoidectomía) y los procedimientos ortopédicos, menos frecuentes, relacionados con la cifosis toracolumbar. En la etapa adulta destaca la cirugía dependiente de la estenosis de canal lumbar.
Merece mención referir los procedimientos ortopédicos relacionados con el alargamiento de miembros y el manejo del genu varo con el objetivo de tratar las comorbilidades esqueléticas propias de la condición.
En relación con el genu varo, la indicación quirúrgica se establece según la gravedad de la deformidad, la sintomatología asociada y la limitación funcional secundaria, habiéndose descrito diferentes técnicas entre las que destacan la osteotomía y el uso de placas de crecimiento guiadas.
En relación con el alargamiento de miembros inferiores (fémur y tibia) y miembros superiores (húmeros) se han descrito diferentes opciones, manteniendo discusión sobre su propia indicación, la edad de inicio, el método empleado y las variables resultado esperadas, destacando su alta tasa de complicaciones9,11. El alargamiento de miembros inferiores, con elongaciones de entre 14 y 40cm, pretendería un incremento de la longitud de los miembros inferiores y con ello de la talla en bipedestación, mejorando así las variables de proporcionalidad corporal, funcionalidad y calidad de vida, dependientes del segmento inferior. De la misma manera, el alargamiento de miembros superiores, con elongaciones de entre 8 y 10cm, permitiría un incremento de longitud de brazos, incrementando la braza y con ello mejorando todas las variables de proporcionalidad corporal, funcionalidad y calidad de vida, dependientes del segmento superior28–31.
Las diferentes opciones quirúrgicas suelen ser descritas de manera independiente sin ser integradas en un protocolo con objetivos claramente definidos. Estos procedimientos deben abordarse de manera integral, ser realizados en centros con experiencia en el abordaje del paciente con acondroplasia, y propuestos mediante un abordaje multidisciplinar antes y después de los mismos, teniendo en cuenta variables antropométricas, radiológicas, funcionales y relacionadas con la calidad de vida.
La ausencia de un tratamiento médico eficaz hasta hace poco tiempo, y a la espera de resultados a largo plazo de las terapias médicas actualmente autorizadas y aquellas en fase de investigación, hacen del alargamiento quirúrgico y corrección de deformidades una opción terapéutica válida en un determinado subgrupo de población con acondroplasia a definir. Queda pendiente valorar los resultados a largo plazo de las terapias médicas para determinar si la opción quirúrgica sigue siendo la única opción disponible, una opción coadyuvante a la terapia médica o queda completamente relegada.
Tratamiento médicoEn Japón está aprobada la hormona de crecimiento11, pero no ha demostrado tener un efecto relevante y no se conocen los efectos a largo plazo en las proporciones corporales1,13. Actualmente se encuentra en desuso.
En la actualidad diferentes dianas terapéuticas se centran en FGFR3, bloqueando su activación, inhibiendo su señal intracelular o aumentando su intercambio5. Entre ellas destaca el hallazgo reciente de los efectos de un análogo del péptido natriurético tipo C (vosoritida) con una vida media más larga que su forma endógena. Al unirse a su receptor, inhibe la vía MAPK (activada por la variante patogénica en el gen FGFR3) a nivel de RAF-1, incrementándose la proliferación y diferenciación de los condrocitos en la epífisis. En 2021 la Agencia Europea del Medicamento (EMA) aprobó vosoritida (voxzogo), para pacientes con acondroplasia confirmada genéticamente de 2 años de edad y mayores cuyas epífisis no se han cerrado, mediante administración subcutánea diaria. El inicio de tratamiento exige la monitorización antropométrica del peso y la talla. El tratamiento debe ser suspendido si la velocidad de crecimiento es <1,5cm/año y/o se han cerrado las epífisis. No ha sido evaluada la seguridad y eficacia de vosoritida en pacientes con fallo renal o hepático.
Inicialmente, tras un ensayo en fase 2 (estudio BMN 111-202; EudraCT number 2013-004137- 32) con 35 pacientes de 5-14 años se concluyó que la dosis que tiene el mejor perfil de eficacia y seguridad es de 15μg/kg/día, sin incrementos significativos del efecto con dosis más altas32. Dicha dosis de 15μg/kg/día ha sido utilizada posteriormente en otro ensayo clínico, aleatorizado, doble ciego en fase 3 (estudio BMN 111-301; EudraCT number, 2015-003836-11) con un total de 121 pacientes de 5-18 años, los cuales fueron aleatorizados 1:1 comparando vosorotida con placebo durante 52 semanas de tratamiento y observando un incremento de la velocidad de crecimiento anual de 1,57cm/año (IC 95% 1,22-1,93, p<0,0001) en el grupo de vosorotida respecto al grupo placebo33. En el ensayo clínico de extensión en fase 3 se han completado hasta 104 semanas (estudio BMN 111-302; EudraCT number 2017-002404- 28; NCT03424018), administrándose en todos los pacientes vosoritida a 15μg/kg/día, manteniéndose la ganancia de la velocidad de crecimiento anual durante el segundo año de tratamiento sin evidencia de taquifilaxia, siendo la velocidad de crecimiento anual en la semana 104 de 5,52 (1,77) cm/año en aquellos pacientes que desde el inicio recibieron vosorotida, y de 5,43 (2,03) cm/año en aquellos pacientes que pasaron de placebo a vosorotida. Al comparar directamente el grupo tratado con vosoritida con el grupo no tratado, el cambio observado de altura fue similar en el primer año de tratamiento, 1,73cm, como en el segundo año de tratamiento, 1,79cm. La ganancia de altura adicional durante el período de tratamiento de 2 años fue de 3,52cm más que los niños no tratados. Parece que una vez que vosoritida alcanza su máximo efecto, este se mantiene constante en el tiempo, siendo el Z-score de altura +0,44 DE (IC 95% 0,25, 0,63) en la semana 104. Además, se evidenció una mejora de las proporciones corporales, disminuyendo el índice segmento superior/segmento inferior –0,05 unidades (IC 95% –0,09, –0,01) comparado con individuos no tratados. El efecto adverso más frecuente fue la presencia de reacciones locales leves y transitorias en la zona de inyección, vómitos y acontecimientos transitorios de presión arterial disminuida, la mayor parte asintomáticos, que se resolvieron sin intervención. Se observaron un total de 14 eventos adversos en el ensayo, pero ninguno relacionado con el fármaco y no hubo eventos adversos relacionados con un crecimiento óseo desproporcionado o patología ósea34.
Al mismo tiempo se están desarrollando otros dos ensayos clínicos en relación con vosorotida. El primero, un ensayo clínico en fase 2 (BMN 111-206, EudraCT number 2016-003826-18, NCT03583697) para evaluar la seguridad y eficacia de vosoritida de 0 a 60 meses. El segundo, un ensayo clínico fase 2 (BMN 111-209, EudraCT number 2020-001055-40, NCT04554940), con 20 pacientes de 0 a 12 meses de edad, que analiza si su administración precoz en pacientes con riesgo de compresión cervicomedular que puedan precisar cirugía es eficaz y seguro35.
Se están desarrollando otras líneas de investigación entre las que destacan otro péptido natriurético (TransCon CNP) de vida media más larga, actualmente en fase 2 (NCT04085523), siendo administrado subcutáneamente de forma semanal, en niños con acondroplasia entre 2 y 10 años36,37.
Otra molécula, FGFR3 soluble (recifercept), usada como ligando de FGF, impidiendo que se una a FGFR3, actualmente en fase 2 (NCT04638153), presenta resultados prometedores tanto a nivel de deformidades esqueléticas como en la obesidad con su uso precoz37,38.
Otra molécula, desarrollada inicialmente como tratamiento oncológico, se encuentra en investigación, es un inhibidor oral selectivo de la tirosina quinasa/FGFR3 (infigratinib), que funciona bloqueando las cascadas de señalización de los FGFR dentro de la célula. En modelos murinos ha mejorado el crecimiento de las cuatro extremidades, las anomalías mandibulares e incrementado el tamaño del foramen magno. Actualmente en fase 2 (NCT04265651), en el ensayo PROPEL 2, estimándose la fecha final del estudio en 202637,39.
Habrá que esperar para valorar la eficacia y seguridad de estas moléculas a largo plazo, así como estudiar la posible acción sinérgica entre las mismas. Recientemente se ha realizado una aproximación a las ventajas y desventajas de cada una de estas líneas de investigación con objeto de analizar el impacto de cada uno de ellos en las comorbilidades asociadas a la acondroplasia37.
ConclusionesLa acondroplasia es considerada una enfermedad rara y representa la displasia esquelética con talla baja más frecuente. Aparte de la talla baja disarmónica, asocia problemas neurológicos, otorrinolaringológicos, traumatológicos, endocrinológicos y psicosociales, por lo que un seguimiento multidisciplinar es necesario, tanto para tratar las complicaciones como para prevenirlas. En estos pacientes es fundamental el apoyo psicológico y soporte entre iguales que mejore su calidad de vida y fomente su autonomía. Nuevas terapias están en estudio y vosoritida, tras los resultados de eficacia y seguridad en ensayos clínicos, ha sido aprobado por la agencia europea EMA para el tratamiento de pacientes con acondroplasia de 2 años de edad y mayores cuyas epífisis no se han cerrado. Estudios en vida real nos permitirán conocer su repercusión/efecto en proporcionalidad, funcionalidad y calidad de vida, entre otros. Adicionalmente, es importante estar atentos a los resultados de eficacia y seguridad de otras moléculas en investigación para tener una perspectiva futura completa de las posibilidades terapéuticas de la acondroplasia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la Fundación ALPE, especialmente a Carmen Alonso y Susana Noval por su inestimable colaboración.

