




. Neonatal screening of glutaric aciduria type 1 (GA-1) has brought radical changes in the course and outcomes of this disease. This study analyses the outcomes of the first 5 years (2015–2019) of the AGA1 neonatal screening programme in our autonomous community.
Material. We conducted an observational, descriptive and retrospective study. All neonates born between January 1, 2015 and December 31, 2019 that participated in the neonatal screening programme were included in the study. The glutarylcarnitine (C5DC) concentration in dry blood spot samples was measured by means of tandem mass spectrometry applying a cut-off point of 0.25 µmol/L.
Results. A total of 30 120 newborns underwent screening. We found differences in the C5DC concentration based on gestational age, type of feeding and hours of life at sample collection. These differences were not relevant for screening purposes. There were no differences between neonates with weights smaller and greater than 1500 g. Screening identified 2 affected patients and there were 3 false positives. There were no false negatives. The diagnosis was confirmed by genetic testing. Patients have been in treatment since diagnosis and have not developed encephalopathic crises in the first 4 years of life.
Conclusions. Screening allowed early diagnosis of two cases of GA-1 in the first 5 years since its introduction in our autonomous community. Although there were differences in C5DC levels based on gestational age, type of feeding and hours of life at blood extraction, they were not relevant for screening.
El cribado neonatal de la aciduria glutárica tipo 1(AG-1) ha supuesto un cambio radical en la evolución y debut de la enfermedad. En este estudio se analizan los resultados de los primeros cinco años (2015–2019) del programa de cribado neonatal (PCN) de AG-1 en nuestra comunidad.
MaterialSe diseñó un estudio observacional, descriptivo y retrospectivo. La población estaba formada por neonatos participantes en el PCN nacidos entre el 1 de enero de 2015 y el 31 de diciembre de 2019. La determinación de glutarilcarnitina (C5DC) en sangre impregnada en papel se realizó mediante espectrometría de masas en tándem con punto de corte en 0,25 µmol/L.
ResultadosEl cribado se llevó a cabo en 30120 recién nacidos. En el análisis del marcador C5DC se encontraron diferencias en la concentración dependiendo de la edad gestacional, del tipo de alimentación y de las horas de vida a la extracción. Estas diferencias no fueron relevantes a efectos del cribado. No se observaron diferencias entre menores y mayores de 1500 g. Se identificaron 2 casos afectos y 3 falsos positivos. No se detectaron falsos negativos. Los diagnósticos fueron confirmados mediante estudio genético. Los pacientes reciben tratamiento desde el diagnóstico y no han presentado crisis encefalopáticas los primeros cuatro años de vida.
ConclusionesEl cribado ha permitido el diagnóstico precoz de dos casos de AG-1 en los cinco primeros años desde su instauración en nuestra comunidad. Aunque existen diferencias en los niveles de C5DC en función de edad gestacional, del tipo de alimentación y de las horas de vida a la extracción, estas no tuvieron trascendencia para el cribado.

Glutaric aciduria type 1 (GA-1) is an inborn error of metabolism with an autosomal recessive pattern of inheritance caused by a deficiency of the enzyme glutaryl-CoA dehydrogenase (GCDH) responsible for the dehydrogenation and decarboxylation of glutaryl-CoA in the lysine, hydroxylysine and tryptophan catabolic pathways. The breakdown of these pathways causes an increase in the levels of glutaryl-CoA and glutaconyl-CoA and a secondary accumulation of glutaric acid and 3-hydroxyglutaric acid, metabolites that have neurotoxic effects.1 Affected patients who are not identified early present chiefly neurologic clinical features, with hypotonia, psychomotor retardation, tremors and dystonias.2 Macrocephaly is a characteristic sign that becomes apparent from age 6 months, although it can be detected in utero.3 In up to 75% of cases, the onset is acute in the form of episodic encephalopathy. This presentation, characterised by decreased level of consciousness, seizures, irritability and extrapyramidal signs, usually develops in the first 6 years of life.1,4 Neuroimaging tests can show features such as fronto-operculo-temporal hypoplasia, basal ganglia degeneration, cystic dilation of the Sylvian fissure, leukoencephalopathy or subdural haematomas; and this disorder is sometimes included in the differential diagnosis of shaken baby syndrome.5
The screening of GA-1 is carried out through the measurement of the levels of glutarylcarnitine (C5-DC) in dry blood spot specimens using tandem mass spectrometry (MS/MS). In the region of Asturias, this test was added to the newborn screening programme (NSP) in late 2014.6,7
In recent years, the performance of the C5-DC marker for detection of GA-1 has been evaluated, especially in patients with low-excretor phenotypes that may have C5-DC levels in the normal range.
The primary objective of our study was to analyse the results of the first 5 years (2015–2019) of newborn screening for GA-1.
Material and methodsWe conducted a retrospective observational and descriptive study. The study universe consisted of all participants in the NSP, who were born between January 1, 2015 and December 31, 2019. We collected the data from the register of the newborn screening laboratory, which is located in the department of clinical biochemistry of the Hospital Universitario Central de Asturias (HUCA) since 2014, and which includes records for all the metabolic screenings performed in public and private hospitals of the autonomous community of Asturias.
For each patient, we retrieved data on the date of birth, the hospital where the specimen was collected, gestational age (GA), birth weight, feeding modality, time of sample collection (hours post birth) and concentration of C5-DC (μmol/L).
The study was approved by the Research Ethics Committee (file 2021.141).
The specimens used in screening were of capillary blood obtained through a heel prick and spotted on Whatman 903 cards. A sample collection protocol was established to obtain these specimens between 48 and 72 h post birth with a sterile automatic or manual lancet. The card had to be spotted with a blood drop and the patient data fields filled out. Specimens were identified by means of a barcode and reference number. Cards were subsequently submitted to the laboratory through specific intra- and inter-hospital pathways.7
The measurement of C5-DC levels was performed by MS/MS following electrospray ionization (ESI). The equipment used to conduct MS/MS was a triple quadrupole mass spectrometer (QTRAP 5500; AB Sciex) connected to liquid chromatography system (Ekspert Ultra LC 100; Eksigent).
The cut-off point set for the C5-DC marker remained constant at 0.25 μmol/L over the 5-year period. This value was adjusted following the analysis of the results obtained in a pilot study in 2014 where the cut-off point had been set at 0.17 μmol/L and was validated annually through the periodic review of percentile distributions.
We conducted a descriptive analysis calculating absolute and relative frequency distributions for qualitative variables and measures of central tendency and dispersion for quantitative variables. We assessed differences in quantitative variables in more than 2 groups by means of the Kruskal-Wallis test after the null was rejected in the normality and homogeneity of variance tests. When the differences were statistically significant, we applied the post hoc Dunn test. To compare variables in 2 groups we used the Student t test or, in the case of unequal variance, the Welch t test. We set the level of statistical significance at 0.05. The statistical analysis was performed with the IBM SPSS Statistics 2023 (IBM Corporation; Armonk New York, USA) and R Development Core Team version 4.1.3R (R Foundation for Statistical Computing, Vienna, Austria) (2022).
To determine the nationwide coverage of the PCN, we retrieved data on the recorded number of births from the webpage Instituto Nacional de Estadística (INE, National Institute of Statistics).8
We applied the GA thresholds established by the World Health Organization to define the following categories: extremely preterm (GA < 28 weeks), very preterm (GA ≥ 28 and < 32 weeks), moderate preterm (GA ≥ 32 and < 34 weeks), late preterm (GA ≥ 34 and < 37 weeks) and term (≥37).9
We defined small for gestational age (SGA) in newborns of either sex as low birth weight below the 10th percentile in the 2013 Fenton growth chart.10
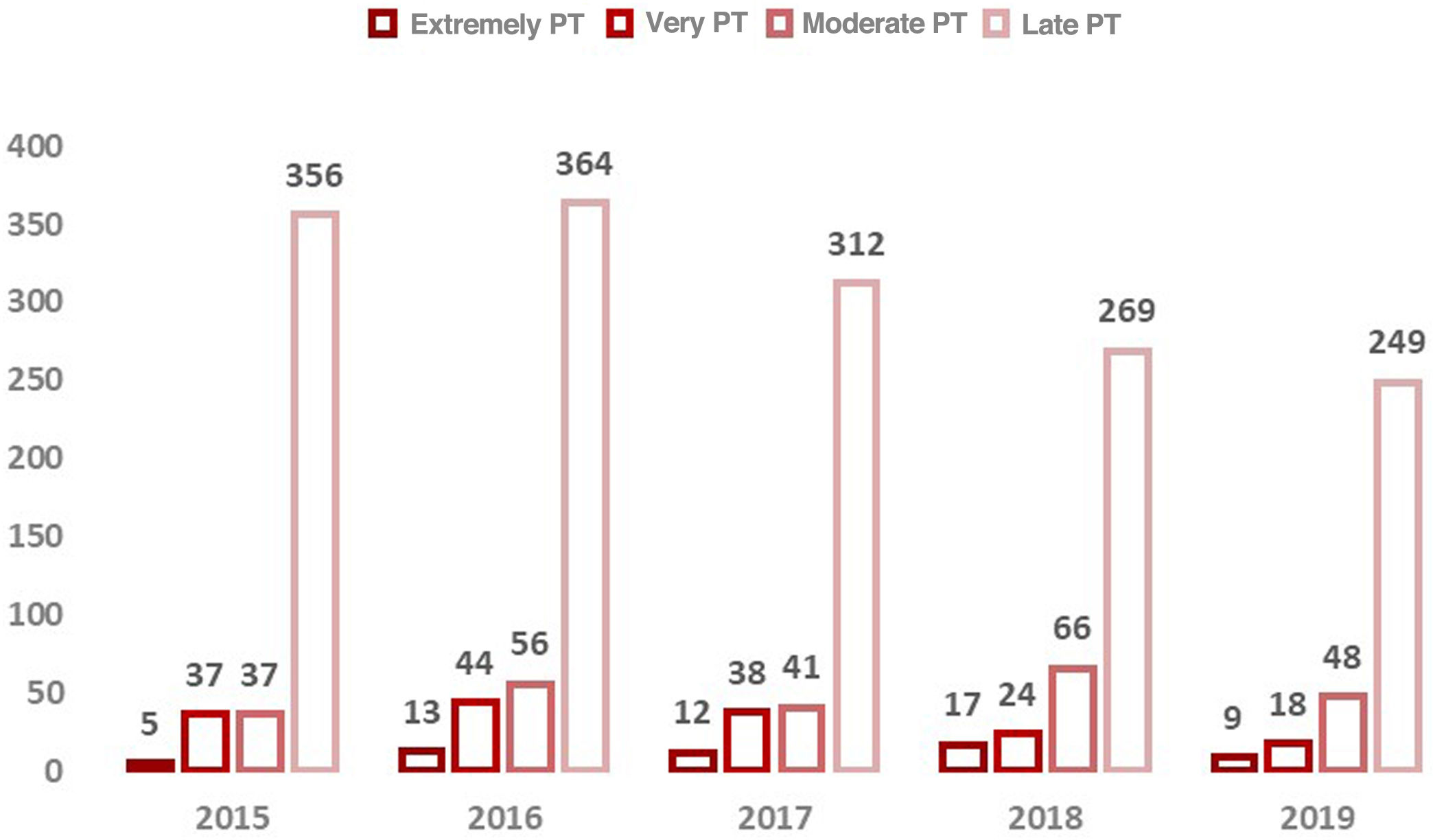
ResultsBetween 2015 and 2019, screening for GA-1 was carried out in 30 120 newborns in the autonomous community of (Fig. 1). The coverage of screening was greater than 99.6% every year. In 2019, all neonates in Asturias and 2 who resided in other autonomous communities were screened, achieving a coverage of 100%.

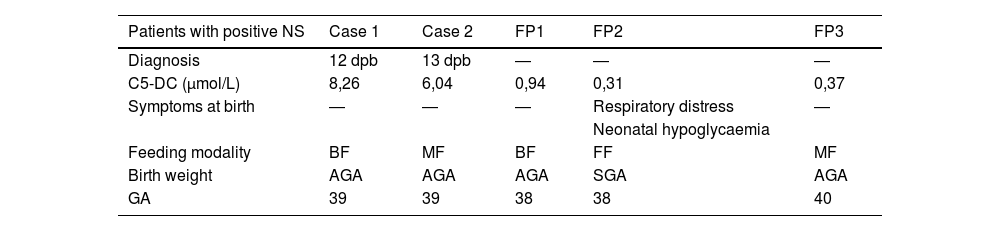
In the sample under study, the 99th percentile for the serum C5-DC level was 0.24 μmol/L, the 99.5th percentile 0.26 μmol/L and the 99.9th percentile 0.33 μmol/L. In the initial measurement of C5-DC levels, 216 samples exceeded the cut-off point (0.25 μmol/L). In a second measurement, 5 remained above the threshold, and the diagnosis was eventually confirmed in 2 patients. Both were term newborns, with birth weights appropriate for gestational age (AGA) and asymptomatic. The feeding modality was breastfeeding (BF) in one and mixed feeding (MF) in the other. During the 5 years of screening, there were 3 false positives (FPs) and 2 true positives (TPs), as described in Table 1. No false negatives (FNs) have been detected to date. The incidence of GA-1 was 1 case per 15 060 births per year.
Description of patients with positive results in newborn screening for glutaric aciduria type 1.
| Patients with positive NS | Case 1 | Case 2 | FP1 | FP2 | FP3 |
|---|---|---|---|---|---|
| Diagnosis | 12 dpb | 13 dpb | ― | ― | ― |
| C5-DC (μmol/L) | 8,26 | 6,04 | 0,94 | 0,31 | 0,37 |
| Symptoms at birth | ― | ― | ― | Respiratory distress | ― |
| Neonatal hypoglycaemia | |||||
| Feeding modality | BF | MF | BF | FF | MF |
| Birth weight | AGA | AGA | AGA | SGA | AGA |
| GA | 39 | 39 | 38 | 38 | 40 |
AGA, appropriate for gestational age; BF, breastfeeding; C5-DC, glutarylcarnitine; dpb, days post birth; FF, formula feeding; FP, false positive; GA, gestational age; MF, mixed feeding; NS, newborn screening; SGA, small for gestational age.
Both patients underwent genetic testing, which detected the heterozygous variants c.374 T > C (p.Leu125Pro) and c.1157 G > C (p.Arg386Pro) in the GCDH gene, both considered of uncertain significance, in one of them, and the heterozygous pathogenic variants c.877 G > A (p.Ala293Thr) and c.1204C > T (p.Arg402Trp) compatible with GA-1 in the other.11
From birth to present, the 2 patients have exhibited normal psychomotor development and they have reached age 4 years without experiencing encephalopathic crises.
We found birth data for 30 106 newborns (99.95%). Of the total births, 93.3% were at term and 6.7% were preterm, of which the category distribution is shown in Fig. 2.

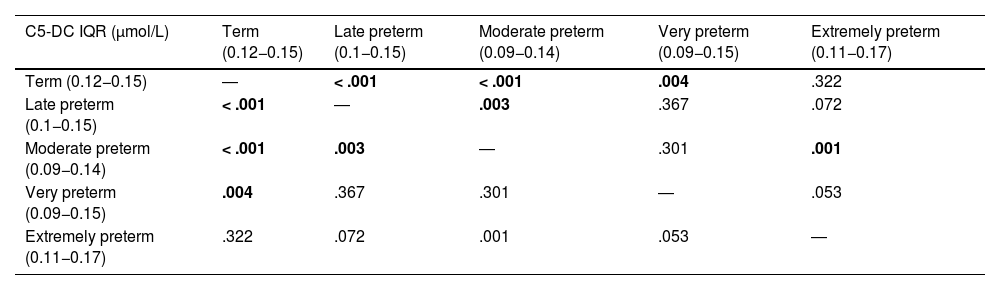
The analysis evinced differences in the concentration of C5-DC based on gestational age (P < .001). In the analysis by preterm category, we found significant differences between term and late preterm newborns, term and moderate preterm newborns, term and very preterm newborns, late and moderate preterm newborns and moderate and extremely preterm newborns (Table 2).
Results of the statistical analysis (P values) comparing C5-DC values based on gestational age.
| C5-DC IQR (μmol/L) | Term (0.12−0.15) | Late preterm (0.1−0.15) | Moderate preterm (0.09−0.14) | Very preterm (0.09−0.15) | Extremely preterm (0.11−0.17) |
|---|---|---|---|---|---|
| Term (0.12−0.15) | ― | < .001 | < .001 | .004 | .322 |
| Late preterm (0.1−0.15) | < .001 | ― | .003 | .367 | .072 |
| Moderate preterm (0.09−0.14) | < .001 | .003 | ― | .301 | .001 |
| Very preterm (0.09−0.15) | .004 | .367 | .301 | ― | .053 |
| Extremely preterm (0.11−0.17) | .322 | .072 | .001 | .053 | ― |
C5-DC, glutarylcarnitine.
We found significant differences between term and late preterm newborns, term and moderate preterm newborns, term and very preterm newborns, late and moderate preterm newborns, and moderate and extremely preterm newborns (P < .05).
Statistically significant results (P < .05) are presented in boldface.
The mean birth weight was 3211 g, while the 50th percentile was 2240 g. The minimum was 500 g and the maximum 5960 g. Of the total newborns, 0.8% (235) weighed less than 1500 g. The proportion of newborns who were SGA was 12.6%. When we compared the C5-DC concentration based on whether the birth weight was less or more than 1500 g, we did not find statistically significant differences (Table 3). However, we did find significant differences between SGA and AGA infants.
Values of C5-DC (interquartile range) in newborns small for gestational age (SGA) versus appropriate for gestational age (AGA) and with low versus normal birth weights (< 1500 g versus ≥ 1500 g). We found significant differences between the SGA and AGA groups.
| SGA | AGA | P | |
|---|---|---|---|
| C5-DC IQR (μmol/L) | 0.1−0.15 | 0.11−0.15 | < .001 |
| <1500 g | ≥1500 g | P | |
| C5-DC IQR (μmol/L) | 0.1−0.16 | 0.11−0.15 | .7 |
AGA, appropriate for gestational age; C5-DC, glutarylcarnitine; SGA, small for gestational age.
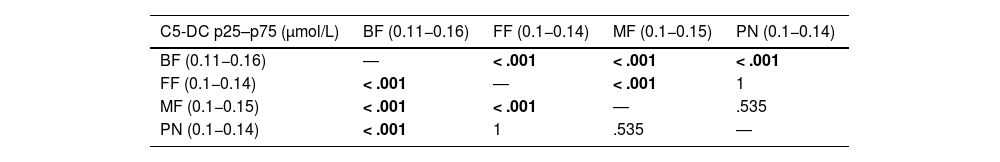
The most frequent feeding modality was breastfeeding (57.3%), followed by formula feeding (FF) (21.1%) and mixed feeding (MF) (20.9%); 101 specimens (0.4%) corresponded to newborns who had received parenteral nutrition (PN). The analysis showed differences in C5-DC values based on the feeding modality. The comparison of the C5-DC values between the groups evinced significant differences between the BF vs FF, the BF vs the MF, the FF vs the MF and the PN vs BF groups (Table 4).
Results of the statistical analysis (P values) comparing C5-DC values based on feeding modality.
| C5-DC p25–p75 (μmol/L) | BF (0.11−0.16) | FF (0.1−0.14) | MF (0.1−0.15) | PN (0.1−0.14) |
|---|---|---|---|---|
| BF (0.11−0.16) | ― | < .001 | < .001 | < .001 |
| FF (0.1−0.14) | < .001 | ― | < .001 | 1 |
| MF (0.1−0.15) | < .001 | < .001 | ― | .535 |
| PN (0.1−0.14) | < .001 | 1 | .535 | ― |
BF, breastfeeding; C5-DC, glutarylcarnitine; FF, formula feeding; MF, mixed feeding; PN, parenteral nutrition.
Statistically significant results (P < .05) are presented in boldface.
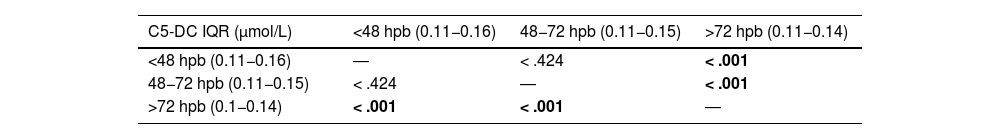
The median time elapsed from birth to the collection of the capillary blood specimen was 52 h. Most specimens were collected at 48 h post birth. When we analysed the trends in C5-DC values based on the timing of specimen collection we found differences between newborns in whom the specimens were collected after 72 h post birth compared to those in whom collection took place between 48 and 72 h or before 48 h (Table 5).
Results of the statistical analysis (P values) comparing C5-DC values based on the timing (hours post birth) of collection of specimens for screening.
| C5-DC IQR (μmol/L) | <48 hpb (0.11−0.16) | 48−72 hpb (0.11−0.15) | >72 hpb (0.11−0.14) |
|---|---|---|---|
| <48 hpb (0.11−0.16) | ― | < .424 | < .001 |
| 48−72 hpb (0.11−0.15) | < .424 | ― | < .001 |
| >72 hpb (0.1−0.14) | < .001 | < .001 | ― |
C5-DC, glutarylcarnitine; hpb, hours post birth.
Statistically significant results (P < .05) are presented in boldface.
Screening for GA-1 was added to the NSP in Asturias in late 2014 in adherence with the resolution of the Directorate General of Public Health published in the Boletín Oficial del Estado (BOE, Official State Bulletin) on November 6, 2014.
In these first 5 years of screening, 2 cases of GA-1 were detected and confirmed. We ought to highlight the high coverage of the NSP in the population and the alarming progressive decrease in the birth rate.
The incidence of GA-1 was greater than currently estimated in Europe (1 in 120 000 births). In 2021, the Revista Española de Salud Pública published a study that assessed the overall prevalence of metabolic diseases based on the reports of the Asociación Española de Cribado Neonatal (AECNE, Spanish Association of Newborn Screening) and the Spanish Ministry of Health for years 2016 to 2018.6,12 All autonomous communities in Spain with the exception of Aragon screened for GA-1, with diagnosis of a total of 57 cases and the highest prevalences found in the autonomous communities of Andalusia, Madrid and Galicia (lower than 1 case per 55 000 births).
In the Community of Madrid, 12 cases of GA-1 were diagnosed through screening between 2011 and 2019, all asymptomatic at the time of diagnosis. The estimated incidence was 1 case in 49 402 births. There were no false negatives.13 In Galicia, over a 20-year period of screening (2000–2019), 7 cases of GA-1 were detected, and a case with low-excretor phenotype subsequently diagnosed in a patient with a negative screening result (FN). The estimated incidence was 1 case in 57 802 births.14 In this period, most autonomous communities established the threshold at a serum level at or above the 99.5th percentile.
False negatives have not been detected in Asturias. The 3 cases that turned out to be FPs corresponded to healthy term newborns who were enterally fed with different feeding modalities (BF, FF and MF), one of whom was SGA. A recently published case series found that renal failure was associated with elevation of C5-DC in newborns, although none of the FPs in the study had this health condition.15 It is important to consider that the C5-DC marker has the same mass-to-charge ratio as 3-OH hexanoylcarnitine (C6-OH). False negatives have been reported in the previous literature in samples collected more than 7 days post birth from newborns who were low excretors and turned out to have C5-DC values in the normal range. However, other case series in the literature have not found differences in the serum concentration of C5-DC between low and high excretors.16,17
As regards genetic testing, the detected A293 T and R402W variants are the most frequent variants in Spain, based on previous case series.13,14,18 R402W is also the most prevalent variant in Portugal and Italy.19 On the other hand, the variants of uncertain significance (L125 P and R386 P) had not been reported before in our region.
Our study suggests that there may be differences in the serum levels of C5-DC based on GA, feeding modality and timing of sample collection, although these differences had not a clinically relevant impact on the results of screening. A study conducted in the United States found that socioeconomic level and SGA were associated, independently and in combination, with the metabolites used for screening, evincing an interaction between these variables and the C5-DC marker.20
The clinical outcomes of patients with GA-1 have changed drastically since the disorder started to be diagnosed through the NSP.21,22 When affected patients were not identified and treated early, 90% had onset with acute encephalopathic crises and motor impairment. The neurologic impairment caused by the GA-1 is progressive and irreversible, leading to cerebral atrophy and cognitive impairment. The cornerstone of treatment is a low-lysine diet. This diet is implemented through the use lysine-free, tryptophan-reduced, arginine-fortified amino acid formulas. Lifelong carnitine supplementation is indicated to reduce oxidative stress and the potential CNS complications secondary to carnitine depletion.23
Comparative studies have shown that when treatment is initiated in the neonatal period, 90% of patients remain asymptomatic and the proportion of symptomatic patients who develop encephalopathic crises decreases to 10%–20%.24 In addition, there is evidence of a 75% reduction in the occurrence of impaired oral motor function, impaired ambulatory capacity and dystonic movements.25 The two patients identified in this study have not developed encephalopathic crises to date.
Among the strengths of the study, we highlight that it is the first to present data on the screening of GA-1 in the region of Asturias since its inclusion in the NSP and that it allowed the detection in our autonomous community of 2 cases of a rare disease. Furthermore, the markers used for this NSP are certified by the standard UNE EN-ISO 15189, which also provides quality assurance measures.
The main limitation of the study is the duration of follow-up, as it was restricted to a 5-year period (2015–2019) due to the need to retrieve data for complete years. In February 2020, there was a change in the reagent manufacturer, so we opted to evaluate this initial period to avoid introducing bias.
This study provides a baseline that will allow comparison to the following 5-year period (2020–2024), at which point it will be possible to assess the screening algorithm from a more experienced perspective and in a greater number of cases, and to consider whether it would be useful to introduce potential improvements, such as the inclusion of additional markers in the screening programme.
ConclusionThe early diagnosis of GA-1 through newborn screening with the C5-DC test is key to modify the natural history of the disease and prevent neurologic damage in patients. The NSP has allowed the early diagnosis of 2 cases of GA-1 in the first 5 years since its introduction, with no evidence to date of any false negatives.
Conflicts of interestThe authors have no conflicts of interest to declare.

