

El síndrome de persistencia de conductos de Müller (SPCM) es un trastorno de la diferenciación sexual que se caracteriza por la persistencia de derivados müllerianos (útero y trompas de Falopio), en varones con cariotipo XY y con virilización normal1.
En un 85% de los casos es debido a mutaciones en el gen de la hormona antimülleriana (AMH), produciendo alteraciones en su secreción o actividad, o a mutaciones en su receptor tipo ii(AMHR2), provocando un cuadro clínico de resistencia hormonal2. Se transmite de forma autosómica recesiva. La determinación de la AMH es útil (indetectable si hay mutaciones en AMH o normal-elevada si hay mutaciones en AMHR23,4).
Presentamos un varón de 2 años y 3 meses por hallazgo de estructuras compatibles con restos müllerianos en cirugía por criptorquidia. Fruto de embarazo gemelar por fecundación in vitro, nació por cesárea a las 37 semanas de gestación con peso de 2.100g (−1,8DE) y longitud de 45cm (−1,85DE). Presentó criptorquidia bilateral desde el nacimiento e hidrocele izquierdo desde el año de vida. Sin antecedentes familiares de interés.
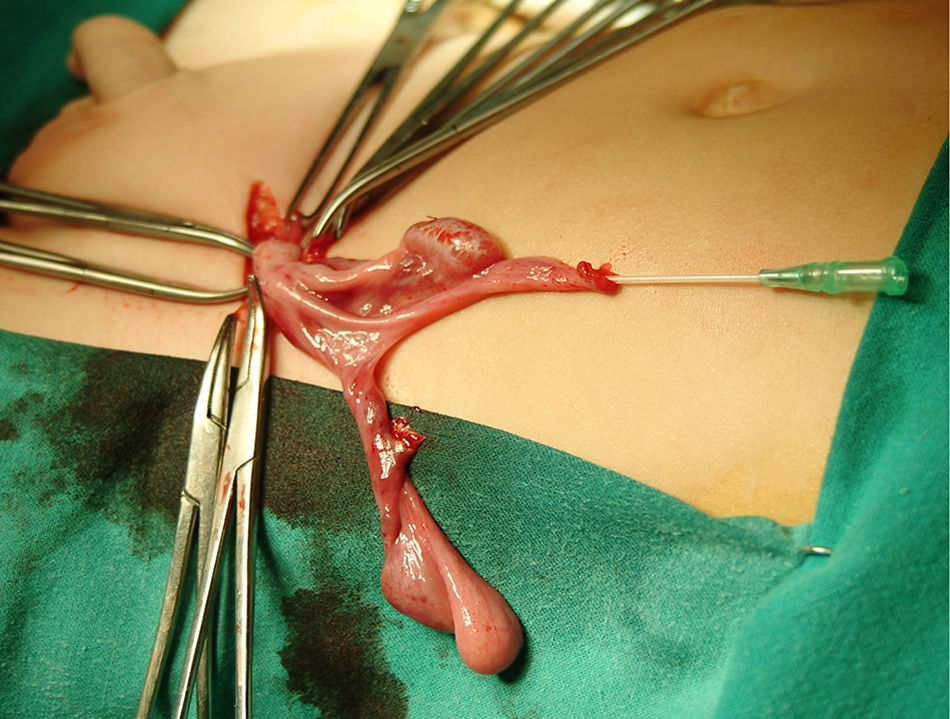
Tras inguinotomía izquierda, se objetivó un conducto permeable que contenía gónada en relación de proximidad con estructura tubular de luz permeable y aspecto tubárico. Se amplió el abordaje observando gónada contralateral en contigüidad con lo que aparentaba ser un útero y trompas hipoplásicos (fig. 1). Por ureterocistoscopia se visualizó uretra posterior masculina con veru montanum y colículos seminales de morfología normal. Tras exéresis de restos müllerianos y biopsia de ambas gónadas, se efectuó orquidopexia y herniotomía izquierda. Ulteriormente se realizó orquidopexia derecha.

La anatomía patológica de las estructuras extirpadas confirmó el hallazgo de restos de conducto de Müller y se describieron ambas gónadas como testículos con cambios de disgenesia con diámetro tubular medio e índice de fertilidad disminuidos, sin apreciarse células de Leydig en intersticio y con índice de células de Sertoli normal.
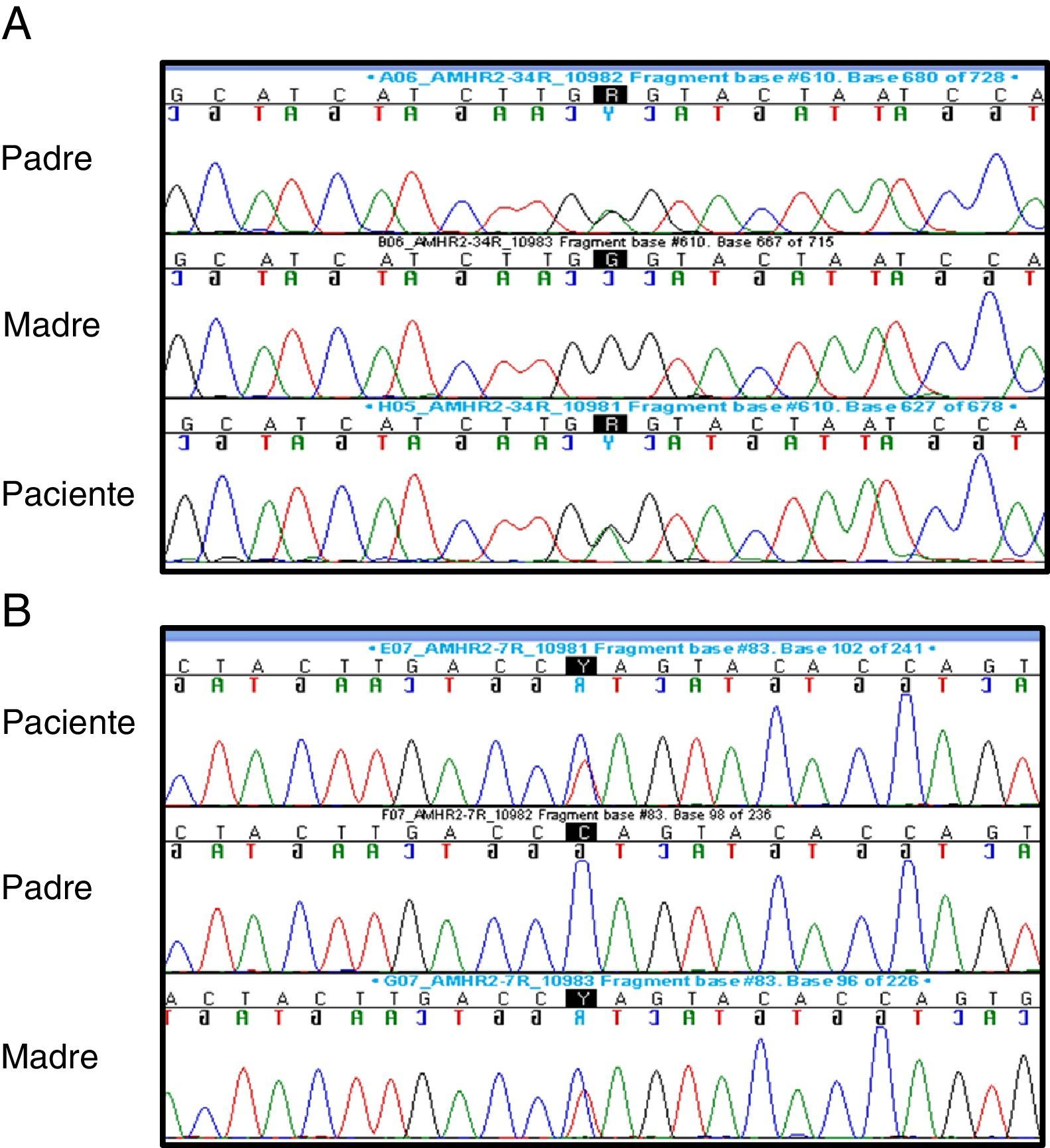
Ante sospecha de SPCM se solicitó: cariotipo: 46,XY; FSH (1,2mUI/mL–normalidad: 0,2-1,4), LH (0,04ng/mL–normalidad: 0,01-0,5-) y testosterona (0,04ng/ml–normalidad: 0,01-0,04-: basales sin alteraciones y AMH: 816pg/L (VN:360-668pg/L). La secuenciación del gen AMHR2 reveló la existencia de una variante alélica previamente descrita en el exón 4 (c.502G>A; p.Ala168Thr, rs374601719) y una nueva variante (c.877C>T; p.Gln 293*) en exón 7, ambas en heterocigosis. La variante del exón 4 estaba presente en el padre y la variante del exón 7 en la madre, ambas en heterocigosis, indicando que el paciente es un heterocigoto compuesto (fig. 2).
 en exón 4 (A) y la variante c.877C>T (p.Gln293*) en exón 7 (B).")
Con 8 años y 7 meses, el paciente presenta peso y talla en percentil 50, testes de 2-3ml, con un pene de 4cm y aspecto escrotal normal (Tanner I) con microcalcificaciones en las últimas revisiones que obligan a un seguimiento clínico y ecográfico estrechos. Tras test de LHRH, los niveles de FSH subieron hasta 5,88mUI/mL y los de LH a 3,88mUI/mL, siendo los niveles de testosterona de 0,18ng/mL.
Los pacientes con SPCM presentan fenotipo masculino al nacimiento y el diagnóstico se realiza normalmente por hallazgo casual durante la cirugía por criptorquidia con hidrocele o hernia inguinal.
El gen de AMHR2, localizado en el cromosoma 12q13, tiene una longitud de 8,7kb y contiene 11 exones4,5. Codifica una proteína transmembrana con un dominio extracelular que se une a AMH y un dominio intracelular con actividad cinasa seronina/treonina que se une al receptor tipo i, activando por fosforilación la transcripción de los genes diana que permiten la acción de la AMH. Se han descrito varias mutaciones en el mismo, siendo la más frecuente la deleción de 27 pares de bases en el exón 10.
El análisis in silico de las variantes c.877C>T y c.502G>A predijo la enfermedad causante empleando «Mutation Taster prediction site». La variante c.877C>T conduce a un stop codon prematuro, que genera una proteína truncada, que pierde parte del dominio cinasa serina/treonina. La substitución c.502G>A se localiza en la última base del exón 4 y parece afectar al proceso de splicing. Además, c.502G>A es una variante rara, con una frecuencia del 0,01% en la base de datos ExAC. Todo ello indica que el SPCM es debido a ambas variantes en AMHR2.
Los testículos en el SPCM presentan diferenciación normal con presencia de células germinales; sin embargo, la fertilidad puede verse comprometida por la degeneración secundaria a la criptorquidia y la alteración de la comunicación con los conductos deferentes, que son en muchos casos aplásicos y pueden estar desconectados de los testes por la presencia de los derivados de Müller.
Se ha descrito un riesgo aumentado de neoplasia testicular (15%), similar al descrito en pacientes con criptorquidia abdominal5,6; este hecho, junto con las microcalcificaciones halladas en nuestro paciente, hace necesario un seguimiento estrecho clínico y ecográfico.
Se trata de un caso clínico excepcional, habiéndose descrito escaso número de pacientes en la literatura internacional.

