






La trombocitopenia inmune primaria, anteriormente conocida como púrpura trombocitopénica inmune, es una enfermedad cuyo manejo diagnóstico y terapéutico ha sido siempre controvertido. La Sociedad Española de Hematología y Oncología Pediátricas, a través del grupo de trabajo de la PTI, ha actualizado el documento con las recomendaciones protocolizadas para el diagnóstico y tratamiento de esta enfermedad, basándose en las guías clínicas disponibles actualmente, revisiones bibliográficas, ensayos clínicos y el consenso de sus miembros. El objetivo principal es disminuir la variabilidad clínica en los procedimientos diagnósticos y terapéuticos con el fin de obtener los mejores resultados clínicos, los mínimos efectos adversos y preservar la calidad de vida.
Primary immune thrombocytopenia, formerly known as immune thrombocytopenic purpura, is a disease for which the clinical and therapeutic management has always been controversial. The ITP working group of the Spanish Society of Paediatric Haematology and Oncology has updated its guidelines for diagnosis and treatment of primary immune thrombocytopenia in children, based on current guidelines, bibliographic review, clinical assays, and member consensus. The main objective is to reduce clinical variability in diagnostic and therapeutic procedures, in order to obtain best clinical results with minimal adverse events and good quality of life.
La trombocitopenia inmune primaria (PTI) es una enfermedad caracterizada por la presentación habitualmente aguda de una trombocitopenia esencialmente periférica, con valores inferiores a 100.000 plaquetas/μL, en niños que carecen de antecedente u otra patología que explique dicha citopenia, con manifestaciones hemorrágicas exclusivamente (generalmente púrpura) o asintomáticos, que se debe a un mecanismo autoinmune1. La incidencia anual es de 1/10.000 niños, con un pico entre los 2 y 4 años; no parece haber diferencias debidas al sexo. Suele existir antecedente de infección viral o vacunación 1-3 semanas antes.
El eje central en la etiopatogenia de la PTI es la pérdida de la tolerancia inmunológica hacia los antígenos específicos de las plaquetas. Inicialmente se documentó y estaba asentado el papel del brazo efector humoral (autoanticuerpos específicos) sobre las plaquetas en la patogénesis de la enfermedad, pero con el avance en los conocimientos de la inmunología y las aplicaciones al conocimiento de la PTI se ha observado la importancia que tiene la inmunidad celular de forma directa e indirecta a través de la cooperación T-B. Igualmente, siempre se ha considerado que la trombocitopenia era consecuencia de la destrucción periférica exclusiva de plaquetas, pero en la actualidad se cree que, por lo menos en un porcentaje de casos, existe afectación en la megacariocitopoyesis, lo que explicaría la ausencia de hiperplasia megacariocítica que se observa en algunos niños y la respuesta al tratamiento con análogos de la trombopoyetina.
En marzo de 2009 se publicaron las recomendaciones de estandarización de la terminología, definiciones y criterios de respuesta de la PTI para adultos y niños. Se elimina el término de púrpura porque el sangrado cutáneo o mucoso está ausente o es mínimo en algunos pacientes. Se mantienen los acrónimos ITP (Immune ThrombocytoPenia) y PTI en castellano, por su amplia difusión y utilización previa1.
En este consenso también se establece el nivel de corte de la cifra de plaquetas en 100.000/μL para el diagnóstico de PTI, por los recuentos frecuentes entre 100.000 y 150.000/μL en individuos sanos y en embarazadas. La clasificación se modifica atendiendo a la historia natural de la PTI en la infancia, en la que aproximadamente dos tercios se recuperan espontáneamente en los primeros 6 meses y la posibilidad de remisión es alta entre los 3 y 12 meses2, e incluso puede ser más tardía.
Posteriormente, en enero de 2010 se publicó el consenso internacional para el diagnóstico y manejo de la PTI3. El diagnóstico sigue siendo de exclusión. El principal problema es el riesgo aumentado de hemorragia. No existe una correlación exacta entre la cifra de plaquetas y las manifestaciones hemorrágicas, aunque estas son más frecuentes por debajo de 10.000/μL. La mayoría de los pacientes están asintomáticos o tienen petequias, hematomas o equimosis aislados en piel o mucosas4,5. Sin embargo, algunos casos pueden presentar hemorragias más graves a niveles cutáneo, mucoso, gastrointestinal o incluso intracraneal (0,1-0,5%)6-9.
En este nuevo protocolo hemos centrado el objetivo del tratamiento en tratar las hemorragias con relevancia clínica, no en corregir las cifras de plaquetas hasta valores normales. Hay que evitar tratamientos innecesarios, potencialmente tóxicos, en pacientes paucisintomáticos y conseguir una adecuada calidad de vida con la mínima toxicidad asociada a la terapia. El tratamiento de las PTI persistentes o crónicas es complejo y se plantean diferentes alternativas.
En este protocolo se actualiza la versión del protocolo PTI-201010.
ObjetivosUnificar criterios diagnósticos, protocolos de seguimiento y tratamiento de la PTI.
Clasificación diagnóstica, criterios de evaluación clínica y respuesta al tratamientoClasificación diagnóstica1PTI de reciente diagnósticoDesde el momento del diagnóstico hasta los 3 meses de evolución.
PTI persistenteDe duración entre los 3 y 12 meses desde el diagnóstico, incluye a:
- •
Pacientes que no alcanzan la remisión completa de forma espontánea.
- •
Pacientes que no mantienen la remisión completa después de suspender el tratamiento instaurado.
Pacientes que continúan con trombocitopenia después de 12 meses desde el diagnóstico.
Criterios de evaluación clínica10Ver la tabla 1.
Criterios de evaluación clínica
| Asintomático |
| Clínica cutánea |
| Clínica cutáneo-mucosa |
| Sangrado activo |
| Epistaxis que precisa taponamiento |
| Hematuria macroscópica |
| Hemorragia digestiva macroscópica |
| Menorragia |
| Gingivorragia importante |
| Cualquier hemorragia con riesgo razonable de precisar trasfusión de hematíes o que condicione un daño orgánico grave |
Fuente: Monteagudo et al.10.
Ver la tabla 2.
Criterios de respuesta al tratamiento10Ver la tabla 3.
Criterios de respuesta al tratamiento
| Remisión completa. Recuento igual o superior a 100.000/μL mantenido más de 6 semanas tras la supresión del tratamiento |
| Remisión parcial. Elevación sobre la cifra inicial con recuento entre 30.000 y 100.000/μL mantenido más de 6 semanas tras la supresión del tratamiento |
| Ausencia de respuesta. No se modifica clínica ni biológicamente |
| Respuesta transitoria. Mejoría inicial (clínica o biológica) con nueva clínica o recuento inferior a 30.000/μL antes de 6 semanas de haber finalizado el tratamiento |
| Recaída. Recuento inferior a 30.000/μL después de 6 semanas de haber finalizado el tratamiento, habiéndose obtenido previamente una remisión completa o parcial |
Fuente: Monteagudo et al.10.
En todos los pacientes con trombocitopenia se debe realizar una historia clínica detallada y una exploración física completa que permitan descartar otras enfermedades hematológicas o situaciones que de forma secundaria puedan producir trombocitopenia.
Los estudios detallados en la tabla 4 son los recomendados por considerarse básicos3,10-12.
Estudios recomendados al diagnóstico
| Hemograma y recuento de reticulocitos, se observa trombocitopenia aislada |
| Morfología en sangre periférica con revisión por persona experta, normal |
| Estudio de hemostasia: tiempo de protrombina, tiempo de tromboplastina parcial activado, fibrinógeno |
| Grupo, Rh y Coombs directo |
| Inmunoglobulinas |
| Estudio microbiológico de citomegalovirus, virus de Epstein Barr, parvovirus B19, herpes simple, herpes 6, VIH, hepatitis B y C |
| Bioquímica hemática: GOT, GPT, LDH, glucosa, urea, creatinina |
| Sedimento urinario |
| Estudio morfológico de médula ósea por punción aspirativa. Indicado en todos los niños que presenten alguna de las siguientes condiciones: clínica que no sea la típica, si hay otras citopenias en el hemograma, aquellos que no responden al tratamiento de primera línea y en los pacientes no tratados que no remiten espontáneamente |
Indicadas en pacientes en los que no remite espontáneamente o no responden al tratamiento. Se recomienda:
- •
Estudio morfológico de médula ósea por punción aspirativa si no se hizo previamente. Valorar la realización de biopsia, inmunofenotipo y citogenética para completar el estudio.
- •
Poblaciones linfocitarias.
- •
Anticuerpos antinucleares y opcionalmente otros estudios de autoinmunidad.
Ver la tabla 5.
Recomendaciones generales
| Al diagnóstico, considerar el ingreso hospitalario en pacientes con sangrado activo, factores de riesgo hemorrágico o con recuento de plaquetas igual o inferior a 20.000/μL |
| Evitar inyectables intramusculares y punciones vasculares en vasos de difícil compresión |
| Contraindicado el empleo de ácido acetilsalicílico o sus derivados; administrar solamente en caso de ser estrictamente necesario otros fármacos que puedan alterar la agregación plaquetaria (antihistamínicos, antiinflamatorios no esteroideos) |
| Deportes: restricción en función de la clínica y el riesgo traumático |
| Antifibrinolíticos: ácido tranexámico13. Especialmente en pacientes con hemorragia activa y en hemorragia de mucosas. Contraindicado si hay hematuria. Se puede administrar por vía oral a dosis de 20mg/kg/8-12h o bien por vía intravenosa 10mg/kg/8-12h |
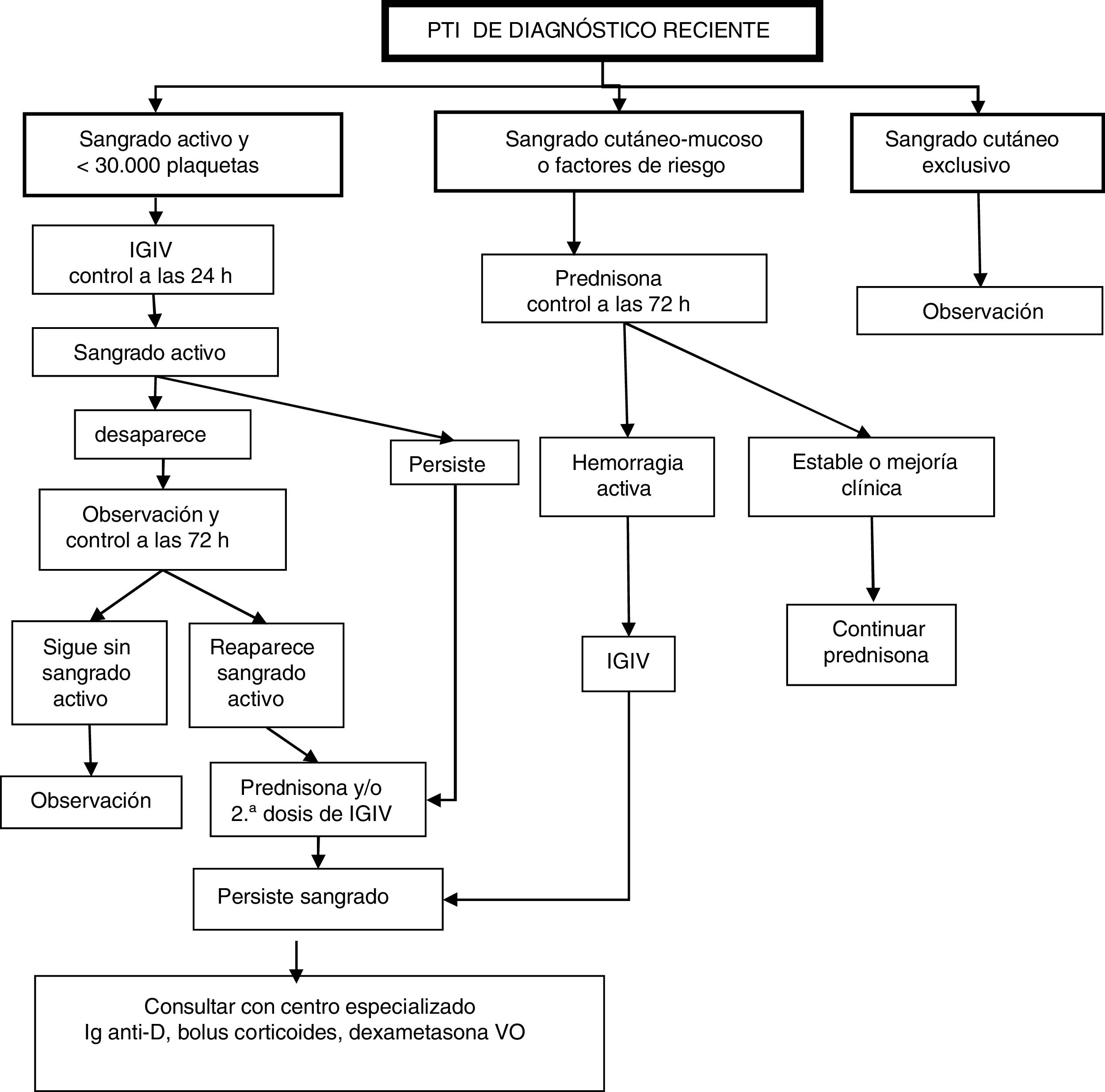
El paciente con PTI de diagnóstico reciente1 puede presentar unas manifestaciones hemorrágicas de gravedad variable en general en función de la cifra de plaquetas, actividad habitual y presencia de otros factores que pueden influir en la hemostasia. Hay que valorar siempre el conjunto de datos clínicos y biológicos para un adecuado enfoque terapéutico. Se ha decidido clasificar a los pacientes en diversos grupos en función de las manifestaciones clínicas y factores de riesgo hemorrágico, con la finalidad de establecer la opción de tratamiento más adecuada3,10,11,14-19.
Clasificación de pacientes y pauta de actuación (fig. 1)Pacientes con sangrado activo y recuento inferior a 30.000 plaquetas/μLSe propone administrar una dosis de inmunoglobulinas intravenosas (IGIV) y nueva valoración a las 24h:
- •
Si persiste el sangrado activo se añaden corticoides (prednisona o metilprednisolona) y/o una segunda dosis de IGIV.
- •
Si desaparece la clínica se vuelve a valorar a las 72h y,
- •
si persiste la mejora clínica entonces pasa a observación;
- •
pero si aparece de nuevo sangrado activo, se inicia tratamiento con corticoides (prednisona o metilprednisolona) o se administra una segunda dosis de IGIV.

Si a pesar de lo anterior sigue con sangrado activo, se recomienda consultar con unidad especializada de Hematología Pediátrica la administración de Ig anti-D (en caso de ser Rh+)17, bolus de corticoides o dexametasona a altas dosis vía oral20.
Pacientes con sangrado cutáneo-mucoso o factores de riesgoSe propone administrar de entrada corticoides (prednisona o metilprednisolona). Si hay evolución clínica a hemorragia activa se administra una dosis de IGIV y se sigue como en el supuesto del grupo anterior.
Pacientes con sangrado cutáneo exclusivoSe propone una actitud expectante y controles periódicos con actuación posterior en función de la evolución.
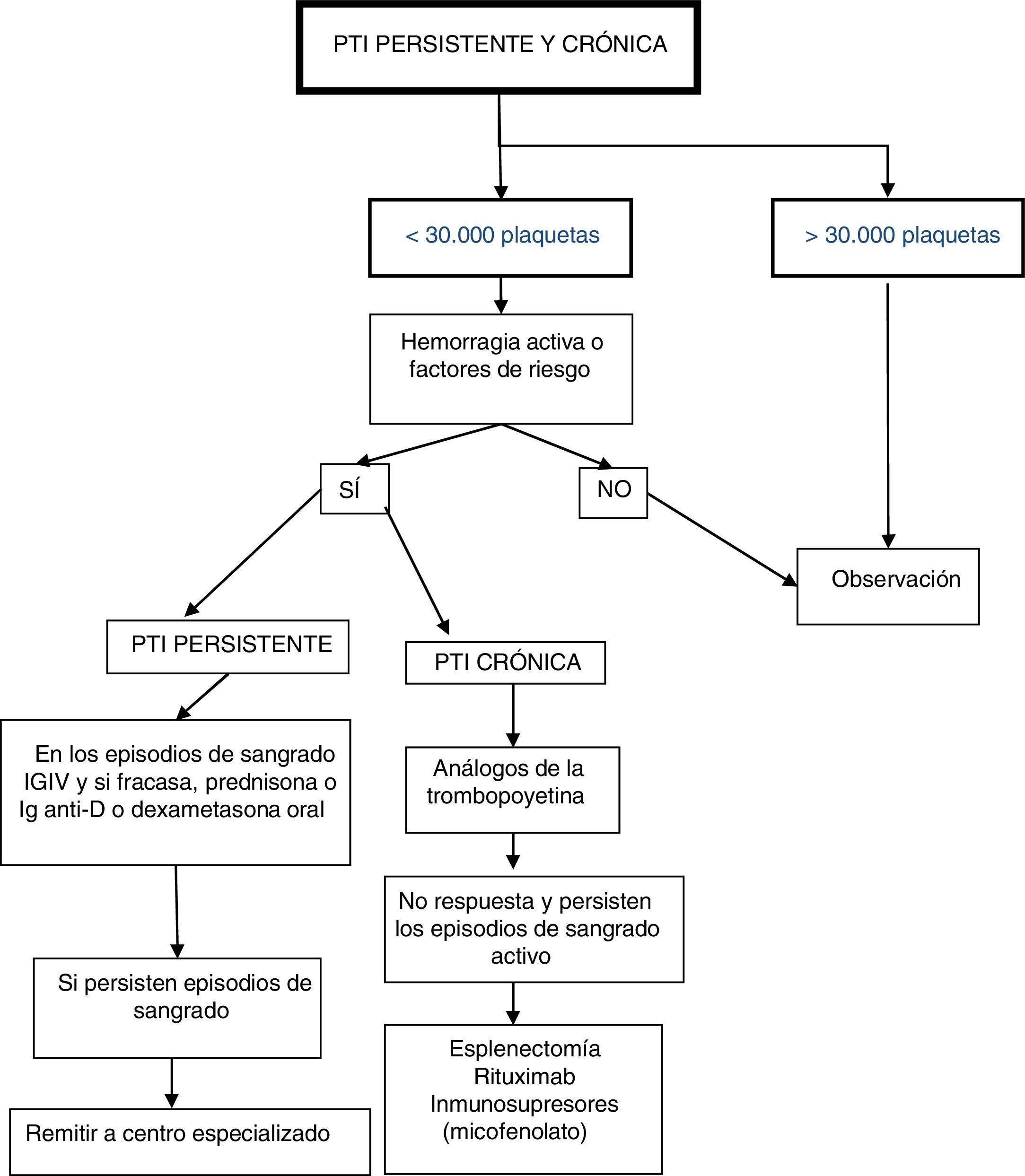
PTI persistente y crónicaLos pacientes afectos de PTI crónica sintomáticos, que requieren el empleo secuencial o permanente de tratamiento, deben ser controlados totalmente o de forma coordinada por centros con unidades especializadas de Hematología Pediátrica.
El paciente con PTI persistente y crónica puede presentar unas manifestaciones hemorrágicas de gravedad variable en general en función de la cifra de plaquetas, actividad habitual y presencia de otros factores que pueden influir en la hemostasia. Hay que valorar siempre el conjunto de datos clínicos y biológicos para un adecuado enfoque terapéutico y procurar que el paciente desarrolle una vida lo más cercana a la normalidad con los mínimos efectos adversos derivados del tratamiento19,21, en espera de que la enfermedad entre en remisión3,10,11,22,23. Está extendido en la literatura y se ha considerado razonablemente segura para el desarrollo de una vida cotidiana normal, el mantenimiento de recuentos por encima de 30.000 plaquetas, por ello, se ha elegido como factor determinante en el análisis de decisión inicial. No obstante, el estilo de vida, la actividad habitual, las manifestaciones clínicas y los factores de riesgo hemorrágico son también determinantes, fundamentalmente si se indica algún tipo de intervención.
Clasificación de pacientes y pauta de actuación (fig. 2)Pacientes con más de 30.000 plaquetas/μL mantenidas de forma estableSe recomienda mantenerlos solamente con observación, con los controles necesarios a juicio del clínico.
Pacientes con menos de 30.000 plaquetas/μL
Para una orientación adecuada hemos de tener en cuenta la existencia de factores de riesgo ligados a su edad, actividad deportiva, entorno familiar y accesibilidad a la atención sanitaria. Aquellos que no presenten episodios de hemorragia activa o factores de riesgo hemorrágico, se aconseja mantenerlos exclusivamente en observación. En caso contrario se recomienda:
- •
En pacientes con PTI persistente: en los episodios de sangrado administrar IGIV3,10,11,14,15,23, Ig anti-D3,15,17, prednisona o dexametasona oral24-27. Si a pesar de ello permanece con episodios de sangrado, se aconseja remitir a centro especializado para revisión y valorar la administración de otros tratamientos.
- •
En pacientes con PTI crónica con clínica mantenida: se recomienda en primera línea la administración de análogos de la trombopoyetina28-36, reservando corticoides o IGIV para episodios hemorrágicos puntuales. Solamente ante el fracaso de lo anterior, se pueden considerar tratamientos de segunda línea como la esplenectomía3, rituximab11,22,33,37-39 y otras opciones de inmunosupresores como el micofenolato33,40,41.
Ver la tabla 6.
Recomendaciones para el manejo de las urgencias con riesgo vital, situaciones de riesgo especial y esplenectomía programada
| Urgencias con riesgo vital |
| Hemorragias del SNC |
| Otras hemorragias que comprometan la vida del paciente |
| Se administran sucesivamente: |
| 1.o metilprednisolona i.v. 10mg/kg |
| 2.o gammaglobulina i.v. 400mg/kg |
| 3.o plaquetas 1 unidad/5-10kg/6-8h |
| 4.o gammaglobulina i.v. 400mg/kg |
| 5.o esplenectomía urgente: valorar según cada caso |
| Situaciones con riesgo especial |
| TCE, politraumatizados y cirugía urgente |
| Administrar IGIV 0,8-1g/kg si plaquetas <50.000/μL y plaquetas si recuento <10.000/μL |
| Cirugía programada (valorar riesgo hemorrágico según intervención) |
| IGIV 0,8-1g/kg si plaquetas <50.000/μL |
| Esplenectomía programada |
| IGIV 0,8-1g/kg si plaquetas <20.000/μL |
| Pinzamiento precoz de la arteria esplénica |
Los corticoides constituyen el tratamiento inicial clásico de la PTI.
Posología- •
Prednisona vía oral o metilprednisolona vía intravenosa, repartida en 3 dosis tras desayuno, comida y cena. Dosis: 4mg/kg/día (dosis máxima 180mg/día) durante 4 días, luego pasar a 2mg/kg durante 3 días y suspender.
- •
Bolus de corticoides: metilprednisolona 30mg/kg/día, dosis máxima 1g, 3 días, infusión en 2h. Requiere especial control de presión arterial y glucosuria.
- •
Dexametasona oral: 0,6mg/kg/día en una dosis, máximo 40mg, durante 4 días. Suele emplearse en ciclos mensuales.
La toxicidad principal de los corticoides es la hiperglucemia y la hipertensión, alteraciones del humor y del sueño; en tratamientos prolongados también síndrome de Cushing, osteoporosis y afectación del crecimiento. Como inmunosupresores aumentan la susceptibilidad a las infecciones.
Inmunoglobulinas intravenosas (IGIV)Posología- •
0,8-1g/kg/dosis única i.v. en perfusión continua, tiempo de infusión de 6-8h, al inicio de la infusión la velocidad es más lenta; se recomienda seguir la pauta de velocidad de infusión indicada en cada preparado.
- •
Anafilaxia, en pacientes con déficit de IgA: se recomienda tener preparado para uso inmediato el tratamiento específico y equipo de reanimación (adrenalina).
- •
Cefalea, náuseas, vómitos (se recomienda disminuir la velocidad de infusión).
- •
Febrícula/fiebre.
- •
Hemólisis aloinmune.
- •
Meningitis aséptica.
Bloqueo de receptores Fc macrofágicos con hematíes recubiertos por Ac anti-D.
Posología- •
50-75μg/kg/día, i.v., dosis única. Perfusión durante 1h diluida en suero fisiológico. Se recomienda premedicar con paracetamol.
Anemia hemolítica inmune y puesto que es un hemoderivado no están exentas del riesgo de transmisión de enfermedades infecciosas. Se ha comprobado la transmisión de hepatitis C.
Se recomiendan los siguientes controles: Hb, Coombs directo, recuento reticulocitario y bilirrubina indirecta. Se aconseja no repetir dosis (a las 2-4 semanas) si presenta bilirrubina I. >1,5mg% y reticulocitos >5% con subictericia o coluria, o descenso de Hb superior a 2g.
Agonistas del receptor de la trombopoyetinaLos análogos de la trombopoyetina, eltrombopag y romiplostim, son dos moléculas de diversa naturaleza, caracterizadas por su acción trombopoyética mediante estímulo del receptor correspondiente. Se han desarrollado los últimos años, con estudios que muestran evidencia de respuesta mantenida en un porcentaje alto de casos refractarios o dependientes de otros tratamientos (corticoides, IGIV). Actualmente tienen indicación en ficha para tratamiento en PTI crónica en niños mayores de un año y en adultos.
Su introducción ha supuesto una mejoría importante en la clínica y en la calidad de vida de los pacientes, con un perfil de seguridad aceptable22,28,29,32,36.
Su lugar en la terapéutica de la PTI crónica ha pasado a estar ubicado por delante de tratamientos que tienen un perfil de riesgos potencialmente mayor, como la esplenectomía y el rituximab, más aún en niños, en los que por la historia natural conocemos casos de remisiones tardías de la enfermedad. Se ha observado en los estudios clínicos que pacientes que son o se hacen no respondedores a uno u otro fármaco pueden mostrar respuesta con el otro.
Es conveniente que la indicación y el control de su manejo sean realizados por unidades pediátricas especializadas de Hematología Pediátrica.
Mecanismo de acción- •
Romiplostim es una proteína recombinante compuesta por dos dominios. Romiplostim estimula el crecimiento y la maduración de los megacariocitos mediante su unión al receptor Mpl de la misma manera que la TPOe, vía JAK2, STAT5, P38 MAPK y AKT.
- •
Eltrombopag es una pequeña molécula no peptídica, activa por vía oral, que se une a la porción transmembrana del receptor de la TPO en un sitio diferente al de la TPOe. Eltrombopag, al unirse al receptor, activa la vía de señalización JAK2/STAT estimulando la producción de plaquetas.
Eltrombopag y romiplostim están indicados en niños mayores de un año de edad afectos de PTI crónica que son refractarios a otros tratamientos de la PTI.
PosologíaRomiplostim- •
Se administra por vía subcutánea una vez por semana.
- •
Dosis de inicio: 1μg/kg/s.
- •
Ajuste de dosis: incremento de 1μg/kg semanalmente hasta que se alcance un recuento plaquetario >50×109/L, sin exceder la dosis máxima de 10μg/kg/s.
- •
La mediana de dosis semanal necesaria del fármaco viene a ser de 5μg/kg. La máxima respuesta se alcanza a las 2 semanas de la primera dosis.
- •
Si la cifra de plaquetas en 2 semanas consecutivas es >150×109/L, debe bajarse la dosis en 1μg/kg. Si la cifra de plaquetas es >250×109/L, debe suspenderse el tratamiento temporalmente, para volver a iniciarlo con una dosis inferior en 1μg/kg cuando la cifra de plaquetas sea <150×109/L.
- •
Hay que evaluar los recuentos plaquetarios semanalmente hasta alcanzar un recuento estable. Luego mensualmente.
- •
Administración por vía oral, una vez al día.
- •
Dosis de inicio:
- •
Niños de 1-5 años: 25mg/d.
- •
Niños de 6-17 años: 50mg/d.
- •
En pacientes de ascendencia del este asiático o aquellos que presenten un daño hepático de moderado a severo: 12,5-25mg/d.
- •
Ajuste de dosis: si a las 2 semanas de iniciado el tratamiento el recuento plaquetario es <50×109/L, se aumenta la dosis en 12,5mg/d (en <6 años) o en 25mg/d (en ≥6 años). Debe esperarse al menos 2 semanas antes de realizar un nuevo ajuste de dosis, de igual manera, hasta conseguir >50×109/L, sin sobrepasar la dosis máxima de 75mg/d.
- •
Para facilitar su absorción, eltrombopag debe tomarse con el estómago vacío y como además quela cationes polivalentes, hay que considerar que, 4h previas a su administración y las 2h posteriores, se debe evitar la toma de antiácidos, calcio (derivados lácteos.) o suplementos que contengan hierro, magnesio, selenio, zinc, aluminio. La falta de esta precaución es causa frecuente de no respuesta al tratamiento.
Ambos han demostrado ser eficaces incrementando los recuentos de plaquetas en alrededor del 80% de pacientes diagnosticados de PTI refractarios a otros tratamientos, aunque la respuesta suele ser estable con mínimos cambios de dosis en la mitad de los casos.
Hay varios estudios que demuestran la eficacia y la seguridad en la utilización de un segundo agonista del receptor de la trombopoyetina cuando el primero ha fallado.
A pesar de que estos fármacos presentan un inicio rápido del efecto terapéutico (1-2 semanas), el recuento plaquetario suele caer a las 2 semanas de la interrupción del tratamiento, de manera que deben ser administrados de forma continuada con la consiguiente elevación del coste, sin olvidar la falta de experiencia en su seguridad a largo plazo. Se van comunicando casos en los que se ha podido retirar el agonista del receptor de la trombopoyetina manteniéndose estable el recuento de plaquetas.
Interrupción del tratamientoDebe interrumpirse su administración si tras 4 semanas de tratamiento a las dosis máximas (romiplostim: 10μg/kg/semana y eltrombopag: 75mg/día), el recuento plaquetario no aumenta hasta un nivel suficiente que evite hemorragias clínicamente relevantes.
Efectos adversosLos acontecimientos adversos más frecuentes descritos en los pacientes tratados son leves, predominando la cefalea e infección respiratoria superior.
En el caso de eltrombopag se ha observado aumento de transaminasas y bilirrubina (reversible tras la retirada del fármaco), por lo que debe monitorizarse su determinación antes de iniciar el tratamiento, cada 2 semanas durante el ajuste de la dosis y mensualmente en pacientes con recuentos estables de plaquetas.
En ambos agonistas se han descrito efectos adversos poco frecuentes, como el aumento de reticulina en la médula ósea (reversible tras la retirada del fármaco), complicaciones trombóticas (más en adultos, aunque también hay algún caso pediátrico), por lo que se debe tener precaución cuando se administren a pacientes con factores de riesgo conocidos de tromboembolismo, así como la rara posibilidad de progresión de neoplasias hematológicas y síndrome mielodisplásico.
También se han comunicado 2 casos de cataratas que llevaban tratamiento concomitante con eltrombopag y corticoides y otros 2 casos que desarrollaron anticuerpos anti romiplostim sin reacción cruzada con la TPO endógena.
EsplenectomíaIndicaciones- •
PTI de diagnóstico reciente o persistente: ante urgencia hemorrágica con riesgo vital que no responde a tratamiento previo.
- •
PTI crónica:
- •
Ante urgencia hemorrágica con riesgo vital.
- •
Valorar en mayores de 5 años, sintomáticos, refractarios a tratamientos previos, que presenta interferencia con su vida normal, con más de 2 años de evolución.
- •
Vacunación antineumocócica, antimeningocócica y frente a hemofilus.
- •
Penicilina oral diaria o amoxicilina: hasta un mínimo de 2 años tras la intervención.
- •
Ante síndrome febril sin foco iniciar antibioterapia con cobertura para neumococo, hemofilus y meningococo.
Su experiencia en la PTI en Pediatría es limitada, aunque puede resultar eficaz en pacientes con formas menos graves de trombocitopenia inmune crónica o en cuadros de citopenia inmune (síndrome de Evans o síndrome linfoproliferativo autoinmune). La dosis recomendada es de 20-40mg/kg/día, repartidos en 2 dosis. La tasa de respuesta global puede llegar hasta un 50-60%, con un tiempo de respuesta entre 4 y 6 semanas. Es bien tolerado, y los principales efectos secundarios son cefalea y molestias gastrointestinales33,40,41.
Rituximab. Anticuerpo monoclonal anti-CD20Ha sido empleado en adultos y también en niños, aunque en estos últimos existe menos experiencia33,37,42. Se obtiene una tasa de respuesta entre el 30 y el 60% en función del tiempo de análisis. Su infusión requiere la vigilancia de problemas inmunoalérgicos agudos ocasionalmente graves. Existe riesgo infeccioso por depleción prolongada de linfocitos B y actualmente sigue bajo vigilancia por la descripción de cuadros de leucoencefalopatía multifocal progresiva, comunicada tras la administración en otras enfermedades43. Su administración debe indicarse por uso compasivo, al no estar incluida esta indicación en ficha técnica.
Conflicto de interesesEmilio Monteagudo: asistencia a Advisory Board de Amgen.
Itziar Astigarraga: asistencia al Advisory Board de Nplate-Amgen.
Áurea Cervera: asistencia a 2 Advisory Boards de Nplate-Amgen. Honorarios de Novartis por ponencia en reunión de PTI.
M. Angeles Dasí: asistencia a Advisory Board de Revolade – Novartis. Asistencia al Advisory Board de Nplate-Amgen.
Ana Sastre: asistencia a Advisory Board de Revolade – Novartis. Asistencia al Advisory Board de Nplate-Amgen. Participación en programa de formación online sobre PTI pediátrica patrocinado por Novartis. Participación como ponente en diversas reuniones formativas patrocinadas por Novartis.
Rubén Berrueco: asistencia a Advisory Board de Revolade – Novartis. Participación en programa de formación online sobre PTI pediátrica patrocinado por Novartis.
José Luis Dapena: asistencia a Advisory Board de Revolade – Novartis. Participación en programa de formación online sobre PTI pediátrica patrocinado por Novartis. Asistencia al Advisory Board de Nplate-Amgen.
Presentado y aprobado en el XI Congreso Nacional de la Sociedad Española de Hematología y Oncología Pediátricas, celebrado en Alicante del 31 de mayo al 2 de junio de 2018.

