


La retinosis pigmentaria es una de las principales causas de discapacidad visual infantil, se caracteriza por una degeneración óptica progresiva que conduce a una disfunción visual grave bilateral. Puede ser causada por diversas afecciones oculares y/o sistémicas, entre las que destacan el síndrome de Bardet-Biedl (SBB) y la lipofuscinosis neuronal ceroidea (LNC)1,2.
El SBB y la LNC son enfermedades hereditarias autosómicas recesivas, caracterizadas por un fenotipo de degeneración retiniana bilateral. Además, el SBB se manifiesta por retraso mental, hipogonadismo, obesidad, alteraciones renales, paraparesia espástica y dismorfia en extremidades; y la LNC por encefalopatía progresiva infantil de inicio precoz (entre los 2 y 11 años) con epilepsia y deterioro de la capacidad mental y motora3,4.
El diagnóstico de 2 enfermedades genéticas en un mismo paciente presenta una incidencia muy baja, siendo la consanguinidad el factor principal de riesgo5.
Se expone un caso de un niño con padres no consanguíneos y un hermano mayor sanos. Presentó polidactilia al nacer, dedo completo que salía de la primera falange del 5.° dedo del pie izquierdo, amputado a los 2 años y alteraciones visuales de inicio precoz. A los 3 años es diagnosticado de retinosis pigmentaria con ceguera completa bilateral y se realiza estudio molecular mediante microarray de genotipado (AsperBiotech®), para cribado de 347 mutaciones de 16 genes (BBS1 a BBS13, PHF6, ALMS1, GNAS1), implicados en la distrofia de retina sindrómica. Los resultados de este análisis mostraron la presencia de la variante c.1169T>G (p.Met390Arg) en homocigosis en el gen BBS1, descrita como mutación patogénica en la base de datos ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). La mutación encontrada, al estar en homocigosis, es causa de enfermedad.
El paciente evoluciona con un retraso psicomotor e intelectual de predominio en el lenguaje. A los 8 años, se aprecia una regresión en tareas motoras habituales acompañada de alteraciones del equilibrio, marcha atáxica y pérdida de fuerza. Coincide en el tiempo con la aparición de crisis atónicas y mioclónicas en el contexto de una epilepsia mioclónica progresiva. Clínicamente se comporta como una enfermedad neurodegenerativa, por lo que se solicita estudio molecular de enfermedades lisosomales.
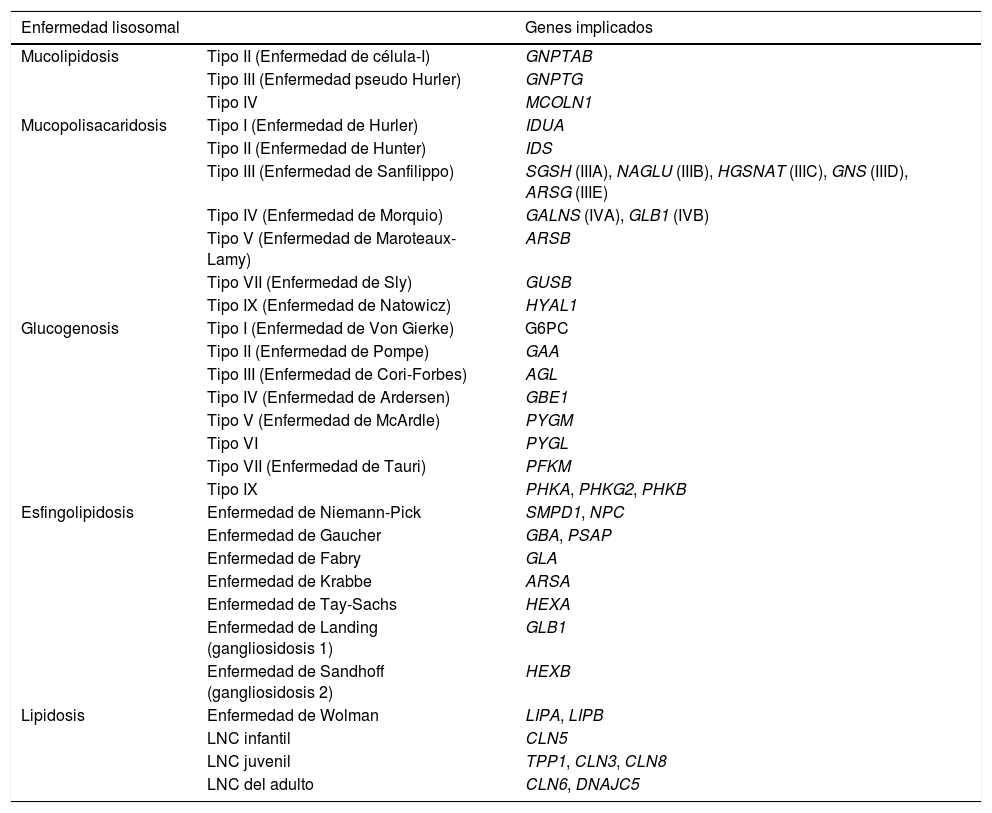
Se realizó secuenciación masiva en la plataforma S5 (Ion Torrent®) de los principales genes implicados en las enfermedades lisosomales (tabla 1). Los resultados muestran la presencia de las siguientes variantes en heterocigosis en el gen CLN5 asociadas a LNC: cambio c.335G>C (p.Arg112Pro) descrito como mutación patogénica en ClinVar; cambio c.835G>A (p.Asp279Asn) descrito como mutación posiblemente patogénica en ClinVar; y la inserción c.291_292insC (p.Ser98fs) que causa un desplazamiento del marco de lectura o frameshift insertion siendo potencialmente patogénica.
Principales genes asociados a las enfermedades lisosomales
| Enfermedad lisosomal | Genes implicados | |
|---|---|---|
| Mucolipidosis | Tipo II (Enfermedad de célula-I) | GNPTAB |
| Tipo III (Enfermedad pseudo Hurler) | GNPTG | |
| Tipo IV | MCOLN1 | |
| Mucopolisacaridosis | Tipo I (Enfermedad de Hurler) | IDUA |
| Tipo II (Enfermedad de Hunter) | IDS | |
| Tipo III (Enfermedad de Sanfilippo) | SGSH (IIIA), NAGLU (IIIB), HGSNAT (IIIC), GNS (IIID), ARSG (IIIE) | |
| Tipo IV (Enfermedad de Morquio) | GALNS (IVA), GLB1 (IVB) | |
| Tipo V (Enfermedad de Maroteaux-Lamy) | ARSB | |
| Tipo VII (Enfermedad de Sly) | GUSB | |
| Tipo IX (Enfermedad de Natowicz) | HYAL1 | |
| Glucogenosis | Tipo I (Enfermedad de Von Gierke) | G6PC |
| Tipo II (Enfermedad de Pompe) | GAA | |
| Tipo III (Enfermedad de Cori-Forbes) | AGL | |
| Tipo IV (Enfermedad de Ardersen) | GBE1 | |
| Tipo V (Enfermedad de McArdle) | PYGM | |
| Tipo VI | PYGL | |
| Tipo VII (Enfermedad de Tauri) | PFKM | |
| Tipo IX | PHKA, PHKG2, PHKB | |
| Esfingolipidosis | Enfermedad de Niemann-Pick | SMPD1, NPC |
| Enfermedad de Gaucher | GBA, PSAP | |
| Enfermedad de Fabry | GLA | |
| Enfermedad de Krabbe | ARSA | |
| Enfermedad de Tay-Sachs | HEXA | |
| Enfermedad de Landing (gangliosidosis 1) | GLB1 | |
| Enfermedad de Sandhoff (gangliosidosis 2) | HEXB | |
| Lipidosis | Enfermedad de Wolman | LIPA, LIPB |
| LNC infantil | CLN5 | |
| LNC juvenil | TPP1, CLN3, CLN8 | |
| LNC del adulto | CLN6, DNAJC5 | |
LNC: lipofuscinosis neuronal ceroidea.
La LNC es autosómica recesiva, por lo que las mutaciones en heterocigosis no serían causa suficiente de enfermedad, a no ser que afecten a los 2 alelos, lo que se denomina heterocigosis compuesta. Para demostrar la heterocigosis compuesta es necesario que una de las mutaciones esté presente en el alelo materno y la otra en el paterno, por lo que se realizó estudio genético de sus progenitores. Las mutaciones c.835G>A (p.Asp279Asn) y c.335G>C (p.Arg112Pro) se encontraron en heterocigosis en la madre y la inserción c.291_292insC (p.Ser98fs) en heterocigosis en el padre. El estudio de sus progenitores confirma la heterocigosis compuesta, al comprobar que las mutaciones detectadas en el gen CLN5 afectan a los 2 alelos del probandus. Las 2 variantes encontradas en heterocigosis en la madre han sido trasmitidas a su hijo, lo que demuestra que ambas mutaciones se encuentran en el mismo alelo y descarta la heterocigosis compuesta en la madre. También se realizó estudio genético a su hermano, encontrándose la inserción c.291_292insC (p.Ser98fs) en heterocigosis (tabla 2).
Mutaciones familiares encontradas en el gen CLN5
| Mutaciones | Probandus | Padre | Madre | Hermano |
|---|---|---|---|---|
| c.335G>C (p.Arg112Pro) | Heterocigosis | Ausente | Heterocigosis | Ausente |
| c.835G>A (p.Asp279Asn) | Heterocigosis | Ausente | Heterocigosis | Ausente |
| c.291_292insC (p.Ser98fs) | Heterocigosis | Heterocigosis | Ausente | Heterocigosis |
Con estos resultados, se concluye que el paciente diagnosticado de SBB con la mutación c.1169T>G (p.Met390Arg) en homocigosis en el gen BBS1, es además, heterocigoto compuesto de las mutaciones c.335G>C (p.Arg112Pro), c.835G>A (p.Asp279Asn) y c.291_292insC (p.Ser98fs) en el gen CLN5, siendo la causa molecular de la LNC. Su madre, padre y hermano son portadores de alguna de las mutaciones familiares.
La presencia de distrofia retiniana grave de inicio temprano junto con el desarrollo de manifestaciones neurodegenerativas en el paciente, no se ajustaban al diagnóstico de SBB de manera exclusiva, esto derivó en la ampliación del estudio molecular hacia la búsqueda de una segunda enfermedad genética, demostrándose el diagnóstico de la LNC con fenotipos clínicos solapados. La concomitancia del SBB y la LNC en un mismo paciente es un hallazgo excepcional, siendo este caso el primero que se describe en la literatura científica.

