

Autoimmune hemolytic anemia (AIHA) is a rare and generally self-limiting disease in children.
Material and methodsA descriptive cross-sectional study was performed in children under 18 years diagnosed with AIHA from January 1997 to July 2019. Clinical variables were collected and AIHA was classified according to the direct antiglobulin test (DAT) in warm AIHA (IgG+/-C3d) and cold AIHA (C3d). Response to treatment and evolution were analyzed.
Results25 patients were included and 72% were males. The median age at diagnosis was 2 years (range 0.4–9). Fever (72%), pallor (68%), jaundice (64%), hepatosplenomegaly and coluria (48%) were the most common presenting symptoms. The median hemoglobin at diagnosis was 5.4g/dl. DAT was positive in 96%, with detection of IgG antibodies in 76%. A single case presented negative DAT. 20% of the patients associated another cytopenia, one of which was subsequently diagnosed with common variable immunodeficiency. Concomitant viral infection was suspected or documented in 32%. Most of the cases were self-limiting and responded to corticosteroid treatment (72%). Those with partial response (24%), mainly those associated with other cytopenias, required other lines of treatment (rituximab, mycophenolate, immunoglobulins,…). Complications (32%) and relapses (26%) were detected only in warm AIHA.
ConclusionsOur case series confirms that AIHA is a very rare disease in childhood. Most cases evolve favorably, although up to a quarter of them require second lines of treatment and, in exceptional cases, they need very aggressive treatments. These latter cases generally correspond to patients who present more than one cytopenia in the course of the disease.
La anemia hemolítica autoinmune (AHAI) es una enfermedad rara en niños, generalmente autolimitada.
Material y métodosEstudio descriptivo transversal en menores de 18 años diagnosticados de AHAI desde enero 1997 a julio 2019. Se recogieron variables clínicas y se clasificaron según el test de Coombs directo (TCD) en AHAI por anticuerpos calientes (IgG+/-C3d) y fríos (C3d). Se analizó la respuesta al tratamiento y su evolución.
ResultadosSe incluyeron 25 pacientes, siendo el 72% varones. La mediada de edad al diagnóstico fue 2 años (rango 0,4–9). Los síntomas predominantes fueron fiebre (72%), palidez (68%), ictericia (64%), hepatoesplenomegalia y coluria (48%). La mediana de hemoglobina al diagnóstico fue 5,4gr/dl. En el 96% el TCD fue positivo, con detección de anticuerpos IgG en el 76%. Un solo caso presentó TCD negativo. Un 20% asociaron otra citopenia, uno de ellos fue diagnosticado posteriormente de inmunodeficiencia variable común. En un 32% se sospechó o documentó una infección viral concomitante. La mayoría de los casos fueron autolimitados y respondieron a tratamiento con corticoides (72%). Aquellos con respuesta parcial (24%), principalmente los que asociaban otras citopenias, precisaron otras líneas de tratamiento (rituximab, micofenolato, inmunoglobulinas,…). Se detectaron complicaciones (32%) y recaídas (26%) únicamente en AHAI por anticuerpos calientes.
ConclusionesNuestra serie confirma que la AHAI es una enfermedad muy poco frecuente en la infancia. La mayoría de los casos evolucionan favorablemente, aunque hasta una cuarta parte precisan segundas líneas de tratamiento y casos excepcionales tratamientos muy agresivos. Estos últimos suelen corresponder a pacientes con más de una citopenia en la evolución de la enfermedad.
Autoimmune hemolytic anaemia (AIHA) is an immune disorder characterised by the presence of autoantibodies that attack the membrane of red blood cells, shortening the mean life span of these cells. It is a rare disease in the paediatric age group, with an estimated incidence of 0.8–1.25 cases per 100 000 children,1,2 although it is the leading cause of extracorpuscular haemolytic anaemia.
The onset can occur at any age, with the incidence peaking at around 3 or 4 years. From an etiological perspective, cases of AIHA can be classified as secondary or primary/idiopathic. In the paediatric population, more than half of the cases of AIHA are associated with another disease, and 40%–50% are idiopathic.3,4
Contrary to cases in adults, pediatric cases most frequently present as a self-limiting disease associated with viral infection. However, in children aged less than 2 years and adolescents, there are more chronic cases that may or may not be associated with systemic diseases, chiefly immunodeficiencies or autoimmune disorders.5,6
Due to the low prevalence of AIHA in the paediatric population, the studies published to date have had a retrospective design and have been conducted in small samples that were not obtained through randomization. As a result, the scientific evidence available for management of AIHA in this population is very limited.7
The aim of our study was to describe the different clinical forms and diagnosis of AIHA in the paediatric catchment population of a tertiary care hospital in the past 22 years and to analyse the approach and response to treatment and patient outcomes.
Material and methodsWe conducted a cross-sectional descriptive study at the Niño Jesús University Children’s Hospital, a tertiary care hospital with a specific department of paediatric haematology and oncology. The study period ranged from January 1997 to July 2019.
Through the coding service, we obtained a list of the patients given a diagnosis of AIHA during this period, and then reviewed their health records. We selected patients aged less than 18 years that met the following criteria for definition of AIHA: a) presence of anemia defined as a low haemoglobin (Hb) concentration more than 2 standard deviations below the population mean adjusted for age; b) evidence of hemolysis (reticulocytosis, elevation of indirect bilirubin, lactate dehydrogenase [LDH], and/or decreased haptoglobin level); c) positive serological/immunological tests; d) negative serological/immunological tests in the presence of laboratory parameters compatible with hemolysis, exclusion of hereditary forms of haemolytic anemia and favourable response to standard treatment.
We collected data on the following variables: sex, age at diagnosis, year of onset, clinical presentation, diagnostic tests performed, treatment received and response to treatment, duration of follow-up and development of complications.
We classified AIHA based on the results of the direct antiglobulin test (DAT, also known as direct Coombs) as: a) warm antibody AIHA, in case of positive IgG results independently of complement activation (C3d); b) cold AIHA in cases caused by cold-reacting antibodies with activation of complement alone (C3d-positive) or c) DAT-negative AIHA.
We also classified response to treatment in 3 categories: a) complete response (CR) to refer to achievement of a Hb concentration of 11g/dL or greater with a reticulocyte count of less than 120×109/μL, allowing discontinuation of treatment; b) partial response (PR) to refer to achievement of a Hb concentration of 7–11g/dL or a reticulocyte count greater than 120×109/μL; c) lack of response (NR) to refer to a Hb concentration remaining under 7g/dL or any other situation requiring sustained treatment.5
We performed a descriptive and inferential statistical analysis with the software SPSS version 2.0.
ResultsWe identified 40 patients that met the inclusion criteria. Of this total, we excluded 15 for the following reasons: 13 due to development of AIHA following hematopoietic stem cell transplantation (HSCT), and 2 because we were unable to access the health records of the patients. The final sample included 25 patients. More than half (72%) were male, and 28% were female. The median age at diagnosis was 2 years (range, 0.4–9).
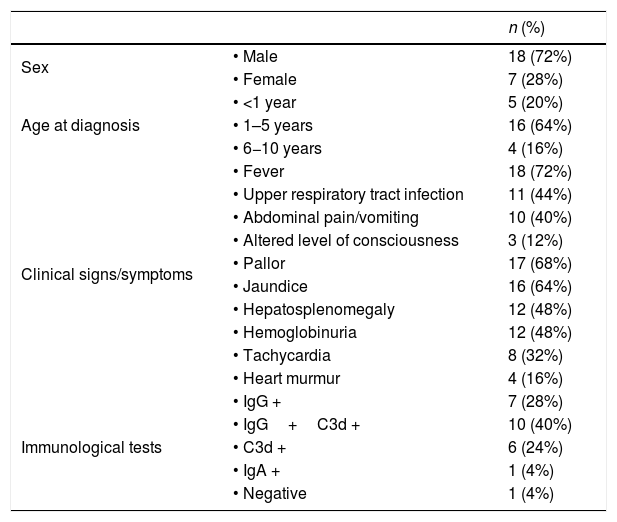
In the sample under study, 72% of patients had a fever 3–10 days before the onset of AIHA and only 44% had other symptoms suggestive of infection. Other frequent symptoms were vomiting and/or abdominal pain (40%). Three patients (12%) had onset with altered level of consciousness. The most frequent presenting symptom was a change in the colour of the skin and mucosae in the form of pallor (68%) or jaundice (64%), followed by hepatosplenomegaly (48%) and choluria (48%). Tachycardia was detected in 32% and heart murmurs in 16% (Table 1). Of the 25 patients, 14 (56%) required admission to the intensive care unit for initial management due to the risk of haemodynamic instability.
Characteristics of the 25 paediatric patients with AIHA.
| n (%) | ||
|---|---|---|
| Sex | • Male | 18 (72%) |
| • Female | 7 (28%) | |
| Age at diagnosis | • <1 year | 5 (20%) |
| • 1–5 years | 16 (64%) | |
| • 6−10 years | 4 (16%) | |
| Clinical signs/symptoms | • Fever | 18 (72%) |
| • Upper respiratory tract infection | 11 (44%) | |
| • Abdominal pain/vomiting | 10 (40%) | |
| • Altered level of consciousness | 3 (12%) | |
| • Pallor | 17 (68%) | |
| • Jaundice | 16 (64%) | |
| • Hepatosplenomegaly | 12 (48%) | |
| • Hemoglobinuria | 12 (48%) | |
| • Tachycardia | 8 (32%) | |
| • Heart murmur | 4 (16%) | |
| Immunological tests | • IgG + | 7 (28%) |
| • IgG+C3d + | 10 (40%) | |
| • C3d + | 6 (24%) | |
| • IgA + | 1 (4%) | |
| • Negative | 1 (4%) |
The median Hb concentration at diagnosis was 5.4g/dL (range, 2.3–10.7). All patients had a high reticulocyte count save for one that had reticulocytopenia. There were high proportions of patients with elevated indirect bilirubin (92%), undetectable haptoglobin levels (88%) and elevated LDH (84%). Other cytopenias were detected during the evaluation in 5 patients. Two of them had an initial presentation of AIHA and experienced episodes of immune thrombocytopenia (ITP) during the follow-up; other patients had concomitant AIHA and ITP at the time of diagnosis, and the remaining patient had ITP first and developed AIHA 5 years later.
In most cases (76%) AIHA was caused by warm antibodies (IgG or IgA), while the remaining 24% were caused by cold-reacting antibodies. Two patients had a negative polyspecific DAT. In one of them, a monospecific irregular antibody test (Panocell Immucor®) performed in a reference laboratory had a strong positive result for IgA as the causative antibody. In the other, the evaluation was expanded with more complex immunological and serological tests, ruling out hereditary forms of haemolytic anaemia (normal results of haemoglobin electrophoresis, osmotic fragility test, endomysial antibodies, and enzymatic determination of glucose-6-phosphate dehydrogenase), and eventually the patient responded to the prescribed treatment with steroids (Table 1).
Most patients underwent immunologic and autoimmunity tests at diagnosis (80%). These studies included measurement of immunoglobulins, antinuclear antibodies and complement activation. In patients that experienced relapses, developed cytopenia or were refractory to steroid therapy (10/25), additional tests were performed before initiation of second-line treatment, including lymphocyte subset tests, apoptosis assays, tests for detection of other antibodies (anti-thyroid, anti-DNA, anti-extractable nuclear antibodies [ENA], antineutrophil cytoplasmic antibodies [ANCA], antiphospholipid) and vaccine response test with measurement of the titres of antibodies against tetanus, diphtheria or pneumococcus. In a single patient (4%) that also had intestinal disease and IgA deficiency, these tests led to diagnosis of common variable immunodeficiency (CVID) during the follow-up. Five patients (20%), all of them with warm AIHA caused by IgG, had concomitant thrombocytopenia (Evans syndrome [ES]).
The records of 8 patients (32%), all of them with warm AIHA, documented the suspicion of viral infection at the time of diagnosis. The main microorganisms isolated in serological tests were Epstein-Barr virus (EBV, n=2), cytomegalovirus (CMV, n=3), parvovirus B19 (n=1), varicella-zoster virus (VZV, n=1) and Mycoplasma pneumoniae (n=1).
Steroid therapy was the first-line treatment in 24 patients (96%). The only exception was a patient that received intravenous antibiotherapy to treat the concurrent infection and a blood transfusion because it was a case of cold AIHA. Treatment achieved a CR in 18 patients (72%) and a PR in 6 (24%). The remaining patient had an initial response to steroid therapy during the acute phase of disease, but we do not know the definitive outcome because the patient was lost to follow-up due to transfer to a different facility after the initial stabilization. Patients with a PR received alternative treatments (rituximab, mycophenolate mofetil [MPM], intravenous immunoglobulin [IVIG], ciclosporin); 23 patients (92%) required transfusion of blood products in the context of anemia
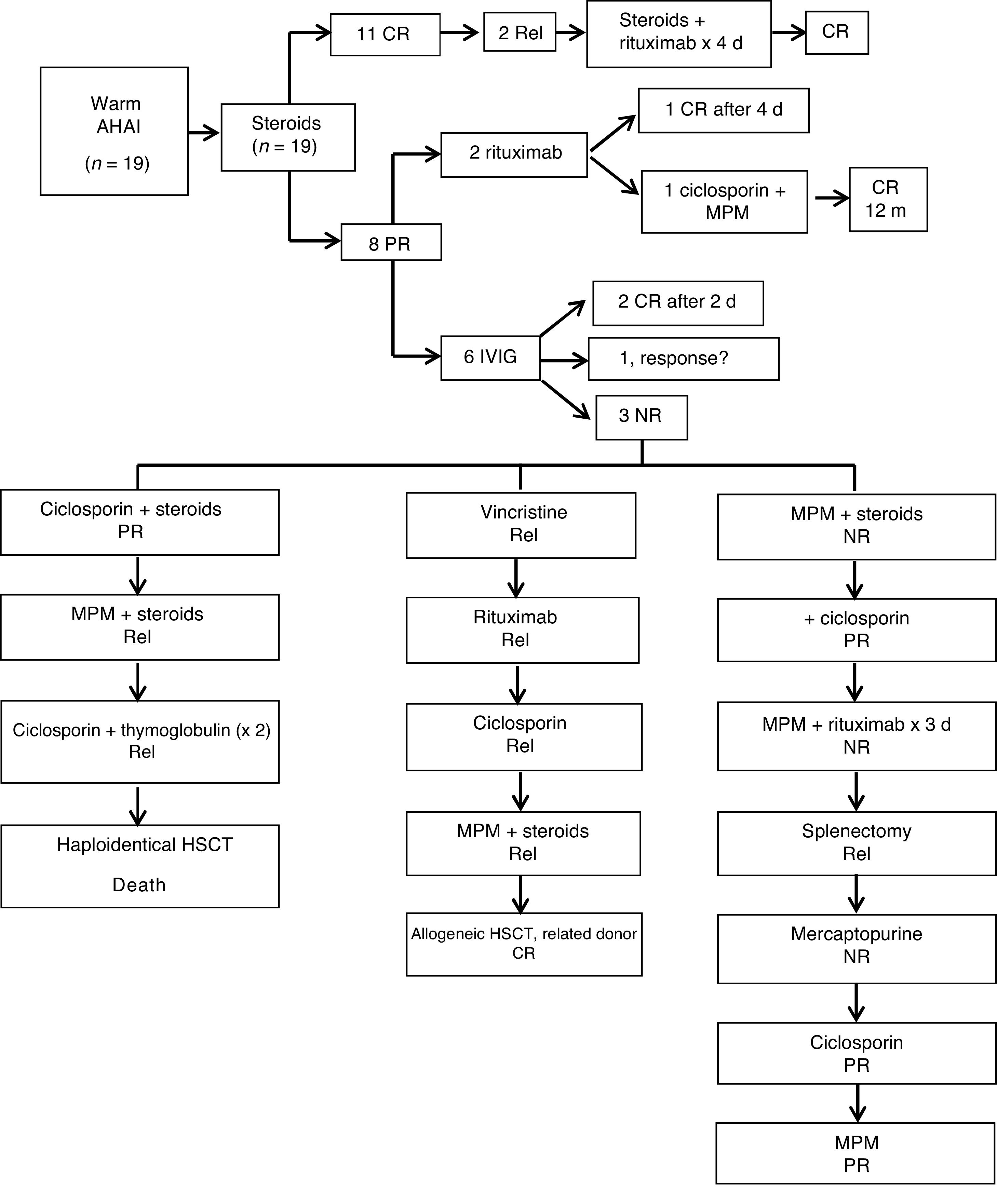
In the subset of patients with warm AIHA (Fig. 1), the first-line treatment was steroid therapy in all (19/19; 2mg/kg/day administered orally in 3 doses). Of this total, 11 (58%) achieved CR after a median duration of steroid therapy of 3 months (range, 0–5 months). The remaining 8 (42%) had a partial response and required other treatments to achieve a CR. Two of these patients received intravenous rituximab (375mg/m2/week). One of the patients with a negative DAT result who responded and achieved a CR after completing a cycle of 4 doses of rituximab. The other patient with a negative DAT result had a serious allergic reaction to the third dose and was switched to ciclosporin (90mg/12h orally for 4 months). Since this coincided with a new infection, in addition to treatment with ciclosporin (50mg/12h), MPM was added (400mg/8h) for a duration of 5 months, and the patient finally achieved a CR 12 months after the diagnosis. In 5 of the 8 patients with PR, AIHA was associated with ITP (ES), which required treatment with IVIG (1g/kg) as adjuvant therapy in addition to the steroids. Three of the 5 experienced a protracted course of disease, requiring various additional treatments: MPM (500mg/12h), mercaptopurine (50mg/m2/day), ciclosporin (−50–150mg/12h), thymoglobulin (3.75mg/kg/day for 5 days), vincristine (1−2mg/day, IV), splenectomy, and 2 required HSCT. The median duration of follow-up was 3.5 years (range, 1–17). Twenty-six percent (5/19) experienced relapses during the follow-up. Six (32%) had complications, mainly recurrent infection, 2 developed gallstones and 1 had significant growth delay secondary to prolonged steroid therapy. There was only 1 death, in a patient that died after HSCT.

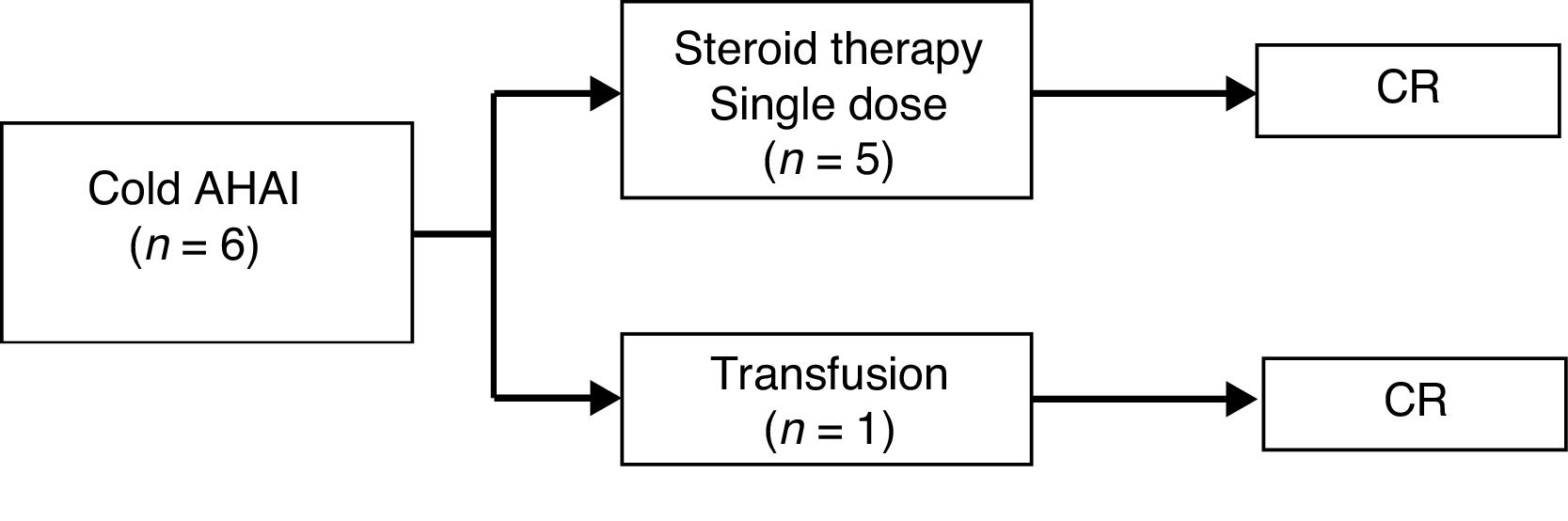
As for the patients with cold AIHA (caused by IgM antibodies), 5 out of the 6 (83%) received a single dose of steroid therapy. In the remaining patient, the anemia resolved after treating the concomitant infection and a blood transfusion, as previously noted. All achieved CR. The median duration of follow-up was 3 months (range, 0–7 months). There were no complications or relapses during follow-up (Fig. 2).
Discussion
Autoimmune hemolytic anemia is a rare disease in the pediatric age group, as demonstrated by the few cases identified in our retrospective series over a time span of more than 20 years. Most of the cases in this series of AIHA were self-limited and responded to steroid therapy (72%) and therefore, despite the initial severity of AIHA, it could be considered a benign disease in a high proportion of patients.
In both children and adults, AIHA is most frequently caused by warm antibodies (70%–80%),8,9 which was corroborated in by our findings (76%). When hemolytic anemia is diagnosed, it is important to exclude the possibility of it being secondary to other conditions (lymphoproliferative diseases, autoimmune diseases, primary immunodeficiencies, pharmacotherapy or infection), as in most cases it results from an underlying condition.10,11 At present, most diagnostic algorithms2,5 recommend investigation of the immune response and autoimmunity in all patients at the time of diagnosis (antiphospholipid and antinuclear antibodies, determination of immunoglobulins, lymphocyte subsets and complement) to be repeated yearly even in case of normal results, as the underlying condition may only become apparent after the diagnosis of AIHA. In our opinion, these tests should be performed in the initial evaluation of all atypical cases, in refractory cases and whenever a relapse occurs. Also, depending on the medical history, the findings of the physical examination and the results of diagnostic tests, additional tests may be indicated, such as bone marrow biopsy and aspiration, imaging tests or flow cytometry, based on the suspected diagnosis.
A high percentage of the patients in our experienced symptoms of infection prior to the onset of AIHA, but a positive microbial isolation was only achieved in half of these cases. The isolated pathogens were predominantly viral (CMV, EBV, parvovirus B19, VZV) and there was also a case of Mycoplasma pneumoniae, which was consistent with the previous literature.12,13 Most of these infections are associated with IgM cold agglutinins.14 Although viral infections are a frequent cause of AIHA in the pediatric population, it is difficult to determine whether the infection is the trigger or primary cause of the anemia.
Five patients in our cohort received a diagnosis of ES. Compared to patients with isolated AIHA, patients with ES usually have chronic AIHA that is recurrent and refractory to treatment.15,16 In our series, none of these patients achieved a CR with steroid treatment alone, and they all required additional treatment, which supports the poorer outcomes described in the previous literature. A significant proportion of patients with associated ES have underlying autoimmune lymphoproliferative syndrome (ALPS),17,18 and therefore it is important to rule out this disease in these patients as well as other autoimmune diseases and immunodeficiencies, as discussed above. In our cohort ALPS was not confirmed in any of the cases. There may be other rheumatologic and immunologic comorbidities, such as common variable immunodeficiency (CVID).19 In our study, one of the patients with ES received a diagnosis of CVID during the follow-up.
A positive DAT confirms the diagnosis of AIHA in the investigation of acquired hemolytic anemia. However, up to 3%–11% of patients, depending on the case series, have negative DAT results.20–22 The involvement of a low-affinity IgG antibodies, IgG sensitization below the threshold of detection of the commercial reagent or the presence of IgA antibodies that are not detected in routine DATs are some of the main reasons that patients with AIHA may have a negative DAT.23–25 When the clinical suspicion of AIHA is strong despite a negative DAT, performance of additional, more complex immunological or serological tests in a reference laboratory should be contemplated,26 as well as performance of the Donath-Landsteiner test to rule out involvement of bithermal hemolysins. Two patients in our cohort had a negative DAT and the workup was expanded in a reference laboratory. In one of these patients, IgA was identified as the causative antibody. In the other, all diagnostic tests were negative, but a clinical diagnosis of warm AIHA was made on account on the clinical manifestations and the response to treatment, with a tendency to relapse following discontinuation of steroid therapy. When other tests cannot be performed or the results are inconclusive but the clinical and laboratory features are compatible with AIHA, once hereditary causes of hemolytic anemia are ruled out, the approach to management should be the same as in cases with a positive DAT.26
Due to the lack of randomised controlled trials, the approach to the management of pediatric patients with AIHA is based on evidence from case series and expert opinion.27
There is general agreement that steroids should be the first line of treatment for warm AIHA. Between 70% and 85% of cases respond with exclusive steroid therapy, with a median time to detection of a response of 15 days (range, 1–3 weeks).28 It is important to do a slow and gradual tapering off, in many cases over as many as 6 months. Approximately 20%–30% of patients require second-line treatment, which is initiated in refractory or relapsed cases.28 Some studies support the use of rituximab as the drug of choice for second-line treatment, achieving a response within a median of 1.4 months (range, 0.5–5 months).29,30 Other authors propose splenectomy as the second step of treatment, which is successful in 60%–90% of patients.31 However, no studies in the literature have compared the efficacy of the available second-line drugs and splenectomy. In addition, performance of splenectomy is not recommended in patients aged less than 6 years. The use of other drugs, such as MPM, azathioprine, ciclosporin, immunoglobulins or alemtuzumab, is reserved for refractory cases. There is no conclusive evidence on the response achieve by these drugs, so there is no consensus as to the superiority of any given drug over the rest.32 Thus, the selection of treatment is made on a case-by-case basis based on the hematological response achieved and the potential adverse effects of the drug. In cases of cold AIHA, symptomatic treatment and treatment of the underlying cause is recommended.33 Transfusion should be reserved for cases of severe anemia or abnormal vital signs.34 In our sample, most patients received at least 1 transfusion. The response rates were consistent with those reported in other series. We ought to highlight that the first signs of a response do not appear until at least 2 weeks from initiation in AIHA cases caused by IgG, so it is recommended that the treatment not be switched until at least that much time has elapsed. Patients with a diagnosis of ES require treatment with several drugs and, also in agreement with the previous literature, a more complicated course of disease is expected in this subset.
The main limitations of our study are the small sample size, the difficulty in determining the time some of the tests were performed and the losses to follow-up. Nevertheless, our findings were similar to those of previous studies in the pediatric population. Thus, we consider that our study contributes relevant information on the clinical course of AIHA in children.
In conclusion, we once again highlight that AIHA is a rare disease in children, with a favourable outcome in most cases on account of an infectious etiology. However, in some cases it can have a protracted course. In these cases, it is important to rule out potential underlying diseases that are more severe, such as immunodeficiencies or inflammatory diseases, and second-line drugs may be needed. Therefore, we would recommend that at least this subset of patients with poorer outcomes be referred to hospitals with experience in the management of this disease.
FundingThe study did not receive any form of funding.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Sánchez MN, Zubicaray J, Sebastián E, Gálvez E, Sevilla J. Autoimmune hemolytic anemia: Case review. An Pediatr (Barc). 2021;94:206–212.

