

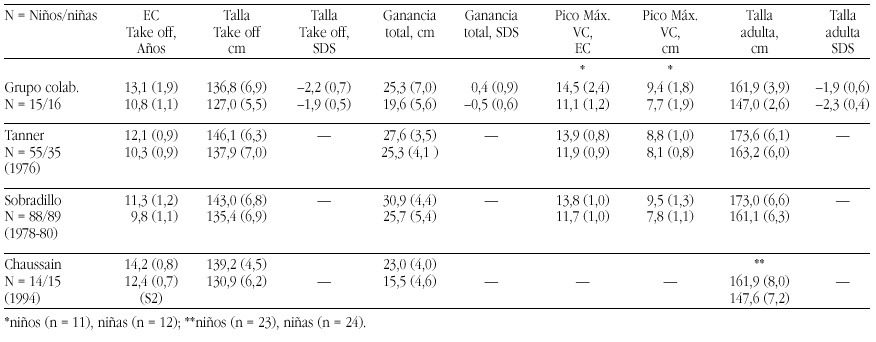
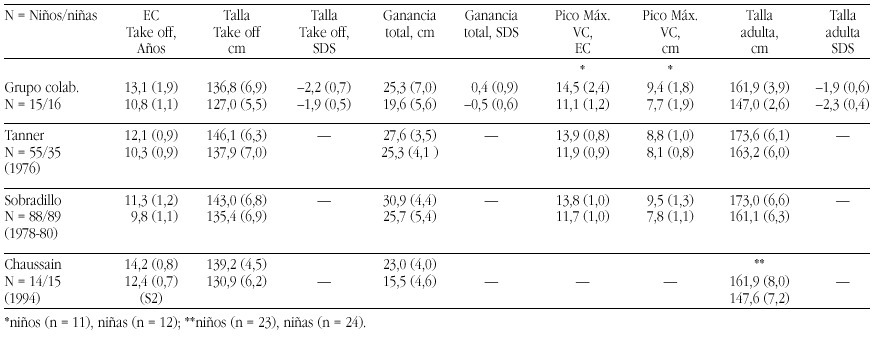
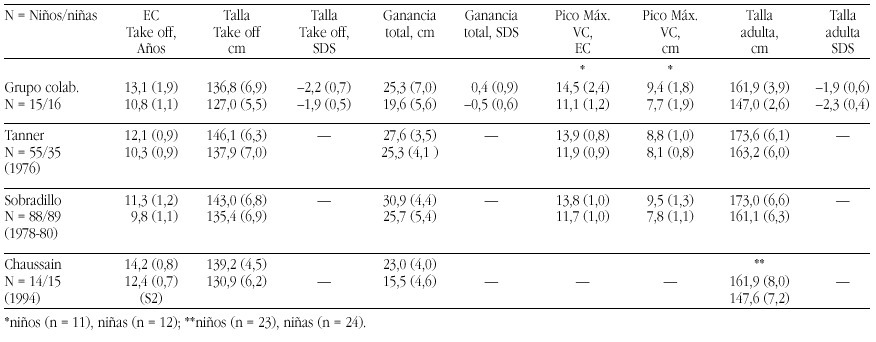
The recent advances in molecular genetic diagnostics have led to the identification of two so far undescribed congenital endocrine syndromes. The patients had been followed in the department for pediatric endocrinology at the University Children's hospital Berlin and their clinical symptoms had been observed for a long time. However, only the possibility of molecular genetic study made it possible to clarify the cause of the complex syndromes.
The POMC-Deficiency Syndrome
Two patients were identified with isolated ACTH-deficiency. The diagnosis was made in the girl in the first month, because her older brother had died at the age of 4 months from liver failure caused by cirrhosis following prolonged jaundice and cholestasis. Postmortem adrenal hypoplasia was recognized and immunostaining of his pituitary revealed the absence of ACTH staining cells, while immunostaining for all the other pituitary hormones was normal. In the girl jaundice and cholestasis developed in the third week of life and Cortisol and ACTH measurements resulted in levels below the detection limits. With hydrocortisone replacement jaundice and cholestasis resolved rapidly and no adrenal crisis was observed. Thus the diagnosis of isolated ACTH-deficiency was made. The second patient, a boy, presented at the age of 1 year with hypoglycemia during febrile convulsions, after a period of postnatal hypoglycemia had resolved after one week with glucose infusions. The endocrine work-up of this patient surprisingly revealed complete ACTH-and cortisol deficiency and he was substituted with hydrocortisone subse quently. During the follow-up at the department for pediatric endocrinology a dramatic and early onset increase in body weight leading to a BMI of 30 and 29 at the ages of 4 and 6 years. Height in both children was above the 97th percentile. Moreover it was recognized that both children had red hair (fig. 1a and b). The etiology of the this syndrome of ACTH deficiency, red hair pigmentation and obesity remained obscure, until the molecular basis of the obesity in the agouti overexpressing mice was clarified. These mice overexpress the agouti protein which is an antagonist to the natural POMC- cleavage peptid alpha-MSH at the MC1 and MC4 receptor, but not at the MC2 (ACTH)-Receptor of the adrenal gland (fig. 2).


Figura 2. The POMC -Melanocortinreceptor- System
Since alpha MSH and ACTH, the natural ligand to the MC2 receptor derive from the POMC molecule, we put for ward the hypothesis that the association of symptoms observed in the two children was caused by a mutation of the POMC-gene. Sequencing of the gene identified a mutations in both patients: The boy was homozygous for a C 3804 A mutation in exon2 and the girl was compound heterozygous for a G 7013 T and a C 7133 del mutation in exon3. Since all mutations abolish the production of alpha MSH and ACTH the symptoms of obesity (MC4 receptor), red hair pigmentation (MC 1 receptor) and ACTH deficiency (MC2 receptor) could be explained. Since the description we were able to identify homozygosity or compund heterozygosity for mutations (C3804 A, C 7133 del, 7102 ins GG) in 3 other patients with exactly the same association of symptoms. Moreover we were able to perform a prenatal diagnosis of heterozgosity in a subsequent pregnancy of the mother of the female patient. This girl is now one year old and had no adrenal insufficiency or red hair, neither did she develop obesity. Moreover the clarification of the molecular cause of their obesity enables the pharmacological intervention
The LHX3 Syndrome
Three children of five children of consanginous parents suffering from combined pituitary hormone deficiencies had been followed for several years at our department. The first child, a daughter was diagnosed with central hypothyroidism and growth hormone deficiency at the age of nine months after the family had immigrated to Germany and in the second female child the diagnosis of bTSH and growth hormone deficiency was established by clinical diagnosis at the age of three months. Both girls also developed hypogonadotropic hypogonadism. The third child as well as the fifth child- is healthy, while diagnosis of central hypothyroidism and growth hormone deficiency was made at the age of two weeks in the fourth child, a boy. All three affected children were substituted with l-thyroxine, growth hormone and at the age of puberty the two girls obtained estrogen replacement, while the boy is still prepubertal with an age of 9 years. The peculiar finding in all affected children was a short neck and an impaired ability to rotate the head (fig. 3). MRI imaging studies had revealed small hypoplastic pituitary remnants in all affected children, while no abnormalities of the spine, bones and neck muscles could be visualized on X-ray or MRI.

Targeted disruption of LHX3, a member factor of the lim-domain containing transcription factor family, had led to a similar phenotype-GH, gonadotropin and TSH deficiency- and moreover LHX3 was shown to be expressed in the ventral proximal motor neurons. LHX3 is a transcription factor expressed in the early embryonic pituitary development.
Since pit-1 and prop-1 mutations had been ruled out in the familial cases, we collaborated with I. Netchine and s. Amselem, who had cloned the human LHX3 gene. Sequencing of the LHX3 gene in the familial cases and in the male patient revealed homozygosity for two different in the affected members of the two families: A missense mutation (Tyr 116 Cys) in the first family and a 23 bp mutation in the homeodoamin leading to a frameshift in the boy of the second family, while the parents and unaffected siblings of family one were hetereozygotes. Thus a new syndrome of combined pituitary hormone deficiencies and impaired neck rotation could be attributed ton LHX3 deficiency in early embryonic development.

