

La seudoacondroplasia (PSACH; MIM #177170) es una osteocondrodisplasia autosómica dominante rara con una prevalencia estimada de 1/30.000 individuos. Las manifestaciones son marcha con base de sustentación amplia («marcha de pato») desde el inicio de la deambulación y estancamiento de la talla desde los 2 años, que evoluciona a talla baja desproporcionada con extremidades cortas, braquidactilia y degeneración articular progresiva precoz. Destaca la normalidad de la longitud al nacer y la ausencia de dismorfias faciales y discapacidad intelectual. Es debida a mutaciones del gen COMP, localizado en 19p13.11, que codifica la proteína oligomérica de la matriz del cartílago (COMP)1. Las mutaciones producen mal plegamiento de la proteína COMP y acumulación de esta en el retículo endoplasmático de condrocitos metafisarios, estrés oxidativo y muerte celular con pérdida de condrocitos en la placa de crecimiento. La displasia epifisiaria múltiple (MED) autosómica dominante también se asocia a mutaciones del gen COMP, aunque es genéticamente heterogénea con mutaciones en los genes MATN3, COL9A1, COL9A2 y COL9A3.
Se presentan un caso de novo y otro familiar.
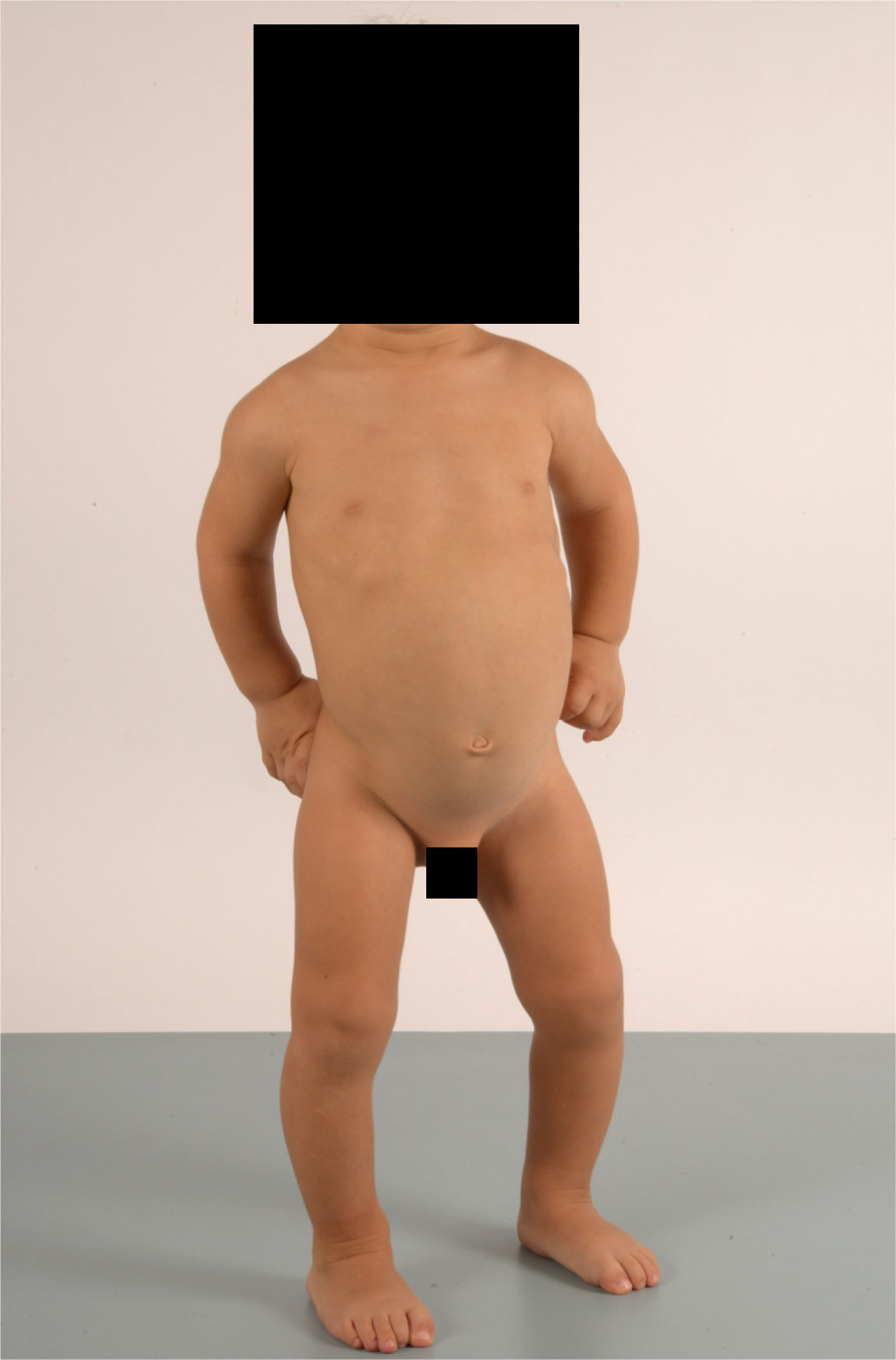
Niño de 2 años (caso1) derivado a consultas externas de Pediatría por cojera desde el inicio de la deambulación. Nacido a las 41 semanas de gestación con peso de 3.510g (0,07 DE), longitud de 53cm (1,29 DE) y perímetro craneal de 36cm (0,38 DE). Niña de 4 años (caso2) derivada por estancamiento de la talla desde los 2 años. Nacida a las 42 semanas de gestación con peso de 3.360g (–0,29 DE), longitud de 50cm (–0,38 DE) y perímetro craneal de 33cm (–1,5 DE). Ambos aportan ecografías prenatales normales y refieren antecedentes familiares sin interés. En el momento del diagnóstico, el caso1 presenta peso de 11,9kg (–0,91 DE) y talla de 82,5cm (–2,36 DE); el caso2, peso de 14,5kg (–1,32 DE) y talla de 92,2cm (–3,44 DE). La exploración física de ambos muestra talla baja, rizomelia de extremidades superiores, braquidactilia, tibias varas, lordosis lumbar y escoliosis leve (fig. 1). También destaca hiperlaxitud ligamentosa y «marcha de pato».
 con talla baja, braquidactilia con rizomelia y tibias varas.")
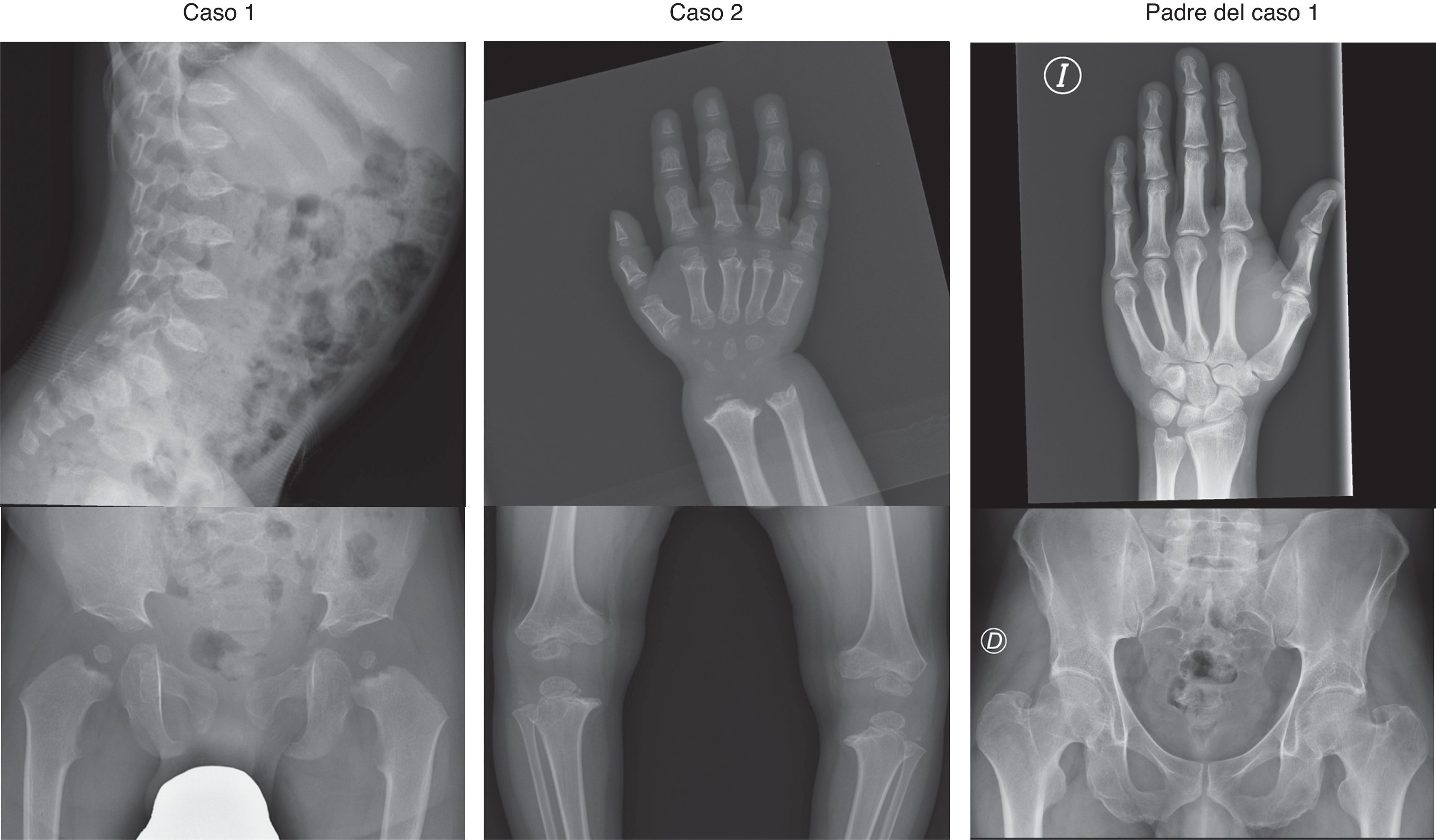
La serie esquelética muestra en ambos osificación retardada de epífisis y metáfisis e irregularidad a nivel de huesos largos, cabeza femoral y acetábulo (fig. 2). En la mano se observan huesos del carpo y metacarpo cortos, falanges con epífisis en forma de cono y metáfisis irregulares. Destacan cuerpos vertebrales con «lengüetas» anteriores. Se realiza analítica sanguínea que resulta sin alteraciones. Se considera el diagnóstico diferencial entre PSACH, acondroplasia, MED y mucopolisacaridosis tipo IV.

Casos 1 y 2: cuerpos vertebrales con «lengüetas» anteriores; huesos del carpo y metacarpo cortos e irregulares, falanges con epífisis en forma de cono; irregularidad de huesos largos, cabeza femoral y acetábulo. Padre del caso1: radiografías de mano y pelvis compatibles con la normalidad.
El estudio de exones y regiones intrónicas adyacentes del gen COMP (NM_000095.2) mostró en el caso1 una mutación en heterocigosis c.1552G>C en el exón 14, que produce la sustitución de aminoácidos p.Asp518His en el dominio T38, ya descrita en asociación con PSACH por Deere et al.2. En el caso2, mostró la mutación en heterocigosis c.868G>T en el exón 9, que produce la sustitución de aminoácidos p.Asp290Tyr en el dominio T31. Dicha mutación no ha sido descrita anteriormente, pero otras dos mutaciones que afectan este codón (p.Asp290Gly y p.Asp290Asn) se han asociado con PSACH3,4. Cuatro predicciones in silico indican que se trata de una mutación patogénica. Se realiza secuenciación del gen COMP en progenitores, que resulta positiva para el padre del caso1, mostrando la misma mutación que en el caso1. Dicho padre presenta talla de 165cm (–1,95 DE, acorde con el contexto familiar) y dismetría de extremidades inferiores (1,5cm), con serie esquelética normal excepto pelvis algo estrecha e hiperlordosis lumbar.
El padre del caso1 supone un reto diagnóstico, al no presentar clínica ni radiología compatibles con el diagnóstico de PSACH ni de MED pero ser portador de mutación patogénica en el gen COMP ya descrita en relación con PSACH3. A pesar de que existe un caso descrito con posible penetrancia incompleta5, en esta enfermedad la penetrancia se considera completa, por lo que sería más probable pensar en expresividad variable con fenotipo muy leve o mosaicismo somático, no habiendo sido posible descartar esta opción.
La talla final media es de 120cm en hombres y de 116cm en mujeres. La complicación más importante es el dolor articular desde la infancia, por artrosis precoz de rodillas, caderas y columna. Pueden presentar hipoplasia de odontoides. En modelos de ratón se ha obtenido una mejoría histológica con tratamiento precoz con agentes antioxidantes y antiinflamatorios, así como con tecnología de oligonucleótidos antisentido6. Precisan seguimiento en un centro especializado, evitar actividades físicas intensas, analgesia, fisioterapia, ocasionalmente cirugía correctora y siempre apoyo psicosocial.

