

El retraso global del desarrollo (RGD) y la discapacidad intelectual (DI) constituyen una enfermedad muy frecuente, con notable impacto social y etiología variada. El estudio de los cromosomas mediante array-CGH ha incrementado, en más de un 20%, el número de diagnósticos etiológicos en nuestra consulta de neuropediatría1.
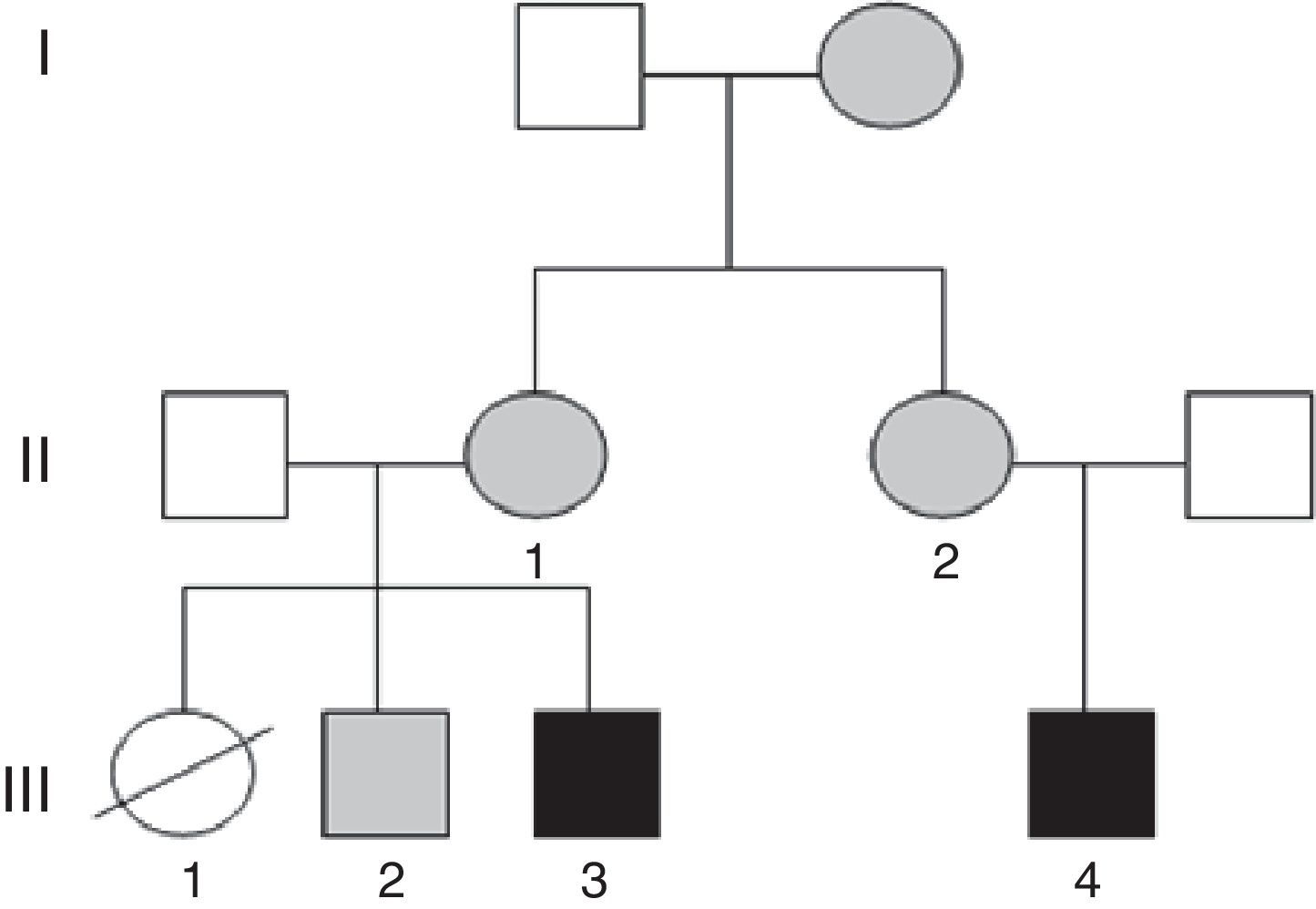
Presentamos los casos clínicos de una familia con varios miembros afectos de DI, en los que se detectan reordenamientos cromosómicos equilibrados y no equilibrados, secundario a un proceso de segregación cromosómica diferente en cada uno de ellos.
Caso 1 (fig. 1, individuo III.4): Varón de 5 años, hijo único, afecto de DI, conductas oposicionistas, escaso lenguaje propositivo, y ausencia de control de esfínteres. Presenta microcefalia desde el nacimiento (PC–4,21 DE), con peso y talla en percentil 75, fenotipo dismórfico, hexadactilia de ambos pies y comunicación interauricular amplia que precisó cirugía. Se realiza array-CGH en el que se detecta una duplicación a nivel 15q26.3 junto a una deleción parcial de la región 7p22.
.")
La haploinsuficiencia de los genes MAD1L1, FTSJ2, NUDT1, SNX8, EIF3B, FAM20C presentes en el cromosoma 7, y cuya dosis se encuentra alterada en el paciente. ha sido asociada a defectos a nivel cardiaco y de DI2.
El incremento de dosis de la región distal del cromosoma 15 se ha asociado al síndrome de sobrecrecimiento 15q (tendencia a la macrosomía, polidactilia, DI y trastornos de conducta), que se ha descrito en más de 100 pacientes3. Clásicamente, se ha asociado al gen IGF1R. No obstante, se ha reportado recientemente una familia, en los que la duplicación no afecta a este gen como en el caso 1, y postulan que sería el gen LRRK1 (del que nuestro caso también presenta 3 copias) el que da lugar al fenotipo descrito. Codifica una proteína que se cree implicada en la regulación del crecimiento y proliferación celular4.
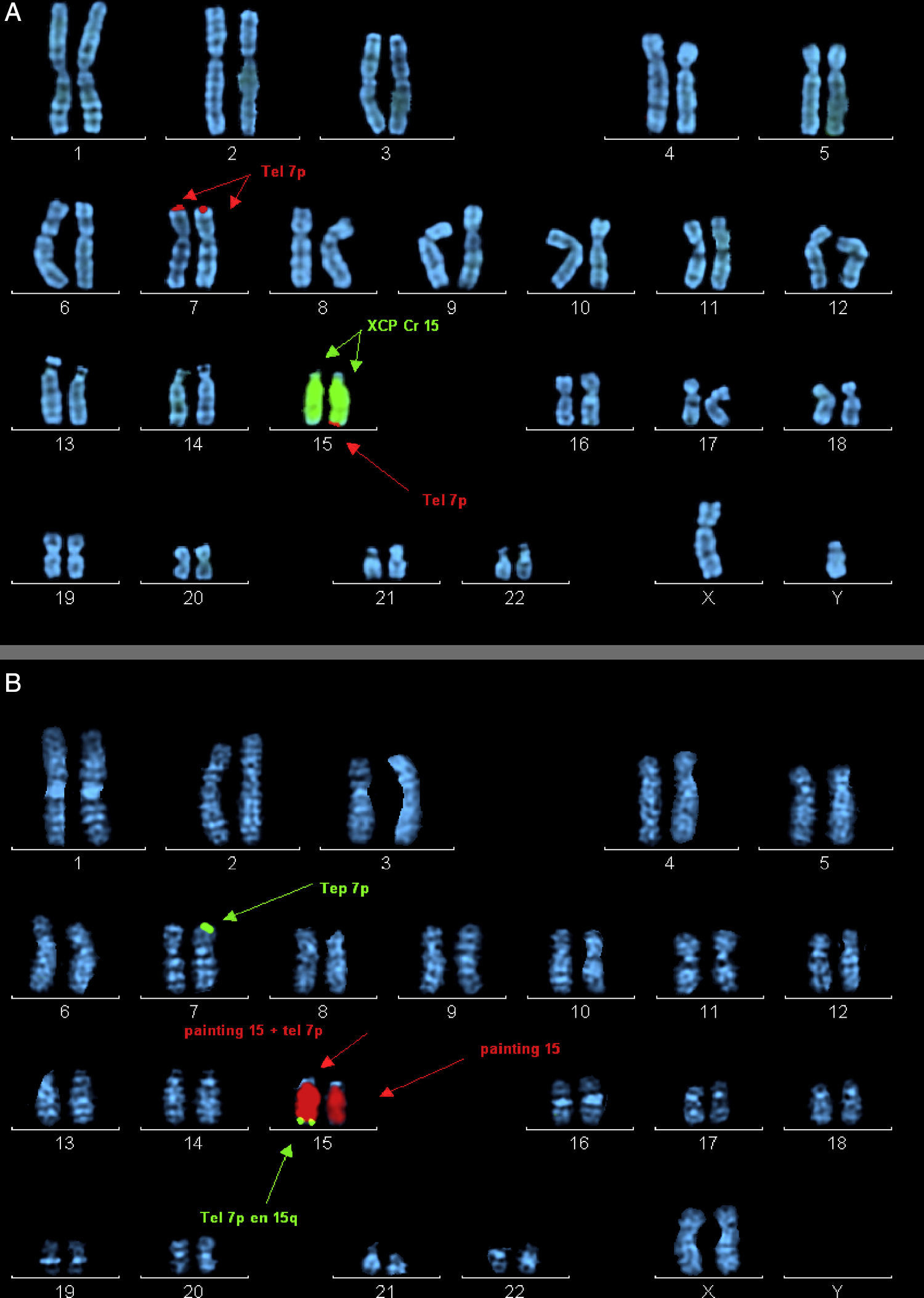
Caso 2 (fig. 1, individuo III.3): Niño de 3 años, primo hermano del caso 1, cuya hermana mayor falleció a los 2 meses de edad, afecta de transposición de grandes vasos, labio leporino y microcefalia (fig. 1, individuo III.1). El caso presenta RGD, macrocefalia, fenotipo dismórfico, cierre de la fontanela tardío, hiperlaxitud articular y pies zambos. En el array se detecta una deleción de la región 15q26.3 y duplicación de 7p22.3p22.1 (fig. 2).
. A) Probando del caso 2. Marcaje mediante la sonda XCP Cr15 (señalada por flechas centrales) y la sonda telomérica 7p (señalada por flechas superiores e inferiores). B) Madre del caso 2. Marcaje mediante la sonda XCP Cr15 (señalada por flechas centrales) y la sonda telomérica 7p (señalada por flechas superiores e inferiores).")
Metafases ordenadas con marcaje por hibridación in situ fluorescente (FISH). A) Probando del caso 2. Marcaje mediante la sonda XCP Cr15 (señalada por flechas centrales) y la sonda telomérica 7p (señalada por flechas superiores e inferiores). B) Madre del caso 2. Marcaje mediante la sonda XCP Cr15 (señalada por flechas centrales) y la sonda telomérica 7p (señalada por flechas superiores e inferiores).
Con respecto a la trisomía segmentaria de 5,1Mb del extremo distal del brazo corto del cromosoma 7 que altera la dosis de más de 40 genes de Refseq, se han descrito más de 40 pacientes con duplicaciones similares. Se caracterizan por un dismorfismo facial específico (macrocefalia, retraso de cierre de las fontanelas, hipertelorismo y orejas de implantación baja), además de DI, trastornos del lenguaje, hipotonía, bajo peso al nacer, criptorquidia y defectos cardiacos. Recientemente, la macrocefalia se ha atribuido a la duplicación del gen RNF216, presente en el caso 2 (III.3)5.
Por otra parte, la deleción terminal del brazo largo del cromosoma 15, de aproximadamente 2,2Mb, afecta a más de una decena de genes RefSeq. A día de hoy se han descrito 12 casos que presentan deleciones distales, que mostraban un retraso de crecimiento pre y posnatal, defectos cardiacos, retraso del desarrollo y rasgos dismórficos6.
Las madres de ambos sujetos (hermanas entres sí) presentan la siguiente fórmula cromosómica 46, XX t(7;15) (p22;q26), así como un hermano sano del caso 2 46, XY t(7;15) (p22;q26) con resultado del array no alterado (fig. 1, individuo III.2). Posteriormente, se comprobó que la abuela materna era también portadora de la traslocación equilibrada.
El asesoramiento genético para todos los miembros de esta familia es esencial, ya que con alta probabilidad la hermana (III.1) del caso 2 (III.3) y prima del caso 1 (III.4) habría heredado una traslocación no equilibrada. La cardiopatía, presente en la paciente, se asocia tanto a deleciones como a duplicaciones del brazo corto del cromosoma 7; sin embargo, las duplicaciones asocian macrocefalia, presente en su hermano (caso 2, III.3), por lo que es más probable que haya presentado la deleción del 7 y duplicación del 15, como su primo (caso 1, III.4), ambos con microcefalia, frecuente en trastornos genéticos del neurodesarrollo.
Aquellos individuos que portan más de una variante del número de copias de significado patológico, presentan fenotipos más severos. Este hecho, añadido a la expresividad variable que presentan muchos de estos desequilibrios cromosómicos hace muy difícil la realización de correlaciones genotipo-fenotipo en estos pacientes.
Por otra parte, hay que añadir los recientes hallazgos mediante las técnicas de secuenciación masiva, en los puntos de rotura de las translocaciones tanto balanceadas como no balanceadas, de reordenamientos y puntos de rotura múltiple con inversiones e inserciones, no detectables por otras técnicas, que conllevan rotura o fusión de genes presentes en estas regiones, con la consecuente implicación clínica7.
Por ello, para llevar a cabo la mejor interpretación de los resultados obtenidos en las pruebas genéticas, es necesario que se formen equipos multidisciplinares, en los que clínicos y genetistas trabajen en estrecha colaboración. De esta forma, además de llegar a la tipificación etiológica, lo que permite optimizar el seguimiento y el asesoramiento genético familiar, se evitan pruebas médicas innecesarias.

