

La trombocitopenia inmune primaria, anteriormente conocida como púrpura trombocitopénica inmune, es una enfermedad cuyo manejo diagnóstico y terapéutico ha sido siempre controvertido. La Sociedad Española de Hematología y Oncología Pediátricas, a través del grupo de trabajo de la PTI, ha actualizado el documento con las recomendaciones protocolizadas para el diagnóstico y tratamiento de esta enfermedad, basándose en las guías clínicas disponibles actualmente, revisiones bibliográficas, ensayos clínicos y el consenso de sus miembros. El objetivo principal es disminuir la variabilidad clínica en los procedimientos diagnósticos y terapéuticos con el fin de obtener los mejores resultados clínicos, con menor incidencia en la calidad de vida y los mínimos efectos adversos.
Primary immune thrombocytopenia (ITP), formerly known as immune thrombocytopenic purpura, is a disease in which clinical and therapeutic management has always been controversial. The ITP working group of the Spanish Society of Paediatric Haematology and Oncology has updated its guidelines for diagnosis and treatment of ITP in children based on current guidelines, literature review, clinical trials and member consensus. The primary objective was to lessen clinical variability in diagnostic and therapeutic procedures in order to obtain best clinical results with minimal adverse events and good quality of life.
La púrpura trombocitopénica inmune (PTI) es una enfermedad caracterizada por una disminución aislada de la cifra de plaquetas por debajo de 100.000/μl, en ausencia de una causa desencadenante de la trombocitopenia. La denominación previa de púrpura trombocitopénica idiopática se sustituyó por inmune debido a la importancia de los mecanismos inmunológicos de destrucción de plaquetas mediada por autoanticuerpos y linfocitos T en su patogenia. Actualmente se recomienda la denominación de trombocitopenia inmune primaria1.
En marzo de 2009 se publicaron las recomendaciones de estandarización de la terminología, definiciones y criterios de respuesta de la PTI para adultos y niños. Este consenso fue establecido por expertos europeos y americanos. Su objetivo es la utilización de un lenguaje común para evitar la heterogeneidad previa de los diferentes estudios y publicaciones. Se elimina el término de púrpura porque el sangrado cutáneo o mucoso está ausente o es mínimo en algunos pacientes. Se mantiene el acrónimo Immune ThrombocytoPenia (ITP) y PTI en castellano, por su amplia difusión y utilización previa1.
En este consenso también se establece el nivel de corte de la cifra de plaquetas en 100.000/μl para el diagnóstico de PTI, por los recuentos frecuentes entre 100 y 150.000/μl en individuos sanos y mujeres embarazadas. La clasificación se modifica atendiendo a la historia natural de la PTI en la infancia, en la que aproximadamente dos tercios se recuperan espontáneamente en los primeros 6 meses y la posibilidad de remisión es alta entre los 3 y 12 meses, e incluso puede ser más tardía.
Posteriormente, en enero de 2010 se publicó el consenso internacional para el diagnóstico y manejo de la PTI2. El diagnóstico sigue siendo de exclusión, ya que no hay ningún parámetro clínico ni analítico que permita establecer este diagnóstico con certeza. Los síntomas y signos clínicos son muy variables. El principal problema es el riesgo aumentado de hemorragia. No siempre hay una correlación exacta entre la cifra de plaquetas y las manifestaciones hemorrágicas, aunque estas son más frecuentes por debajo de 10.000/μl. La mayoría de los pacientes están asintomáticos o tienen petequias, hematomas o equimosis aislados en piel o mucosas3,4. Sin embargo, algunos casos pueden sufrir hemorragias más graves5–9 a nivel cutáneo, mucoso, gastrointestinal o incluso intracraneal (0,1-0,5%). La denominación PTI grave se reserva para los pacientes con manifestaciones hemorrágicas clínicamente relevantes.
El objetivo del tratamiento es prevenir estas hemorragias con relevancia clínica, más que corregir las cifras de plaquetas hasta valores normales. Las decisiones terapéuticas se deben tomar en función de múltiples factores y las recomendaciones basadas exclusivamente en las cifras de plaquetas son muy controvertidas. Muchos expertos consideran que los niños con PTI sin sangrado no requieren tratamiento, independientemente del número de plaquetas, aunque precisan un control y seguimiento estrecho. En caso de sangrado o en circunstancias de riesgo, se aconsejan tratamientos que produzcan un rápido ascenso de los recuentos de plaquetas para prevenir o frenar las hemorragias7. Un concepto importante es evitar tratamientos innecesarios, potencialmente tóxicos, en pacientes asintomáticos o con descensos moderados de las plaquetas y conseguir una adecuada calidad de vida con la mínima toxicidad asociada a la terapia. El tratamiento de las PTI persistentes o crónicas es complejo y se plantean diferentes alternativas médicas o quirúrgicas.
En este protocolo se propone incluir los pacientes con PTI en edades comprendidas entre seis meses y dieciocho años.
ObjetivosUnificar los criterios diagnósticos, protocolos de seguimiento y las pautas terapéuticas de la PTI.
Clasificación diagnóstica, criterios de evaluación clínica y respuesta al tratamientoClasificación diagnósticaPTI de reciente diagnósticoDesde el momento del diagnóstico hasta los 3 meses de evolución.
PTI persistenteDe duración entre los 3 y 12 meses desde el diagnóstico, incluye a:
- •
Pacientes que no alcanzan la remisión completa de forma espontánea.
- •
Pacientes que no mantienen la remisión completa después de suspender el tratamiento instaurado.
Pacientes que continúan con trombocitopenia después de 12 meses desde el diagnóstico.
Criterios de evaluación clínicaClínica cutánea
Clínica cutáneo-mucosa
Sangrado activo
- •
Epistaxis que precisa taponamiento
- •
Hematuria
- •
Hemorragia digestiva macroscópica
- •
Menorragia
- •
Gingivorragia importante
- •
Cualquier hemorragia con riesgo razonable de precisar trasfusión de hematíes
- •
Hematuria
- •
TCE, politraumatismo previo
- •
Antiagregantes hasta 7-10 días antes
- •
Diátesis hemorrágica: coagulopatía, vasculitis
Recuento igual o superior a 100.000/μl mantenido más de seis semanas tras la supresión del tratamiento.
Remisión parcial (RP)Elevación sobre la cifra inicial con recuento entre 30.000 y 100.000/μl mantenido más de seis semanas tras la supresión del tratamiento.
Ausencia de respuesta (AR)No se modifica clínica ni biológicamente.
Respuesta transitoria (RT)Mejoría inicial (clínica o biológica) con nueva clínica o recuento inferior a 30.000/μl antes de seis semanas de haber finalizado el tratamiento.
Recaída (REC)Recuento inferior a 30.000/μl después de seis semanas de haber finalizado el tratamiento, habiéndose obtenido previamente una remisión completa (RC) o parcial (RP).
Exploraciones complementarias al diagnósticoEn todos los pacientes con trombocitopenia se debe realizar una historia clínica detallada y una exploración física completa que permitan descartar otras enfermedades hematológicas o situaciones que de forma secundaria puedan producir trombocitopenia.
Los estudios detallados a continuación son los recomendados, por considerarse básicos para un diagnóstico y seguimiento adecuados. Obviamente esta lista no excluye otras pruebas adicionales que puedan efectuarse o consideren oportunas en los diversos Centros:
- -
Hemograma y recuento de reticulocitos.
- -
Morfología en sangre periférica con revisión por persona experta.
- -
Estudio de hemostasia: tiempo de protrombina (TP), tiempo de tromboplastina parcial activado (TTPA), tiempo de trombina (TT), fibrinógeno.
- -
Grupo, Rh y Coombs directo.
- -
Inmunoglobulinas.
- -
Estudio microbiológico de: citomegalovirus (CMV), virus de Epstein Barr (EBV), parvovirus B19, herpes simple, herpes 6, virus de la inmunodeficiencia humana (VIH), hepatitis B y C.
- -
Bioquímica hemática: GOT, GPT, LDH, glucosa, urea, creatinina.
- -
Control de hematuria microscópica.
- -
Estudio morfológico de médula ósea por punción aspirativa: indicado en todos los niños con clínica que no sea típica, anomalías en el hemograma y en aquellos en los que la morfología en sangre periférica no haya podido ser revisada por una persona experta, especialmente si se inicia tratamiento con corticoides.
Indicadas en pacientes en los que no remite espontáneamente o no responden al tratamiento.
- -
Estudio morfológico de médula ósea por punción aspirativa si no se hizo previamente. Valorar la realización de biopsia, inmunofenotipo y citogenética para completar el estudio.
- -
Poblaciones linfocitarias.
- -
Anticuerpos antinucleares y opcionalmente otros estudios de autoinmunidad.
- -
Otros: detección de H. pylori10, estudio de celiaquía.
Al diagnóstico, considerar el ingreso hospitalario en pacientes con sangrado activo, factores de riesgo hemorrágico o con recuento de plaquetas igual o inferior a 20.000/μl.
Evitar inyectables intramusculares y punciones vasculares en vasos de difícil compresión.
Contraindicado el empleo de ácido acetilsalicílico o sus derivados; administrar solo en caso de ser estrictamente necesario otros fármacos que puedan alterar la agregación plaquetar (antihistamínicos, antiinflamatorios no esteroideos).
Deportes: restricción en función de la clínica y riesgo traumático, evitar deportes de contacto hasta la resolución de la enfermedad.
PTI de diagnóstico recienteEl paciente con PTI de diagnóstico reciente, puede presentar unas manifestaciones hemorrágicas de gravedad variable, en general, en función de la cifra de plaquetas, actividad habitual y presencia de otros factores que pueden influir en la hemostasia. Hay que valorar siempre el conjunto de datos clínicos y biológicos para un adecuado enfoque terapéutico. Se ha decidido clasificar a los pacientes en diversos grupos en función de las manifestaciones clínicas, recuento de plaquetas y factores de riesgo hemorrágico, con la finalidad de establecer la opción de tratamiento más adecuada.
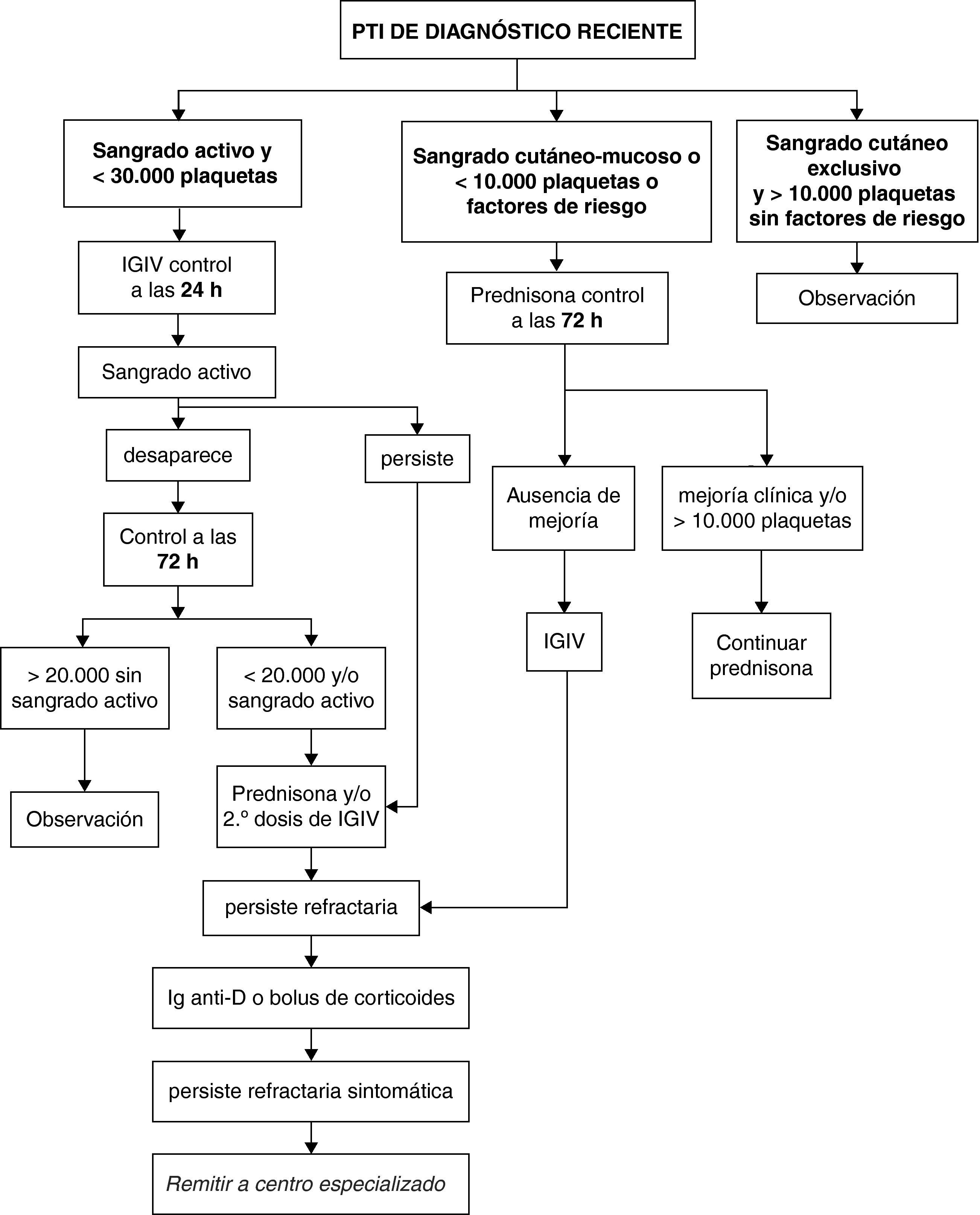
Clasificación de pacientes y pauta de actuación (fig. 1)Pacientes con sangrado activo y recuento inferior a 30.000 plaquetas/μlSe propone administrar 1 dosis de inmunoglobulinas intravenosas (IGIV) y nueva valoración a las 24h:
- •
Si persiste el sangrado activo se añaden corticoides y/o una segunda dosis de IGIV.
- •
Si desaparece la clínica se vuelve a valorar a las 72h y
- ∘
si remonta clínica y analíticamente, entonces pasa a observación,
- ∘
pero si persiste por debajo de 20.000/μl o aparece de nuevo sangrado activo, se inicia tratamiento con corticoides.
- ∘

Si se muestra también refractario, se ensaya tratamiento con inmunoglobulina anti D (Ig-anti-D) en caso que sea Rh+ o bolus de corticoides. Los pacientes que se muestren refractarios al tratamiento anterior y persistan con clínica hemorrágica importante deben ser remitidos a un centro hospitalario especializado para revisión y valoración de tratamientos de tercera línea.
Pacientes con sangrado cutáneo-mucoso o recuento de plaquetas inferior a 10.000 plaquetas/μl, o factores de riesgoSe propone administrar de entrada corticoides. A las 72h de iniciados, si no hay mejoría clínica o biológica, se administra una dosis de inmunoglobulinas intravenosas (IGIV) y se sigue con los corticoides. Si se muestra también refractario se ensaya tratamiento con Ig-anti-D (en caso que sea Rh+) o bolus de corticoides.
Pacientes con sangrado cutáneo exclusivo y recuento mayor de 10.000 plaquetas/μl sin factores de riesgoSe propone una actitud expectante y controles periódicos con actuación posterior en función de la evolución.
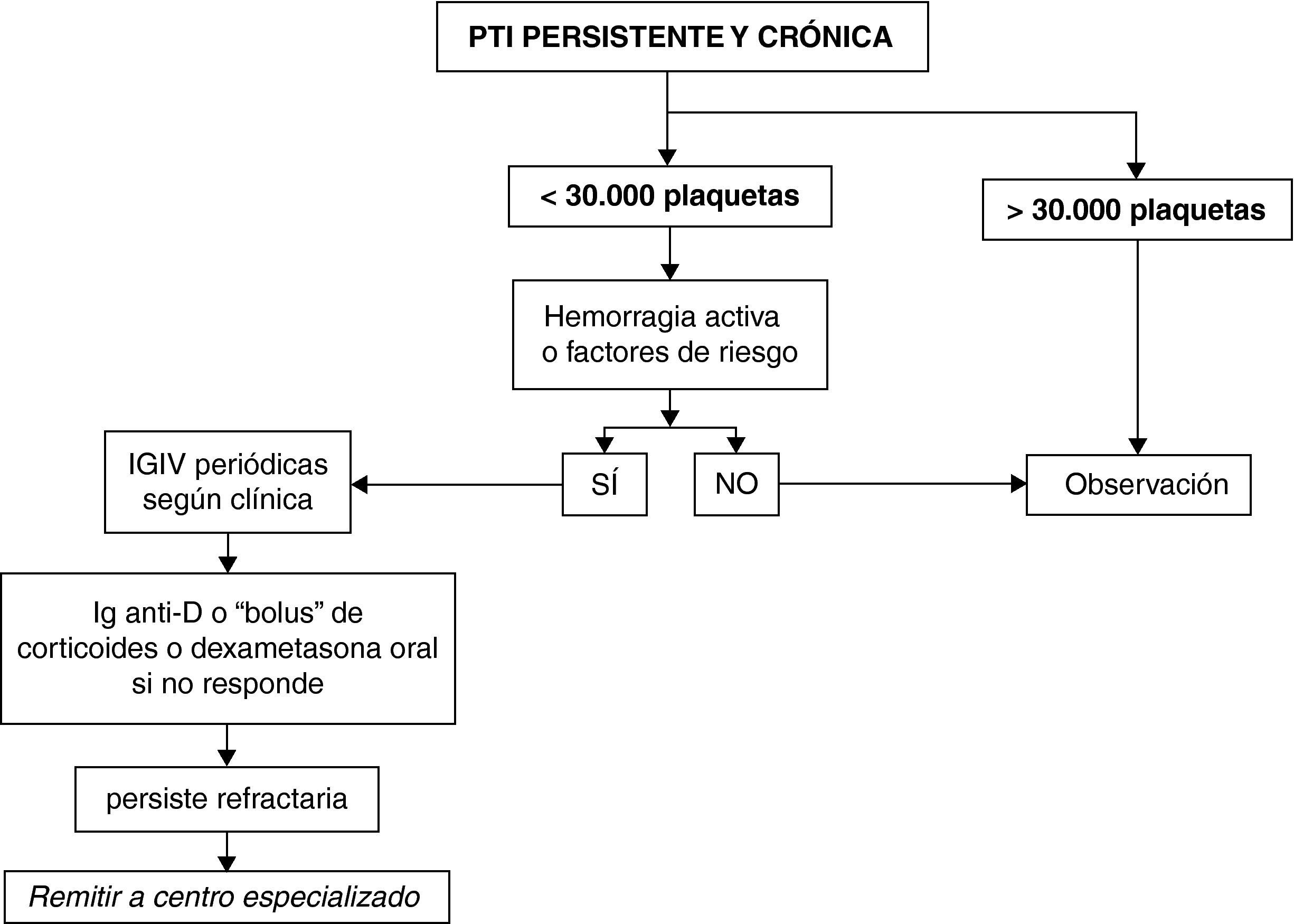
PTI persistente y crónicaEl paciente con PTI persistente y crónica puede presentar unas manifestaciones hemorrágicas de gravedad variable, en general en función de la cifra de plaquetas, actividad habitual y presencia de otros factores que pueden influir en la hemostasia. Hay que valorar siempre el conjunto de datos clínicos y biológicos para un adecuado enfoque terapéutico y procurar que el paciente desarrolle una vida lo más cercana a la normalidad con los mínimos efectos adversos derivados del tratamiento, en espera de que la enfermedad entre en remisión. Está extendido en la literatura y se ha considerado razonablemente seguro para el desarrollo de una vida cotidiana normal el mantenimiento de recuentos por encima de 30.000 plaquetas; por ello, se ha elegido como factor determinante en el análisis de decisión inicial. No obstante, el estilo de vida, la actividad habitual, las manifestaciones clínicas y los factores de riesgo hemorrágico son también determinantes, fundamentalmente si se indica algún tipo de intervención.
Clasificación de pacientes y pauta de actuación (fig. 2)Pacientes con más de 30.000 plaquetas/μl, mantenidas de forma estableSe recomienda mantenerlos en observación, con los controles necesarios a juicio del clínico.
Pacientes con menos de 30.000 plaquetas/μl
A aquellos que no presenten episodios de hemorragia activa o factores de riesgo hemorrágico se aconseja mantenerlos en observación. En caso contrario se recomienda la administración de IGIV periódicas con una frecuencia variable personalizada según las manifestaciones clínicas y/o los factores de riesgo. Si el paciente no responde se aconseja pasar a tratamientos de segunda línea (Ig-anti-D, bolus de corticoides o ciclos de dexametasona oral). Si a pesar de ello permanece refractario, se aconseja remitir a un centro especializado para revisión y valorar la administración de tratamientos de tercera línea.
Tratamiento de las urgencias con riesgo vital y situaciones con riesgo especialUrgencias con riesgo vitalHemorragias del SNCOtras hemorragias que comprometan la vida del paciente
Se administran sucesivamente:
- 1.
metilprednisolona i.v. 10mg/kg
- 2.
gammaglobulina i.v. 400mg/kg
- 3.
plaquetas 1 unidad/5-10kg/6-8h
- 4.
gammaglobulina i.v. 400mg/kg
- 5.
esplenectomía urgente: valorar según cada caso
Administrar IGIV 0,8-1g/kg si plaquetas < 50.000/μl y plaquetas si recuento < 10.000/μl.
Cirugía programada (valorar riesgo hemorrágico según intervención)IGIV 0,8-1g/kg si plaquetas < 50.000/μl.
Esplenectomía programadaIGIV 0,8-1g/kg si plaquetas < 20.000/μl.
Clampaje precoz de la arteria esplénica.
Tratamientos de la PTITratamientos de primera línea11–13Corticoterapia- a)
mecanismo de acción:
- •
estabilización de la pared vascular
- •
disminución de la producción de Ac. antiplaquetarios
- •
disminución del aclaramiento por el SMF de plaquetas con Ac. adheridos
- •
alteración de la unión del Ac. con la superficie plaquetar
- •
inmunosupresión celular
- •
- b)
posología:
- •
prednisona vía oral o metilprednisolona vía i.v., repartida en tres dosis: tras desayuno, comida y cena
- •
4mg/kg/día (dosis máxima 180mg/día) durante 4 días, luego pasar a 2mg/kg durante 3 días y suspender
- •
- a)
mecanismos de acción:
- •
bloqueo de los receptores Fc del SMF
- •
bloqueo y disminución de la síntesis de autoAc por acción de los Ac. antiidiotipo
- •
- b)
posología:
- •
0,8-1g/kg/dosis única i.v. en perfusión continua, tiempo de infusión 6-8 h, al inicio de la infusión la velocidad es más lenta; se recomienda seguir la pauta de velocidad de infusión indicada en cada preparado
- •
- c)
efectos adversos:
- •
anafilaxia, en pacientes con déficit de IgA: se recomienda tener preparado para uso inmediato el tratamiento específico y equipo de reanimación (adrenalina,…)
- •
cefalea, náuseas, vómitos (se recomienda disminuir la velocidad de infusión)
- •
febrícula/fiebre
- •
hemólisis aloinmune
- •
meningitis aséptica
- •
- d)
riesgos biológicos:
- •
Las IGIV son hemoderivados y por tanto, aunque durante el fraccionamiento plasmático y purificación se reduce la posible carga viral, no están exentas del riesgo de transmisión de enfermedades infecciosas. Se ha comprobado la transmisión de hepatitis C
- •
- e)
características recomendadas
Con la finalidad de disminuir los efectos adversos y asumir el menor riesgo biológico se recomienda que el preparado reúna las siguientes características:
- •
mínimo contenido de IgA
- •
mínimo contenido en formas poliméricas
- •
mayor seguridad biológica; para ella se recomienda:
- ∘
“pool” de donantes con adecuados controles clínicos y biológicos eliminando personas de poblaciones consideradas de riesgo
- ∘
control microbiológico sobre plasma previo al fraccionamiento y sobre producto final
- ∘
empleo de un método de inactivación viral
- ∘
- a)
mecanismo de acción:
- •
Bloqueo de receptores Fc macrofágicos con hematíes recubiertos por Ac. anti D
- •
- b)
posología:
- •
50-75 microgramos/kg/día, i.v. dosis única
- •
Perfusión durante 1 h diluido en suero fisiológico. Se recomienda premedicar con paracetamol
- •
- c)
efectos adversos y riesgos biológicos:
- •
Anemia hemolítica inmune y puesto que es un hemoderivado no están exentas del riesgo de transmisión de enfermedades infecciosas. Se ha comprobado la transmisión de hepatitis C
- •
Se recomiendan los siguientes controles: Hb, Coombs directo, recuento reticulocitario y bilirrubina indirecta. Se aconseja no repetir dosis (a las 2-4 semanas) si presenta bilirrubina I. > 1,5 mg% y reticulocitos > 5% con subictericia o coluria, o descenso de Hb superior a 2g
- •
- a)
“Bolus” de corticoides:
- •
metilprednisolona: 30mg/kg/día (máximo 1 g), 3 días
- •
infusión en 2 h
- •
control de T.A. y glucosuria
- •
- b)
Dexametasona oral:
- •
0,6mg/kg/día en 1 dosis, máximo 40mg, durante 4 días, cada mes
- •
- a)
Indicaciones
- •
PTI de diagnóstico reciente o persistente: ante urgencia hemorrágica con riesgo vital que no responde a tratamiento previo
- •
PTI crónica:
- ∘
ante urgencia hemorrágica con riesgo vital
- ∘
valorar en mayores de 5 años sintomáticos refractarios a tratamientos previos, que presenta interferencia con su vida normal, con más de dos años de evolución
- ∘
- •
- b)
Preparación para la intervención:
- •
Ver esquema para tratamiento ante urgencias con riesgo vital y situaciones de riesgo especial
- •
- c)
Fracaso de la esplenectomía:
- •
Descartar bazo accesorio
- •
- d)
Profilaxis y manejo de la infección en el paciente esplenectomizado:
- •
Profilaxis:
- ∘
Vacunación antineumocóccica, antimeningocóccica y frente a hemofilus
- ∘
Penicilina oral diaria o amoxicilina: hasta un mínimo de dos años tras la intervención
- ∘
- •
Tratamiento de la infección: ante síndrome febril sin foco iniciar antibioterapia con cobertura para neumococo, hemofilus y meningococo
- •
Constituyen un conjunto de moléculas de reciente aparición en la clínica19–23, del que no hay experiencia publicada en niños; no obstante, recientemente se comunicó el primer ensayo en fase I-II con Romiplostin. Actualmente hay dos preparados:
- •
Romiplostin: se administra por vía subcutánea con periodicidad semanal. Actualmente aceptado en España para el tratamiento de PTI en adultos esplenectomizados refractarios a otros tratamientos y en aquellos en los que esté contraindicada la esplenectomía, como tratamiento de segunda línea.
- •
Eltrombopag: se administra por vía oral. Hasta el momento, existe menos experiencia con este preparado.
Ha sido empleado en adultos y también en niños24–26, aunque en estos últimos existe menos experiencia. Se obtiene una tasa de respuesta entre el 30 y 60% en función del tiempo de análisis. Su infusión requiere la vigilancia de problemas inmunoalérgicos agudos ocasionalmente graves. Existe riesgo infeccioso por depleción prolongada de linfocitos B y actualmente está bajo vigilancia la descripción de cuadros de leucoencefalopatía multifocal progresiva27, comunicada tras la administración en otras enfermedades. Su administración debe indicarse por uso compasivo, al no estar incluida esta indicación en ficha técnica.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

