

En los países desarrollados, el cáncer infantil representa la primera causa de muerte por enfermedad en la edad pediátrica. Su incidencia ha ido aumentando de manera continuada desde los años 501, como consecuencia de las mejoras de las herramientas diagnósticas y de los registros, acompañándose de una mejora importante en el pronóstico y la supervivencia.
No obstante, la curación del cáncer infantil parece haber alcanzado un tope terapéutico, siendo alrededor del 70% en las últimas décadas2.
Hoy en día sabemos que el cáncer infantil es una enfermedad multifactorial, con base genética aún no totalmente conocida, que presenta una gran implicación del sistema inmunológico y una modulación por la exposición al medioambiente.
En el cáncer infantil, las medidas preventivas son actualmente ineficaces. Sin embargo, la identificación de susceptibilidad hereditaria podría ser muy relevante para el paciente y su familia. En algunos casos, esto podría llevar a instaurar medidas preventivas para la detección precoz del proceso maligno, tanto en el caso índice como en sus familiares.
Genética del cáncerEl cáncer resulta de la acumulación dentro de una célula de diferentes alteraciones genéticas, que a veces se acumulan durante años. Estas alteraciones llevan a una proliferación celular anormal y a la expansión clonal, que en última instancia puede invadir otros tejidos.
En la mayoría de los casos, las lesiones genéticas que promueven la tumorigénesis se producen en células somáticas y no implican alteraciones adquiridas por vía germinal.
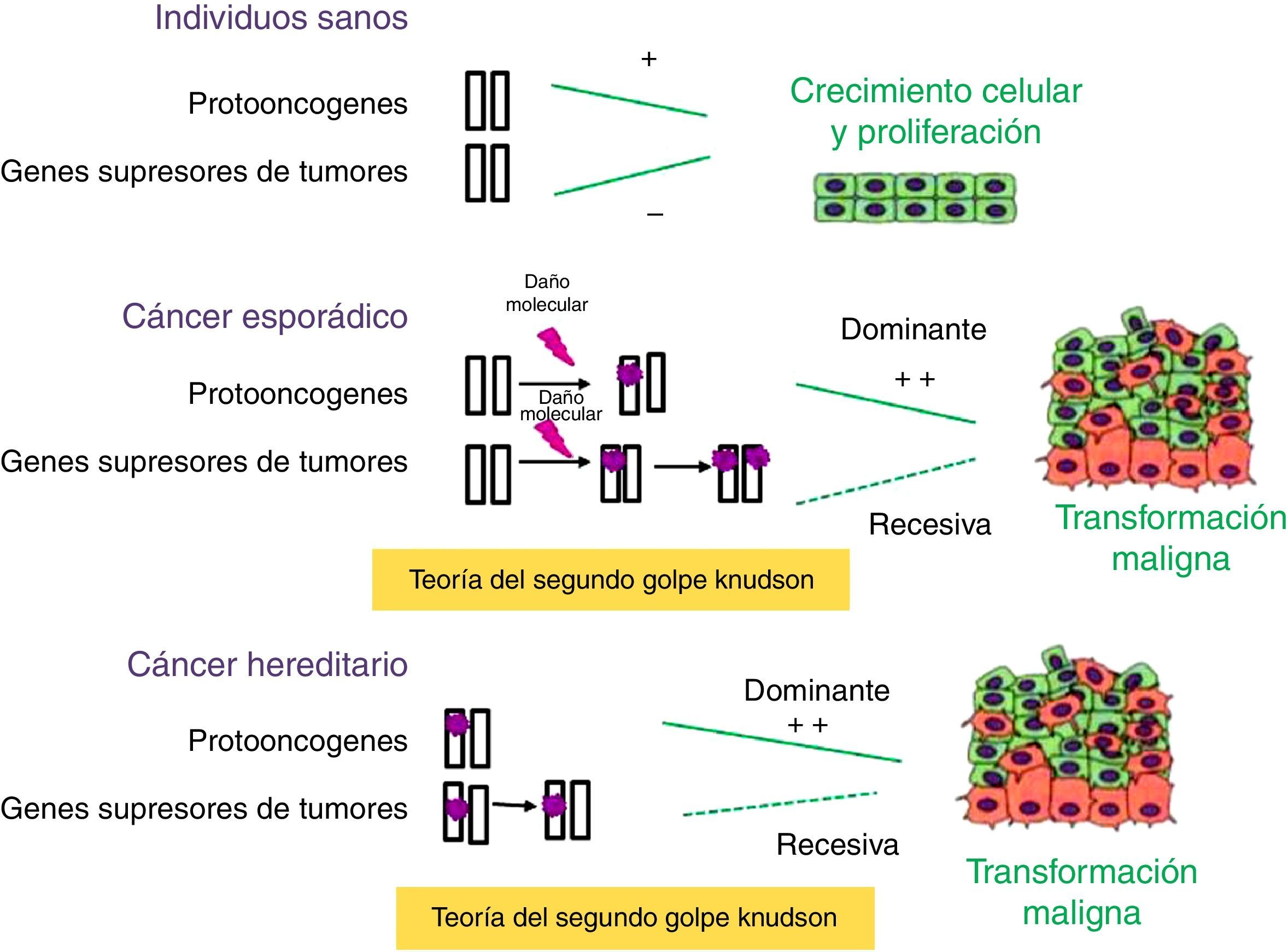
Se han identificado numerosos genes implicados en el desarrollo tumoral, que se encuadran en uno de estos 3 grupos: genes supresores de tumores, protooncogenes y genes implicados en mantener la estabilidad del ADN. Los genes supresores de tumores controlan fisiológicamente la proliferación, inhibiendo la progresión del ciclo celular o promoviendo la apoptosis (fig. 1). Normalmente, una copia funcional del gen es suficiente para ejercer la función. La inactivación de ambos alelos permite la proliferación incontrolada y, por tanto, contribuye a la tumorigénesis. Por el contrario, los protooncogenes tienen fisiológicamente un efecto positivo sobre la proliferación celular y contribuyen a la progresión tumoral cuando sufren mutaciones que los activan de forma permanente. En este caso, mutaciones en uno de los alelos son suficientes para producir un crecimiento celular incontrolado. Los genes que intervienen en la estabilidad del ADN no intervienen directamente en la regulación de la proliferación celular, pero la falta de función contribuye a una mayor tasa de mutaciones y, por tanto, aumenta la posibilidad de desarrollar tumores3.

Papel de los genes supresores de tumores y protooncogenes en la tumorigénesis. Los protooncogenes y los genes supresores de tumores tienen funciones antagónicas: los primeros promueven el crecimiento celular, mientras que los genes supresores de tumores codifican proteínas que inhiben la proliferación celular. Cuando se producen mutaciones esporádicas o heredadas en los protooncogenes se transforman en oncogenes, los cuales son capaces de orquestar la multiplicación anárquica de las células. En los genes supresores de tumores deben producirse 2 eventos mutacionales, según la teoría del segundo golpe de Knudson, para que se inicie el proceso tumoral. En el cáncer hereditario, la primera mutación es heredada y, por tanto, la probabilidad de presentar cáncer es mayor que en la población general.
En general, se sospecha de susceptibilidad heredada al cáncer en familias con: 2 o más familiares de la misma rama con el mismo tipo de cáncer, varias generaciones afectadas, edades tempranas de diagnóstico, pacientes con cánceres primarios múltiples, ocurrencia de distintos tipos de cánceres genéticamente relacionados (como cáncer de mama y ovario, o cáncer de colon y útero), mayor frecuencia de afectación bilateral o multifocal que unilateral y la presencia de alteraciones no malignas y cáncer en la misma persona o en la misma familia (p. ej., hábitos marfanoides y neoplasia endocrina múltiple tipo 2B). Sin embargo, debido a la variabilidad fenotípica y a que la penetrancia o proporción de pacientes que teniendo la mutación desarrollarán cáncer es dependiente de la edad, muchas familias pueden no cumplir estos criterios4.
La mayoría de los genes implicados en la predisposición genética al cáncer son genes supresores de tumores, solo en un 10% de los casos se observan mutaciones con ganancia de función en protooncogenes. En el caso de susceptibilidad por mutaciones en genes supresores de tumores, se hereda una copia defectuosa del gen, pero para el desarrollo tumoral es necesaria la pérdida somática del otro alelo, según la teoría del segundo golpe de Knudson. Por eso, se considera que las mutaciones en genes supresores de tumores son recesivas. Debido a que la probabilidad de que se produzca mutación en un alelo es mayor que la probabilidad de que se produzca en los 2 alelos del gen, la incidencia de cáncer en portadores de alguna mutación germinal en genes supresores es mucho mayor que la de la población general.
El caso de los portadores de mutaciones dominantes o de ganancia de función en protooncogenes es diferente. Aunque solo es necesario tener un alelo mutado del gen para desarrollar cáncer, tener la mutación no significa necesariamente que la persona vaya a desarrollar cáncer. Dependerá de la penetrancia clínica de cada tipo de tumor.
La predisposición genética al cáncer se debe a mutaciones que están presentes en todas las células del organismo. Estas mutaciones, que se denominan mutaciones germinales porque han sido adquiridas por vía germinal, son precigóticas y pueden haberse heredado o pueden ser el resultado de una mutación nueva en una de las células germinales de los progenitores (óvulo o espermatozoide). Por tanto, sería la primera vez que la mutación aparece en la familia. En un individuo sin predisposición genética al cáncer, las mutaciones se producen en las células somáticas y son poscigóticas.
Implicaciones clínicas de la predisposición genética al cáncer infantilLa aplicación de las nuevas técnicas de secuenciación genética en los niños con cáncer está permitiendo ampliar el conocimiento sobre la base molecular de los tumores infantiles. A diferencia de las técnicas genéticas convencionales, las tecnologías de alto rendimiento, como la secuenciación masiva o la secuenciación de nueva generación (NGS, de sus siglas en inglés next-generation sequencing), permite secuenciar de forma paralela millones de fragmentos de ADN a un precio cada vez más reducido y en un tiempo mucho menor. Tiene además el potencial de detectar numerosos tipos de variación genómica en un único experimento.
En un estudio reciente se ha realizado la secuenciación del genoma completo mediante NGS de 1.120 pacientes con cáncer y edades comprendidas entre los 0 y los 19 años. Se ha encontrado que un 8,5% tenía mutaciones germinales predisponentes, frente al 1% de mutaciones encontradas en la población control5. El hecho de que sea significativamente mayor el porcentaje de mutaciones germinales predisponentes en población con cáncer plantea muchos interrogantes: ¿sería ético y costo-efectivo realizar un cribado genético en el período neonatal con el objetivo de detectar aquellos pacientes en riesgo de desarrollar cáncer?, ¿se podría realizar un seguimiento de estos pacientes?, ¿podrían beneficiarse de un diagnóstico precoz?
Por otro lado, en este mismo estudio, la historia familiar de cáncer era similar en los pacientes con cáncer y en los controles utilizados, probablemente debido a que los padres eran jóvenes, a que puede tratarse de mutaciones de novo (no se realizaron estudios de segregación) o a que en muchos casos la historia familiar estaba incompleta. Este dato podría indicar, por un lado, que la historia familiar no debería ser la única indicación para realizar estudios moleculares familiares y, por otro, que en algunas familias los casos índices pueden presentarse en la edad pediátrica, en ausencia de cánceres en la edad adulta.
Sería interesante realizar estudios adicionales que nos permitan conocer si es elevado el porcentaje de mutaciones predisponentes heredadas. Esto indicaría que detectar dichas mutaciones en los probandos no solo podría ser útil para ellos, sino también para sus familiares. La realización de test genéticos en los familiares del paciente podría identificar a otros individuos en riesgo que pudieran beneficiarse de igual forma de una intervención temprana, que mejore la morbimortalidad asociada.
Gracias a la Fundación Cris contra el Cáncer (http://www.criscancer.org/es) y la fundación Uno Entre Cien Mil (http://unoentrecienmil.org/) por su apoyo en la realización de este trabajo.

