

NEFROLOGIA
Zona póster I (planta sótano del Auditorio)
P25910:15 h
ENTEROPATIA AUTOINMUNE ASOCIADA A NEFRITIS TUBULOINTERSTICIAL POR AUTOANTICUERPOS ANTIMEMBRANA BASAL TUBULAR Y A GLOMERULONEFRITIS NECROSANTE
María del Carmen Bravo Laguna, Marta Melgosa Hijosa, M.L. Picazo García, Laura Espinosa Román, R. Álvarez Doforno, Rosa A. Lama More
Hospital Materno-Infantil La Paz, Madrid.
Objetivo: Conocer la evolución y respuesta terapéutica de un paciente con enteropatía autoinmune, nefritis tubulointersticial y glomerulonefritis necrosante. Materiales y métodos: 1 paciente diagnosticado en el servicio de nefrología del hospital infantil La Paz. Caso clínico: Varón de 19 años de edad, diagnosticado de enteropatía autoinmune con anticuerpos antienterocito (+). En su evolución ha presentado 3 brotes diarreicos (último en enero de 2002) tratados con corticoides, CyA (los dos primeros) y tacrolimus (el último). En junio de 2004, en fase de retirada de tacrolimus y sin sintomatología digestiva, el paciente presenta una insuficiencia renal aguda (máxima creatinina = 2,5 mg/dl) que se acompaña de fiebre, anorexia, astenia, aumento de reactantes de fase aguda y anemización. Diuresis y tensiones arteriales normales. Proteínas totales, albúmina y colesterol normales. En el sedimento tenía microhematuria y proteinuria significativa pero no nefrótica. Complemento, ANA, ANCA, anti-MBG (membrana basal glomerular) y anticuerpos antienterocito fueron negativos pero se detectaron anticuerpos antimembrana basal tubular (anti-MBT). La biopsia renal presentaba una nefritis tubulointersticial aguda por anti-MBT y una nefropatía membranosa asociada a una glomerulonefritis necrosante con proliferación extracapilar.
Se inició tratamiento con bolos de metilprednisolona así como ciclofosfamida oral (2 mg/kg/d) que al cabo de un mes fue sustituida por bolos i.v. administrándose un total de 5 con periodicidad mensual. La fiebre desapareció así como los síntomas generales tras el inicio del tratamiento. 7 meses después el paciente está asintomático, los anti-MBT se han negativizado, y el filtrado glomerular y el sedimento se han normalizado (creatinina = 1,2 mg/dl; FG: 102 ml/min/1,73 m2).
P26010:20 h
TROMBOSIS VENOSA RENAL NEONATAL Y ALTERACIONES HEMOSTATICAS
María de la Serna Martínez, María del Carmen Bravo Laguna, Inés Orellana Díaz, Carlota Fernández Camblor, Antonia Peña Carrión, Mercedes Navarro Torres
Hospital Materno-Infantil La Paz, Madrid.
Objetivo: Presentación de la asociación de factores protrombóticos y déficits de coagulación con la trombosis venosa renal neonatal. Material y métodos. Dos pacientes.
Caso 1: Niña de 4 años de edad, hija de madre diabética e hipertensa, que a los 2,5 días de vida presenta hematuria, oliguria, hipertensión arterial, masa renal izquierda y acidosis metabólica. Diagnóstico: trombosis venosa renal izquierda y agenesia renal derecha (mediante eco-Doppler). Canalización de arteria y venas umbilicales, tratamiento con rt-PA a 0,5 mg/kg de choque y a 0,25 mg/kg en perfusión posterior. Heparina i.v. a las 24 h debido a la presencia de sangrado umbilical y hemofiltración 5 días, con diálisis peritoneal posteriormente (12 días). Repermeabilización de la vena renal a los 5 días de vida. Estudio de coagulación: déficit de antitrombina III (37 %), que se normalizó a los 3 años de vida, y mutación 20210 del gen de la protrombina (heterocigota), encontrado también en la madre. Presenta insuficiencia renal crónica moderada con filtrado glomerular de 34 mL/min/1,73 m2 por Cr-EDTA y riñón único izquierdo de 3DE de tamaño con pérdida de la diferenciación corticomedular.
Caso 2: Varón de 18 años que a los 19 días de vida presenta vómitos, hematuria, hipertensión arterial, masa en flanco izquierdo, anemia, acidosis metabólica y niveles de creatinina sérica de 3,4 mg/dl. Diagnóstico: trombosis de venas cava inferior y ambas renales (por ecografía). Tratamiento: plasminógeno y urocinasa local a través de silástico central, continuándose con plasminógeno y estreptocinasa. Recanalización de las venas en ecografías posteriores. Sepsis por Candida albicans, recibiendo anfotericina B durante 3 semanas. Estudio de coagulación: déficit relativo de proteína S libre (27 %) y anticoagulante lúpico positivo bajo. A los 8 años, nefrectomía del riñón izquierdo por hipertensión arterial. Varios familiares (incluida la madre) con antecedentes de trombosis y déficit de dicha proteína. Un hermano nacido en el 2004 fallecido el segundo día de vida por trombosis venosa múltiple secundaria al mismo déficit. Actualmente presenta insuficiencia renal con filtrado glomerular de 28 mL/min/1,73 m2 por Cr-EDTA y riñón único derecho de + 1,6DE de tamaño con aumento de la ecogenicidad grado I y pérdida de la diferenciación corticomedular. Conclusiones. La trombosis venosa renal neonatal es infrecuente. Resaltamos la importancia de la asociación con desórdenes genéticos del sistema hemostásico.
P26110:25 h
TERAPIAS DE DEPURACION EXTRARRENAL EN NIÑOS CRITICOS. ¿NUEVAS PERSPECTIVAS?
Francisco Vela Enríquez, Inés Tofé Valera, Elena García Martínez, Montserrat Antón Gamero, Susana Jaraba Caballero, Ignacio Ibarra de la Rosa, Esther Ulloa Santamaría, M. José Velasco Jabalquinto, Manuel Frías Pérez, Juan Luis Pérez Navero
Hospital Universitario Reina Sofía, Córdoba.
Antecedentes y objetivos: El fracaso renal agudo (FRA) es una causa frecuente de morbimortalidad en niños críticos. Las terapias sustitutivas renales realizan el aclaramiento de las toxinas y el ultrafiltrado a través de una membrana semipermeable hasta que la función renal es restablecida. Esto facilita el manejo de los pacientes y permite una nutrición y balance hídrico adecuados. La elección del tipo de técnica depende de la estabilidad hemodinámica, causa del FRA, indicación del inicio de la técnica y disponibilidad y experiencia personales. La mejora de la tecnología y mayor supervivencia de los niños críticamente enfermos han ampliado y modificado las indicaciones de las terapias de depuración extrarrenal.
Métodos: Estudio retrospectivo de los pacientes ingresados en la Unidad de cuidados intensivos pediátricos que recibieron terapia sustitutiva renal desde octubre del 1993 hasta diciembre del 2003. Se analizó la enfermedad de base, tipo de terapia utilizada e indicación de la misma, complicaciones de la técnica y pronóstico final.
Resultados: Se recogieron 76 pacientes (V/M) con una edad media de (±) años. El postoperatorio de cirugía cardíaca fue la causa más frecuente que motivó el inicio de la técnica (40,8 %). Las terapias lentas continuas fueron las más utilizadas durante todo el estudio (56,6 %). El uso de la diálisis peritoneal disminuyó en los últimos cinco años (51,5 % vs 30,9 %). La sobrecarga de volumen (84,2 %) fue la indicación más frecuente. Las principales complicaciones fueron metabólicas. Sólo 6 pacientes desarrollaron insuficiencia renal crónica y un 56,6 % del total fueron éxitus.
Conclusiones: El desarrollo tecnológico de las terapias lentas continuas permite un control metabólico y de volumen más preciso así como la posibilidad de aclarar mediadores de la inflamación. Esto ha incrementado su utilización en los últimos años. La diálisis peritoneal es más utilizada en recién nacidos y lactantes por la dificultad de acceso vascular y en aquellos pacientes con enfermedades renales primarias.
P26210:30 h
ESTUDIO MOLECULAR DE TRES PACIENTES PEDIATRICOS CON HIPEROXALURIA PRIMARIA TIPO 1
María Mar O'Callaghan Gordo, M. Isabel Luis Yanes, Marisela López Méndez, Víctor Manuel García Nieto, Alfredo Santana, Armando Torres, Eduardo Salido
Hospital Universitario Ntra. Sra. de Candelaria, Santa Cruz de Tenerife y Hospital Universitario de Canarias, La Laguna (Santa Cruz de Tenerife).
Introducción: La hiperoxaluria primaria tipo 1 (HP1) es una rara enfermedad de herencia autosómica recesiva, causada por mutaciones en el gen alanina glioxilato aminotransferasa (AGTX) que codifica un enzima importante en la metabolización del glioxilato. Una actividad insuficiente AGTX conduce a un incremento en la conversión de glioxilato a oxalato.
Pacientes y métodos: la paciente 1 fue estudiada a los 9 meses de edad tras observarse cuatro cálculos en una radiografía simple de abdomen. A los 22 meses padeció una insuficiencia renal aguda por enclavamiento bilateral de los cálculos. El paciente 2, hermano de la anterior, a los 6 años era portador de tres cálculos ecográficos. La oxaluria de ambos hermanos era de 110,8 y 185,1 mg/día/1,73 m2, respectivamente. Los niveles de los ácidos glicólico y L-glicérico fueron normales. El paciente 3 fue remitido a los 6 años de edad, por haber expulsado 5 cálculos. En la ecografía se observó, además, un cálculo coraliforme bilateral. La oxaluria era de 315,2 mg/día/1,73 m2.
Resultados: Los tres pacientes son portadores de una mutación en el gen AGTX consistente en un cambio T-C en el nucleótido 853 (exón 7), que corresponde a un cambio Ile a Thr en el residuo 244 de la proteína AGTX (I244T). Aunque I244T no afecta la actividad AGTX o su localización subcelular, cuando está presente en la misma molécula el polimorfismo común Leu-11 a Pro (L11P), se produce una modificación intragénica que da lugar a una proteína mal plegada que tiende a formar agregados inactivos con una menor actividad enzimática en los extractos celulares solubles.
Conclusiones: La mutación I244T en combinación con el polimorfismo L11P en el gen AGTX demuestra que la HP1 es una enfermedad conformacional que podría ser susceptible de tratamiento con chaperones farmacológicos. Su demostración ha confirmado el diagnóstico de HP1, a pesar de existir niveles normales de ácido glicólico en dos de los pacientes. Es la primera vez que se describe esta mutación en España, en pacientes pediátricos con HP1.
P26310:35 h
SINDROME DE BARTTER Y CELIAQUIA, UNA PUERTA ABIERTA A LA INVESTIGACION
Lucía Díaz de Entresotos Villazán, Carmen Madrigal Díez, M. Reyes Mazas Raba, Mercedes Sánchez Rodríguez, M. Teresa Viadero Ubierna, Susana Vidal Piedra, Beatriz Sangrador Martínez, M.ª Lourdes Jiménez Hernández, Domingo González-Lamuño, Pedro Fernández García
Hospital Universitario Marqués de Valdecilla, Santander (Cantabria).
Introducción: El síndrome de Bartter es una entidad de baja prevalencia caracterizada por una alcalosis metabólica hipopotasémica e hipoclorémica. Se debe a una alteración en el transporte de electrólitos en el túbulo renal, habiéndose descrito defectos generalizados en el transporte de membrana. Puede encontrarse asociado a otras patologías relacionadas con el transporte de membrana, lo cual puede dificultar el diagnóstico inicial o de sus enfermedades asociadas. Presentamos un caso de síndrome de Bartter y celiaquía, asociación no descrita previamente y que puede ser casual o debida a factores patogénicos comunes.
Niña de 19 meses con escasa ganancia ponderal. Peso: 7,480 (< p3), talla: 74 cm (< p3). Discreta anorexia, estreñimiento y vómitos ocasionales. En la exploración destaca aspecto hipotrófico, distensión abdominal, escaso panículo adiposo, piel seca, frente abombada y escasa dentición. Hemograma, bioquímica, urocultivo y cloro en sudor, normales. Iones y gasometría: discreta alcalosis metabólica hipopotasémica e hipoclorémica. Anticuerpos antigliadina y antitransglutaminasa positivos. Giardia lamblia en heces. Con la sospecha de enfermedad celiaca (biopsia no valorable y pendiente de HLA) e infección por Giardia se inició tratamiento con metronidazol y dieta exenta en gluten. En los meses siguientes no se evidenció mejoría en la curva ponderal, ingresando a los 5 meses por vómitos. La exploración era similar a la anterior (peso: 8,330). Gasometría e iones: marcada alcalosis metabólica con hipopotasemia e hipocloremia. Con la sospecha de síndrome de Bartter (aldosterona y renina elevadas) se inicia tratamiento con potasio e inhibidores de las prostaglandinas, objetivándose una mejoría clínica y analítica. La ganancia ponderal a los 20 días fue de 1.300 g, sin incidencias salvo una diarrea por Campylobacter. Se mantiene con dieta sin gluten por su síndrome celiaquiforme con HLA compatible y pendiente de nueva biopsia.
Discusión: Algunas enfermedades pueden ocasionar pérdidas crónicas de iones dando alteraciones metabólicas que simulan un Bartter (Seudobartter); las alteraciones en el transporte de membrana, el aumento en los niveles de prostaglandinas y los defectos en el manejo de determinados péptidos, como la GH, descritas en el Bartter, pueden llevar a alteraciones en la mucosa e infecciones intestinales crónicas manifestadas como un síndrome celiaquiforme.
P26410:40 h
UROPATIAS OBSTRUCTIVAS SILENTES: NUESTRA EXPERIENCIA EN LOS TRES ULTIMOS AÑOS
Joaquín Susmozas Sánchez, Carmen Vicente Calderón, Salvador Gracia Manzano, Dolores Hernández Gil, José M. Olivares Rossell
Hospital Universitario Virgen de la Arrixaca, Murcia.
Introducción: La nefropatía obstructiva es el conjunto de alteraciones renales como consecuencia de la modificación en el flujo de la orina. La estenosis pieloureteral es la más frecuente. En la actualidad la mayoría de los casos se sospecha por medio de la ecografía prenatal, si bien precisará confirmación postnatal. En niños de mayor edad la presentación clínica puede ser en forma de infección del tracto urinario (ITU), dolor abdominal recurrente o hematuria tras un traumatismo.
Casos clínicos: presentamos seis pacientes, de 3 a 10 años (mediana = 3.75 años), con diagnóstico final de estenosis pieloureteral (5) y megauréter estenótico (1).
Los motivos de consulta fueron: dolor abdominal (3), neumonía y hematuria (1), ITU (1), y polidipsia (1). Ninguno presentaba antecedentes de ITU. Sólo uno tenía ecografía prenatal con informe de normalidad. Dos de ellos ingresaron en la sección de Neonatología por causa ajena a la nefrológica (uno por prematuridad y otro por ictericia). Todos los pacientes nos fueron remitidos de otras unidades por el hallazgo en ecografía abdominal de hidronefrosis grado III-V. En dos de ellos se trataba de pacientes con riñón único. La función renal inicialmente fue normal excepto en uno, que presentaba proteinuria. Las tensiones arteriales fueron normales. Se efectuó: CUMS, DMSA, UROGRAFÍA y MAG-3 con diurético confirmando estenosis pieloureteral en cinco de los casos (uno con riñón único) y megauréter estenótico en el restante a su vez con riñón único. Tras tratamiento quirúrgico corrector, la evolución fue favorable en cuatro y dos presentaron complicaciones: litiasis renal en uno de ellos e insuficiencia renal crónica en el otro.
Discusión: Se debería establecer criterios para la realización de screening de uropatías por medio de ecografía renal en el niño. Sugerimos efectuarla en lactantes de uno a tres meses en el que conste la no realización de ecografía prenatal. De este modo se podría obtener un diagnóstico y tratamiento precoz, pudiéndose evitar complicaciones que puedan surgir posteriormente.
P26510:45 h
FACTORES PRONOSTICO DE LESION PARENQUIMATOSA Y AFECTACION FUNCIONAL RENAL ASOCIADOS A REFLUJO VESICOURETERAL DE ALTO GRADO. ESTUDIO PRELIMINAR
Montserrat Gispert-Saüch Puigdevall, Margarida Catalá Puigbó, Verónica Pérez Herrera, Beatriz Balsera Baños, Marta Abad García, Joaquim Bosch Marcet, Xavier Codina Puig, M. Amalia Zuasnabar Cotro
Hospital General de Granollers, Barcelona.
Introducción: El reflujo vesicoureteral (RVU) de alto grado es un factor importante de lesión y alteración funcional renal.
Objetivo: Evaluar los factores pronóstico de afectación parenquimatosa y funcional en las unidades renales de pacientes diagnosticados de RVU de alto grado (grado III, IV y V), controlados en nuestro centro durante los últimos 20 años.
Material y métodos: Estudio retrospectivo de 155 unidades renales diagnosticadas por cistografía miccional seriada de RVU de alto grado. Variables analizadas: sexo, edad al diagnóstico, unilateralidad-bilateralidad, unidad renal afectada, grado de reflujo, motivo del diagnóstico, infecciones previas al diagnóstico, antecedentes familiares, lesión prenatal, infecciones posteriores al diagnóstico. Se valora la afectación parenquimatosa y funcional mediante gammagrafía renal con ácido dimetilmercaptosuccínico (DMSA). Estudio estadístico con análisis bivariante y test Chi-cuadrado.
Resultados: Se ha observado lesión parenquimatosa con afectación funcional renal en un 31,6 % de las unidades renales (función renal relativa en DMSA < 45 %). Las variables: sexo masculino, grado de reflujo (predominio grado V), infecciones urinarias de repetición, ecografías con dilataciones, cicatrices o atrofia, y alteraciones prenatales (hidronefrosis prenatal) se correlacionan con mayor lesión funcional y parenquimatosa (p < 0,05).
Conclusiones: 1) Predominio de sexo femenino en la prevalencia de RVU de grado III y de sexo masculino en grados IV y V. 2) El sexo masculino condiciona un riesgo 2,6 veces mayor en las alteraciones funcionales asociadas a RVU de alto grado respecto al femenino (OD 1,29-5,3). 3) Afectación más severa en RVU de grado V (p < 0,001). 4) Correlación entre la presencia ecográfica de dilataciones, cicatrices o atrofia y afectación funcional renal (p < 0,02). 5) Correlación entre RVU de alto grado diagnosticado a partir de hidronefrosis prenatal y afectación funcional y parenquimatosa (p < 0,05). 6) Predominio de sexo masculino en las lesiones por reflujo diagnosticadas por hidronefrosis prenatal (100 %, p < 0,002).
P26610:50 h
TUBULOPATIAS HIPERCALCIURICAS NO IDIOPATICAS. REVISION DE TRES CASOS
Teresa Molins Castiella, M.ª José Azanza Agorreta, Inmaculada Nadal Lizabe, Eva Rupérez García, Francisco José Gil Sáenz
Hospital Virgen del Camino, Pamplona (Navarra).
Objetivo: Revisión de niños controlados en nuestra unidad con hipercalciuria no idiopática. Diagnóstico, tratamiento y evolución clínica.
Casos clínicos: Lactante de 12 meses, remitido por su pediatra por proteinuria persistente. Balance renal: proteinuria tubular en rango nefrótico, hipercalciuria (Ca/Cr 0,54), hiperuricosuria, hiperfosfaturia sin acidosis metabólica. En ecografía renal: mínima nefrocalcinosis. Diagnóstico: enfermedad de Dent (pendiente confirmación genética). Evolución: buena sin tratamiento, con ecografías renales estables y serie ósea normal.
Lactante de 3 meses que en estudio por estancamiento ponderal, rechazo de tomas y deshidratación se objetiva acidosis metabólica hiperclorémica, con pH urinario inapropiada y persistentemente elevado e hipercalciuria. Eco renal: nefrocalcinosis. Diagnóstico: acidosis tubular distal. Tratamiento: bicarbonato sódico y potásico. Evolución satisfactoria.
Niña de 9 años, remitida para estudio de ITU bajas de repetición y episodios de hematuria macroscópica. Antecedentes familiares de nefrolitiasis. Balance renal: insuficiencia renal leve, hipercalciuria, hipomagnesemia con fósforo y PTH normales. Ecografía renal: nefrocalcinosis. Oftalmología: cicatriz macular y alteraciones retinianas con disminución de agudeza visual. ORL: normal. Diagnóstico: Síndrome hipercalciuria-hipomagnesemia. Tratamiento: sales de magnesio, tiazidas y potasio. Evolución: asintomática, sin crisis de tetania.
Conclusión: Señalar las diferentes formas de presentación, tratamiento y evolución de 3 enfermedades con una base fisiopatológica común.
P26710:55 h
MANIFESTACIONES AUTOINMUNES DE LA INFECCION PORCAMPYLOBACTER JEJUNI
Esther Trillo Bris, Jorge Manuel Vega Fernández, Cristina Maroto García, Lucía Lacruz Pérez, M. Dolores Rodrigo Jiménez, Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca (Baleares).
Introducción: La Púrpura de Schönlein-Henoch (PSH) y la nefropatía IgA (NIgA) están relacionadas patogénicamente, pudiendo considerarse una misma enfermedad con diferente espectro clínico. El Campylobacter jejuni, patógeno fundamentalmente intestinal, se ha asociado a enfermedades autoinmunes. Presentamos un caso de PSH y otro de NIgA asociado a C. jejuni.
Caso 1: Niño, 14 años. Cuadro de fiebre, dolor abdominal, diarrea de 24 h de evolución y hematuria macroscópica en las últimas 12 h. Deterioro progresivo de la función renal con aumento de creatinina y proteinuria. Antecedentes: sin interés. P. complementarias: Hemograma, coagulación: normal. Bioquímica: Cr 1,35 mg/dl, urea 51 mg/dl, uratos 7,4 mg/d, GFR 51 ml/min/1,73m2,VSG 49mm/1.ª h, PCR 10,7 mg/dl, proteinuria 20 mg/m2/h, hematíes dismórficos abundantes en orina. Urocultivo, hemocultivo y frotis faríngeo negativos. En coprocultivos se aísla C. jejuni. ASLO normal, PPD negativo, estudio de imagen e inmunológico normales. Buena evolución clínica con normalización de la función renal en 2 semanas.
Caso 2: Niña, 7 años. Presenta púrpura palpable de 2 días de evolución, asociada a febrícula, artralgias erráticas y artritis de grandes articulaciones en las últimas 24 h. La semana previa presentó tumefacción articular sin traumatismo asociado y gastroenteritis aguda por C. jejuni. No clínica catarral. Antecedentes: sin interés. P. complementarias: hemograma: leucocitosis con neutrofilia, coagulación normal. Bioquímica: Cr: 0,4 mg/dl, urea 20 mg/dl, GFR 152 ml/min/1,73 m2, PCR: 6.4 mg/dl, VSG 41 mm/1.ª h, no proteinuria ni hematuria. Serología negativa. Coprocultivos: se aísla C. jejuni. Estudio de imagen normal. Estudio inmunológico: IgA: 300 mg/dl, ANA positivo (1/160). Durante su ingreso por HTA transitoria, precisa nifedipino oral. Tras mejoría clínica es dada de alta. Una semana después, presenta nuevo brote cutáneo y articular, con persistencia de C. jejuni en coprocultivo. Se inicia antibioterapia remitiendo el cuadro.
Comentarios: La coincidencia en el tiempo de PSH o NIgA y la infección por C. jejuni sugieren una relación causal. Aunque existen pocos casos publicados en la literatura de PSH y NIgA por C. jejuni, probablemente esta asociación sea mayor dada la dificultad de aislamiento de esta bacteria. Sugerimos que ante el diagnóstico de PSH o nefropatía con clínica digestiva, debería ser investigada la presencia de C. jejuni en heces mediante medios de cultivo específicos.
P26811:00 h
EVALUACION COMPARATIVA DE LAS DILATACIONES DE LA VIA EXCRETORA RENAL DETECTADAS PRENATALMENTE Y EN EL PERIODO NEONATAL
Álex Francisco Núñez Adán, María Ángeles Muñoz Muñoz, Guillermo Gómez-Luengo Carreras, Carmen de la Cámara Moraño, Francisca Luisa Gallardo Hernández
Hospital Comarcal Valle de los Pedroches, Pozoblanco (Córdoba).
La dilatación de la vía excretora es la alteración estructural renal más frecuente, pudiéndose detectar prenatalmente.
Objetivos: Evaluar el tipo y evolución de las dilataciones de la vía excretora renal detectadas intraútero y determinar si existen diferencias significativas con las detectadas posnatalmente de forma casual sin sospechar patología renal.
Material y métodos: Estudio retrospectivo de todos los recién nacidos vivos en nuestro Hospital durante un período de 6 años, con diagnóstico prenatal o postnatal de dilatación de la vía excretora renal. Se realiza eco postnatal a todos ellos y posteriores estudios de imagen de control y/o funcionales si procedía.
Resultados: Muestra: Diagnóstico prenatal (grupo I): 46: 24 niños y 16 niñas; incidencia del 0,87 % de los recién nacidos vivos. Diagnóstico postnatal (grupo II): 29: 16 niños y 13 niñas. Edad gestacional al diagnóstico fetal más frecuente entre las 34-36 semanas de gestación. Diagnóstico prenatal: 40 casos: no patológica: 6, dilatación leve: 31, dilatación moderada-severa: 3. Diagnóstico postnatal: grupo I: se confirma el diagnóstico en 31 casos, con 47 unidades renales afectas: no dilatación:10, dilatación grado I: 16, grado II: 11, grado III: 9 y grado IV: 1. Grupo II: 36 unidades: dilatación grado I: 18, grado II: 17 y grado III: 1. Se realizó cums a 19 niños del grupo I, detectándose reflujo vesicoureteral (RVU) en 4 casos, con 5 unidades afectas:1 de grado I, 2 de grado III, 1 de grado IV y 1 de grado V. En el grupo II, se realizó cums a 10 pacientes, encontrándose 2 casos de RVU, con 3 unidades afectas: 1 de grado I y 2 de grado II. Evolución: Grupo I: involución: 22 casos, estable o regresión: 9. Grupo II: involución: 23 casos, estable o regresión: 5. Ningún caso de progresión en ambos grupos.
Conclusiones: No existen diferencias significativas entre las dilataciones de la vía excretora detectadas prenatalmente y las detectadas en el período neonatal en todos los parámetros estudiados.
Ambas poblaciones presenta un comportamiento favorable, con involución en la mayoría de los casos en un período no superior a 12 meses.
Los reflujos vesicoureterales de más alta gradación se observan en las dilataciones detectadas prenatalmente, 1 con dilatación grado IV y 2 con dilatación grado III.
P26911:05 h
SENSIBILIDAD Y ESPECIFIDAD DE CUATRO FORMAS DE ESTIMAR LA FUNCION RENAL PARA DETECTAR ANOMALIAS MORFOLOGICAS EN PACIENTES PEDIATRICOS
Marisela López Méndez, M. Isabel Luis Yanes, Carmen Luz Marrero Pérez, Roque Abián Montesdeoca Melián, Dolores Sabina Romero Ramírez, Carlos Cárdenas, Marta de Sequera, Ana Allende, Víctor Manuel García Nieto
Hospital Universitario Ntra. Sra. de Candelaria, Santa Cruz de Tenerife.
Introducción: En muchos trastornos renales se observa un ascenso de los niveles de creatinina, únicamente, en fases avanzadas de la enfermedad. Hemos estudiado dos parámetros que detectan básicamente anomalías de la función glomerular y otros dos que estudian la función tubular y los hemos relacionado con los hallazgos observados en la gammagrafía renal.
Pacientes y métodos: Se han estudiado retrospectivamente las historias clínicas de 151 niños (75V, 76M) con una edad de 5,68 ± 3,81 años (rango: 1-16). El 64,2 % (n = 97) eran portadores de malformaciones renales. La malformación más frecuente fue el reflujo vesicoureteral (n = 58). Se recogieron los valores de los cocientes calculados entre las concentraciones urinarias de microalbúmina (MAU) y N-acetil-glucosaminidasa (NAG) con respecto a la creatinina (Cr). La osmolalidad urinaria máxima (Uosm) se determinó tras la administración de 20 mg de desmopresina. El GFR se calculó según la fórmula de Schwartz. Las gammagrafías se realizaron tras la administración del ácido Tc-99 dimercaptosuccínico (DMSA). Se excluyeron las gammagrafías realizadas en la fase aguda de una pielonefritis.
Resultados: En 79 niños (52,3 %) el DMSA fue anormal. El 27,1 % (41/151) tenía defecto de concentración. El cociente MAU/Cr fue elevado en el 17,1 % (25/146) de los casos y el cociente NAG/Cr estaba aumentado en el 7,1 % (4/56). El 6,7 % de los pacientes tenía insuficiencia renal (7/104). La sensibilidad de Uosm, MAU/Cr, NAG/Cr y GFR para detectar anomalías morfológicas renales fue de 37,9 %, 27,8 %, 11,5 % y 11,3 %, respectivamente. La especificidad de Uosm, MAU/Cr, NAG/Cr y GFR para detectar anomalías morfológicas renales fue de 86,1 %, 92,9 %, 96,4 % y 100 %, respectivamente. Uosm se correlacionó inversamente con NAG/Cr (r: 0,63; p < 0,001) y con MAU/Cr (r: 0,4; p < 0,001) y directamente con el GFR (r: 0,57; p < 0,001).
Conclusiones: La osmolalidad urinaria máxima es la prueba funcional más sensible para detectar anomalías morfológicas renales. Aunque los otros métodos muestran más especificidad, creemos que la prueba de concentración es la más adecuada, inicialmente, en el estudio funcional de niños con problemas renales.
P27011:10 h
LA ALDOSTERONA EN EL TRATAMIENTO CON FUROSEMIDA Y BALANCE DE POTASIO
M. de las Mercedes Fariñas Salto, Andrés Alcaraz Romero, M.ª del Mar Santos Sebastián, Gema Arriola Pereda, Carlos Romero Román, Bibiana Riaño Méndez, Mónica Hervías, Luis Sancho Pérez
Hospital General Universitario Gregorio Marañón, Madrid.
Objetivo: Estudiar el balance de potasio en niños tratados con furosemida (F) en el postoperatorio de cirugía cardíaca. Analizar diferencias entre administración en perfusión continua (Fpc) o en bolos (Fb). Estudiar el efecto de la aldosterona en el balance de K en estos niños.
Métodos: Estudio prospectivo, observacional y longitudinal, realizado en niños intervenidos con CEC en 2003. Se incluyeron niños que recibieron Fb o Fpc (excluidos si depuración extrarrenal). En orina (24 h) y en sangre se determinaron iones, creatinina (Cr), urea, y osmolaridad los días 2.º a 4.º de postoperatorio, y aldosterona (A) en suero. Se recogieron datos de: diuresis, tratamiento con F, aportes diarios de K y pérdidas (urinarias, drenajes y digestivas), calculando el balance de K (Balance K) (mEq/kg/día). Asimismo, se calculó diariamente el gradiente transtubular de K (GTTK) y el cociente Na/K en orina. En el análisis de los datos se utilizó el test de Mann-Whitney para comparaciones intergrupo, y correlación para estudio de relación entre variables continuas.
Resultados: Se incluyeron 66 niños: 29 tratados con Fb y 37 con Fpc. Las pérdidas de K en orina y los aportes de K fueron mayores en niños con Fpc. El balance diario de K (osciló entre 1,2 y 0,29 mEq/kg/día), el GTTK (mayor de 5 en ambos grupos) y Na/K en orina (mayor de 2 en ambos grupos) fueron similares en niños con Fb o Fpc. Las concentraciones séricas de A fueron mayores en los niños con Fpc vs Fb (2.º día 201 vs 120, 3.º día 316 vs 142 y 4.º día 278 vs 71, P < 0.05). Se encontró correlación de los valores de A con GTTK y pérdidas urinarias de K (P < 0,001). Al estratificar por grupos de tratamiento, las correlaciones se mantuvieron sólo en el grupo de Fpc.
Conclusiones: Las pérdidas de K en orina son mayores cuando la F se administra en perfusión continua y estos niños necesitan aportes mayores de K. La aldosterona influye en las pérdidas de K en orina de estos niños. La aldosterona es mayor en los niños tratados con perfusión continua.
P27111:15 h
URÉTER ECTOPICO: A PROPOSITO DE DOS CASOS
Rebeca Villares Alonso, José M. Avilla Hernández, Francisco Javier Navarro Sebastián, M.ª Luisa Rodríguez Rodil, M.ª Fe Muñoz Bermudo, María Arriaga Redondo
Complejo Hospitalario de Móstoles, Madrid.
Introducción: La ectopia ureteral (EU) consiste en una implantación anómala del meato ureteral. Aunque su manifestación clínica característica es fácilmente reconocible, no siempre se llega a un diagnóstico temprano, hecho descrito en la literatura. Se presentan dos casos clínicos de niñas atendidas en nuestro servicio por presentar incontinencia urinaria.
Caso 1: Niña de 9 años con incontinencia diurna y nocturna desde hace 1 mes, con patrón miccional normal. Previamente continente. E.F: orificio a 1 cm del meato uretral en porción media de pared vaginal. UIV: DPU derecha con dilatación polo superior, no se ve uréter. Renograma isotópico (RI): ectasia no obstructiva pielón superior, función 31,8 %. Cistouretroscopia: meato ureteral eutópico correspondiente a polo inferior RD, no se localiza meato pielón superior. Tratamiento: reimplante ureteral de ambos uréteres derechos. Evolución: continencia urinaria.
Caso 2: Niña de 4 a con ITU recurrente desde lactante con genitales externos femeninos aparentemente normales. Pruebas previas: ECO: ureterohidronefrosis bilateral de ambos grupos caliciales superiores hasta vejiga. UIV (x2): DPU bilateral, dilatación pielones superiores y uréteres en Y con probable abocamiento a vejiga. Refiere en ese momento patrón miccional normal aunque continuamente mojada. ECO: meatos a nivel vesical con drenaje de orina e imágenes tubulares bilaterales posteriores a vejiga con posible drenaje a nivel vaginal. UIV: DPU completa, abocamiento del pielón superior izquierdo en vagina. EF: meatos ectópicos a ambos lados en vestíbulo vaginal. Pruebas isotópicas: pielón superior izquierdo 23 %, pielón superior derecho 12 %. ambos con cicatrices; ectasia no obstructiva. Tratamiento: reimplante ureteral bilateral (Politano-Leadbetter), remodelación uréter derecho. Evolución: logra continencia urinaria, ITU recurrente. UIV: retraso eliminación pielón superior derecho. DMSA: nefropatía cicatricial ambos pielones superiores. RI: dificultad de drenaje del uréter derecho.
Conclusiones: 1) Ante clínica de incontinencia urinaria con patrón miccional normal, incluso de aparición tardía, debe excluirse la presencia de uréter ectópico, agotando todas las pruebas diagnósticas disponibles aunque las iniciales no lo sugieran. 2) Suele ser un diagnóstico tardío, más difícil en período de lactante. 3) El tratamiento quirúrgico logra la continencia, optándose por preservar los hemirriñones en nuestros casos.
P27211:20 h
MARCADORES DE DAÑO TUBULAR RENAL EN EL POSTOPERATORIO DE CIRUGIA CARDIACA
Elena Cidoncha Escobar, Andrés Alcaraz Romero, Ayoub Bennami, Bibiana Riaño Méndez, Itziar Marsinyach Ros, Ángel Carrillo Álvarez, Francisco Cañizares Hernández, Raúl Roberto Borrego Domínguez
Hospital General Universitario Gregorio Marañón, Madrid y Hospital Universitario Virgen de la Arrixaca, Murcia.
Objetivos: Estudiar los valores de marcadores de daño tubular en el daño renal asociado al período perioperatorio de cirugía cardíaca en niños.
Métodos: Estudio prospectivo, observacional, descriptivo y analítico. Se incluyeron los niños ingresados en UCI tras intervención por cardiopatías. Durante los primeros 2 días de ingreso se determinó en plasma creatinina (Cr), Na, osmolaridad y en orina de 24 h Cr, Na, osmolaridad, proteínas, β2-microglobulina (β2mg) y N-acetil-beta-glucosaminidasa (NAG). En la evaluación del daño renal se utilizó el cociente β2mg/Cr y NAG/Cr, la FENa y aclaramiento de Cr (CCr) y agua libre (CH2O), y como disfunción renal se consideró descenso del CCr < 50 % del basal y necesidad de dosis altas de furosemida (F) (> 12 mg/kg/día). Datos presentados como medianas (P25-P75), y analizados con correlación de Spearman y correlaciones parciales; las comparaciones entre grupos se realizaron con el test de Mann-Whitney.
Resultados: Fueron incluidos 70 niños (1 mes a 16 años, mediana 9 meses). Los valores de los parámetros evaluados se asociaron con disfunción renal, y se presentan en la siguiente tabla (P50):

El cociente proteínas/Cr en orina fue mayor en los niños con disfunción renal (P = 0,018). Hubo correlación de los valores de NAG/Cr con los de β2mg, CCr, CH2O y proteínas/Cr. Controlando el efecto del CCr se mantiene la correlación con b2mg/Cr y sobre todo con proteínas/Cr (r = 0,607, P < 0,001).
Conclusiones: El incremento urinario de NAG es un indicador de disfunción renal en el postoperatorio de cirugía cardíaca. Su aumento se relaciona con la alteración de otros parámetros de función tubular. El aumento de la actividad de NAG se relaciona con la eliminación de proteínas en la orina.
P27311:25 h
REVISION CLINICO-EPIDEMIOLOGICA DE LOS PACIENTES CONTROLADOS EN NUESTRO SERVICIO POR INSUFICIENCIA RENAL CRONICA (IRC) DESDE EL 1994-2005
Ana Belén Escobar Izquierdo, M. del Carmen Molina Molina, Pilar Méndez Pérez, Emilia Hidalgo Barquero del Rosal, José M. García Blanco
Hospital Universitario Infanta Cristina, Badajoz.
Objetivos: Descripción clínica y epidemiológica de los pacientes controlados por el Servicio de Nefrología Pediátrica en los 10 últimos años.
Material y métodos: Revisión de las historias clínicas de los pacientes en seguimiento por IRC en el servicio de Nefrología Pediátrica de nuestro Hospital desde el 1994 hasta la actualidad y posterior análisis estadístico de los resultados obtenidos mediante el programa SPSS.
Resultados: Se encontraron 34 pacientes controlados en nuestro servicio por IRC en estos últimos 10 años, de los cuales el 50 % fueron varones y el 50 % mujeres. La media de edad fue de 11,5 años y la mediana de 12. En cuanto a la edad de diagnóstico, la media fue de 2,5 años y la mediana de 1,5. Los diagnósticos principales fueron: 35,3 % uropatías, 26,5 % vasculares (SHU, nefropatía hipóxico-isquémica...), 14,7 % nefropatías hereditarias, 11,8 % glomerulopatías, 5,9 % displasias y 5,6 % restantes de otro origen. En cuanto a las variables clínicas analizadas: ninguno presentó osteodistrofia, el 38,3 % anemia, 20,6 % hipertensión arterial, 23,5 % acidosis metabólica y el 14,7 % retraso del crecimiento. El grado de IRC hasta su derivación a Nefrología de Adultos o hasta la actualidad fue: en el 44,1 % IRC leve, en el 29,4 % IRC moderada y en el 26,5 % IRC severa. En último término analizamos el tratamiento seguido por los pacientes: el 79,4 % se encontraban en tratamiento dietético (dieta hipoproteica e hiposódica), el 26,5 % con bicarbonato, el 58,5 % con quelantes del fósforo, el 35,3 % con vitamina D, el 23,5 % con EPO, el 44,1 % con antihipertensivos y sólo un 3 % se encontraba en tratamiento con GH.
Conclusiones: La IRC en la edad pediátrica tiene una igual distribución por sexos. El factor etiológico más importante son las uropatías y dentro de estas las más importantes son las nefropatías por reflujo. Durante la edad infantil suelen predominar los estadios leves, precisando escaso tratamiento farmacológico.
P27411:30 h
PIELONEFRITIS XANTOGRANULOMATOSA: A PROPOSITO DE UN CASO
Rebeca Villares Alonso, José M. Avilla Hernández, Miguel Mora Durban, M.ª Jesús Fernández Acereño, M.ª Fe Muñoz Bermudo, Javier Blumenfeld Olivares
Complejo Hospitalario de Móstoles, Madrid.
Introducción: La pielonefritis xantogranulomatosa (PNX) es una forma atípica y severa de infección crónica del parénquima renal de evolución subaguda, siendo una variante morfológica de la pielonefritis crónica. Gran imitadora de otras patologías renales, a menudo está clínicamente infradiagnosticada. Se presenta un caso de PNX.
Caso clínico: Niña rumana de 11 años remitida por dolor y palpación de masa en hipocondrio izquierdo de 1 mes de evolución, referida como esplenomegalia dada la presencia de Monotest positivo. A.P: ITU recurrente. Disuria, polaquiuria y piuria "desde siempre", más intensa en últimos meses, con urocultivos negativos. E.F: masa firme en hipocondrio izquierdo, 2 traveses por debajo del nivel del ombligo, de consistencia dura, dolorosa, con contacto lumbar evidente, PPR izquierda+. Analítica: OE: intensa piuria, bacteriuria. RFA, función renal, estudio inmunológico, estudio de litiasis y catecolaminas en orina: normales. Hemocultivo, urocultivo, Mantoux, y serologías negativas. Baar en orina negativos. Citología orina:PMN y células histiocitarias de aspecto espumoso. Pruebas de imagen: Eco: riñón izquierdo (RI) aumentado de tamaño, sin diferenciación corticomedular, lesiones quísticas corticales y lesiones hiperecogénicas con sombra acústica. TC: anulación funcional RI, masas hipodensas en área renal, uréter dilatado con cálculo de 3 cm adyacente a meato vesical. Dx presunción PNX. Tratamiento: nefroureterolitectomía izquierda, Anatomía patológica (AP): macroscopia: parénquima renal sustituido por estructuras quísticas rellenas de material amarillento; microscopia: histiocitos, células gigantes tipo multinucleado, atrofia glomerular. Dx definitivo: PNX. No vuelve a revisión.
Discusión: El diagnóstico diferencial de masas abdominales intrarrenales es amplio, debiendo incluir la PNX. La mayoría de los autores concluyen que el dx preoperatorio no es fácil dado su cuadro clínico abigarrado, similar a otras patologías (Tumor de Wilms, Tbc...), ayudando el hecho de que sea unilateral, que la función del riñón afecto este ausente o muy disminuida y la presencia de numerosos y grandes cálculos, no siempre presentes. Es por ello la necesidad de estudios analíticos y de imagen amplios, debiendo descartar que la inflamación se extienda más allá del riñón. Su diagnóstico definitivo es AP y la cirugía curativa. La clínica bizarra, y sobretodo los hallazgos radiológicos en esta paciente, nos hicieron sospechar una PNX que se confirmó en la AP.
P27511:35 h
ECTASIA PIÉLICA NEONATAL: REVISION DE 98 CASOS
Silvia Martínez García, Raquel Sánchez García, Carmen Vicente Calderón, Salvador Gracia Manzano, Moisés Sorlí García, Amparo Gilabert Úbeda, Francisco Valero, Vicente Bosch Jiménez, Manuel Sánchez-Solís de Querol
Hospital Universitario Virgen de la Arrixaca, Murcia.
Objetivos: Analizar el manejo y diagnóstico de los pacientes con ectasia piélica en la casuística de nuestro hospital.
Pacientes y métodos: Diseñamos un estudio retrospectivo en le que se revisan 98 pacientes, diagnosticados de ectasia piélica en el primer mes de vida desde enero de 1999 a diciembre de 2004. Se recogieron los siguientes datos: sexo, indicación y edad de realización de la 1.ª ecografía, grados en mm (I < 10, II de 11-15, III de 16-20, IV > 20), otras exploraciones complementarias, tratamiento y evolución ecográfica.
Resultados: Utilizamos el programa estadístico SPSS con los siguientes resultados: de los 98 pacientes, 78 fueron varones (79,6 %) y 20 mujeres (20,4 %); tenían dilatación bilateral 43,9 %; de las ectasias unilaterales, fueron izquierdas 69 % y derechas 31 %. Respecto al grado de ectasia en riñón derecho se encontraron grado I 56,6 %, II 28,3 %, III 5 %, IV 10 % y en el riñón izquierdo grado I 53 %, II 29,6 %, III 9,8 %, IV 7,4 %. Motivo de la 1.ª ecografía: alteración ecográfica prenatal 36,7 %, infección urinaria 31,6 %, prematuridad 17,3 %, estudio de síndrome polimalformativo 3 %, otras 11,2 %. Se realizo CUMS en 88,7 %, y se encontraron alteraciones en un 46 %, de los cuales tenían reflujo grado III o mayor en un 45 %, correlacionándose el grado de reflujo con la presencia de la ectasia (p < 0,05). Se realizó renograma diurético en 26,5 %, con alteraciones en el 88 % de los mismos, siendo obstructivas 60,8 % y no obstructivas 39,2 %. Se realizo DMSA en 38,7 %, existiendo alteraciones en 31 %. En cuanto al tratamiento, precisaron cirugía 32,6 % (4 pieloplastias, 11 reimplantaciones ureterales, 11 cirugías endoscópicas, 4 heminefrectomías), según el grado de ectasia siguieron tratamiento antibiótico profiláctico 37,7 % y vigilancia sin tratamiento 29,6 %. Durante su evolución presentaron infección urinaria 42,9 %. Se normalizó la pielectasia sin tratamiento quirúrgico en 43,9 %, siendo la edad media de desaparición de 13 meses.
Conclusión: En la población estudiada, la ectasia piélica predomina en varones (relación 4:1) siendo más frecuente en lado izquierdo. Se asocia a reflujo vesicoureteral en el 37 % y a uropatía obstructiva 14 %, precisando cirugía en un alto porcentaje, todos ellos con dilataciones piélicas mayores de 15 mm. Por todo ello, indicamos el estrecho seguimiento de estos pacientes.
P27611:40 h
CISTINURIA: REVISION DE NUESTRA CASUISTICA. 37 CASOS
Salvador Gracia Manzano, Carmen Vicente Calderón, Antonio Vicente Santos, Encarnación Guillén Navarro, Concha González, Asunción Fernández Sánchez
Hospital Universitario Virgen de la Arrixaca, Murcia.
Introducción: La cistinuria es una enfermedad hereditaria, debida a un defecto en el transporte tubular renal e intestinal de cistina y aminoácidos dibásicos. Heterogeneidad genética y alélica. Causante 5-10 % litiasis en la infancia. Disponible screening metabólico en orina para el diagnóstico precoz.
Objetivo: Revisión de nuestra casuística.
Material y métodos: Desde 1989-2002 diagnosticamos 37 niños afectos de cistinuria detectados por screening neonatal en orina y confirmados a los 12 meses. Se recoge orina de 24 h de los padres y de los pacientes para determinar el fenotipo. Se analiza la incidencia, características fenotípicas, tratamiento y presencia de nefrolitiasis. Se clasifican en homocigotos y heterocigotos según la excreción de cistina (> 250 mg/g creatinina) y tipo I/tipo no I según la excreción de cistina y aminoácidos dibásicos de los padres.
Resultados: Se realizó screening neonatal a 186419 RN, estimándose una incidencia de 1/4236 RN. 37 pacientes (21V/16M). Tipo I homocigoto (HMI) 13, tipo no I homocigoto (HMNI) 4 y 20 tipo no I heterocigoto (HTNI). 4 pacientes presentaron litiasis: 2 HMI y 2 HMNI. Edad media de aparición de litiasis: 3 años y 3 meses; sólo 1 paciente presentó síntomas mientras que en los otros 3 la litiasis se detectó de forma casual en ecografía de control. Todos recibieron tratamiento con hiperhidratación y/o alcalinización y uno precisó tratamiento quirúrgico. Se realizó estudio molecular en 2 pacientes en los que se detectó mutación en el gen SLC3A1, clasificados fenotípicamente como HMI.
Conclusiones: 1) La incidencia de cistinuria en nuestro estudio es más alta, probablemente debido a su detección precoz. 2) El screening metabólico neonatal en orina permite la identificación precoz de esta patología y la posible prevención de litiasis. 3) La cistinuria es una causa poco frecuente de litiasis en la infancia aunque a tener en cuenta como posible etiología. 4) Una correcta identificación fenotípica y genotípica permitirá una mejor profilaxis y tratamiento de esta patología.
NEONATOLOGIA
P27710:15 h
MALFORMACIONES ANORRECTALES CONGÉNITAS: REVISION DE 47 CASOS
Olga Domínguez García, Carlos Orbea Gallardo, Gerardo Bustos Lozano, Alberto Galindo Izquierdo, María López Díaz
Hospital Universitario 12 de Octubre, Madrid.
El objetivo de este trabajo ha sido estudiar las características, evolución y pronóstico neonatal de las malformaciones anorrectales congénitas en nuestro hospital. Para ello hemos realizado un estudio retrospectivo descriptivo en un período de tiempo de 14 años, de enero de 1.990 hasta diciembre del 2.003, en el que se han recogido 47 casos.
El embarazo fue controlado en 46 de los pacientes. Ninguno de ellos estaba diagnosticado prenatalmente. El 60 % de los casos fueron varones y la edad gestacional media al nacimiento fue de 37,9 ± 2,8 semanas (28-42 semanas). El peso medio al nacer fue de 2.874 ± 750 g (800-4.320 g) con un porcentaje de prematuridad (< 37 semanas) del 25,5 %.
Respecto al tipo de malformaciones recogidas, en la mayoría de los casos no consta si eran altas o bajas, pero sí podemos decir que en 27 de los pacientes no se acompañaban de fístulas. En los 20 restantes los tipos de fístulas fueron: 9 rectovaginales, 8 perineales, 2 rectouretrales y 1 rectovesical.
Las cirugías practicadas fueron 0,9 intervenciones/paciente, realizándose 28 colostomías y 14 anoplastias. En 5 de los niños no se practicó ninguna cirugía por limitación del esfuerzo terapéutico (LET). El tiempo medio de estancia hospitalaria fue de 18 días. Como principales complicaciones destacan: sepsis nosocomiales en 4 pacientes y una enterocolitis necrosante (NEC) secundaria a cirugía en 1 de los niños. No se encontró ningún caso de peritonitis poscirugía ni de intestino corto.
Hay que destacar la presencia de patología asociada hallándose en la muestra estudiada: 9 síndromes VACTERL, 2 síndromes de regresión caudal, 1 síndrome de Potter, 2 trisomías 21, 1 trisomía 12, 1 trisomía 8 y 4 síndromes polimalformativos no filiados.
La mortalidad total fue de 6 pacientes (12,7 %), siendo 5 de ellos LET por síndromes polimalformativos severos, y el caso restante a consecuencia de una NEC con sepsis por cándida.
Como conclusiones principales de este estudio podemos destacar: el escaso porcentaje de prematuridad en estas malformaciones en comparación con otras obstrucciones intestinales congénitas, la gran dificultad existente para su diagnóstico prenatal y su asociación frecuente a síndromes polimalformativos.
P27810:20 h
REVISION DE LA CASUISTICA DE MENINGITIS NEONATAL EN NUESTRO HOSPITAL 2000-2004
Silvia García Martínez, Fuensanta Alemán Lorca, Ángel B. Brea Lamas, Juan José Quesada López, Vicente Bosch Jiménez, Juan José Agüera Arenas, M.ª Victoria López Robles
Hospital Universitario Virgen de la Arrixaca, Murcia.
Objetivos: Realizar un estudio descriptivo de la meningitis en el neonato en relación a los mecanismos de transmisión de la infección, factores asociados, microorganismos responsables, complicaciones y secuelas.
Métodos: Se lleva a cabo una revisión retrospectiva de las historias clínicas de los pacientes entre 0-28 días diagnosticados de meningitis en nuestro hospital entre el 1 de enero del 2000 y el 31 de diciembre del 2004. Se asignan los pacientes según el mecanismo de transmisión a tres grupos: meningitis vertical, nosocomial y comunitaria, clasificándose como meningitis microbiológicamente probada [líquido cefalorraquídeo (LCR) (+)], probable [LCR () y hemocultivo (+)] y no probada [LCR y hemocultivo ()].
Resultados: Se diagnosticaron 47 casos de meningitis. Un 32 % eran verticales, 30 % nosocomiales y 36 % comunitarias. De las meningitis verticales, fueron probadas el 80 %, aislándose en el 60 % el estreptococo grupo B (EGB), sólo estas presentaron secuelas neurológicas al alta (55,5 %), sin ningún éxitus. Excluyendo el sexo masculino, el 46,6 % no tenían ningún factor asociado. De las meningitis nosocomiales, el 86 % fueron probadas, identificándose Serratia marcescens en el 30,7 %. Un 86 % fueron prematuros, sometiéndose a técnicas invasivas el 60 %. La mortalidad en este grupo fue del 40 %, la mayoría (60 %) de peso < 1.500 g, si bien no encontramos ningún patógeno asociado a mayor mortalidad. De los supervivientes el 55,5 % presentaron secuelas neurológicas. En las meningitis comunitarias el 70 % fueron no probadas, debutando el 82 % pasados los primeros 7 días de vida. El 60 % de las meningitis verticales fueron tardías, más de 7 días, y un 33 % de las nosocomiales fueron precoces.
Conclusiones: En las meningitis verticales el germen prevalente es el EGB, mientras que en las nosocomiales existe un amplio espectro de patógenos, destacando serratia. En las meningitis no probadas sospechamos etiología viral por su curso benigno, aunque no se solicitaron cultivos específicos para virus. Probablemente el número real de meningitis nosocomiales se encuentra subestimado por el bajo índice de punciones lumbares que se realizaron. La frecuencia de las secuelas en las meningitis por EGB es similar a las nosocomiales, sin embargo la mortalidad fue mucho mayor en estas.
P27910:25 h
TRATAMIENTO CONSERVADOR EN LA PERITONITIS MECONIAL: A PROPOSITO DE UN CASO
Cristina Pérez Aragón, José Manuel Gallego Soto, Simón Pedro Lubián López, Juan Mena Romero, Manuel Mendoza Jiménez, Isabel Benavente Fernández, Francisco Vega Burgos, Raimundo Arnet Jiménez
Hospital Universitario Puerta del Mar, Cádiz.
Introducción: La peritonitis meconial es el resultado del paso de meconio de la luz intestinal a la cavidad peritoneal. A menudo, se asocia con una perforación prenatal del tracto intestinal. Su incidencia aproximada es de 1/35.000 recién nacidos, aunque no es excepcional la regresión espontánea intraútero.
Caso clínico: Recién nacido varón de 37 semanas de gestación que ingresa en Neonatología por hidrocele de gran tamaño. Producto de primera gestación, parto por vacuoextracción. Apgar 8/9. Peso: 2.440 g. Controles ecográficos prenatales normales. Antecedentes familiares sin interés. A las pocas horas de su ingreso presenta vómitos biliosos y coloración pajiza de piel, no constatándose eliminación de meconio en las primeras horas de vida e iniciando distensión abdominal progresiva. Ante la sospecha de obstrucción intestinal se realiza radiografía de abdomen evidenciándose imágenes compatibles con peritonitis meconial. Se decide actitud conservadora. Al segundo día de vida presenta neumoperitoneo por lo que se realiza punción abdominal con colocación de drenaje peritoneal bilateral, observándose en los días sucesivos eliminación de exudado peritoneal amarillento y mejorando el estado general del paciente. A los 19 días de vida, se observa fístula escrotal izquierda con extravasación de líquido purulento meconial, por lo que se realiza drenaje bilateral escrotal. Tres días después inicia deposiciones por ano. Se solicita enema opaco donde se visualiza microcolon con imágenes sugestivas de perlas de meconio sobre colon transverso. Dos días después se retiran los drenajes escrotales y abdominales. Posteriormente, dada la buena evolución, se inicia alimentación enteral permitiendo la retirada progresiva de nutrición parenteral. Al alta, el tránsito intestinal es normal. Test del sudor: 38 mEq/l. Estudio genético de fibrosis quística: negativo.
Conclusiones: Ante cuadro de obstrucción intestinal con tumefacción testicular es necesario descartar peritonitis meconial. Confirmamos la idoneidad del tratamiento expectante no quirúrgico en casos seleccionados de peritonitis meconial.
P28010:30 h
EPIDEMIOLOGIA DEL SINDROME DE DOWN EN EL PERIODO NEONATAL DURANTE 20 AÑOS EN NUESTRO MEDIO
Diego López de Lara, M. Isabel Armadá Maresca, Luis Arruza Gómez, Gemma Villar Villar
Hospital Clínico San Carlos, Madrid.
Antecedentes: El síndrome de Down es la anomalía cromosómica más frecuente. Es causa del 8-10 % de las oligofrenias constituyendo un problema social importante con una incidencia de 1/600-800 RN vivos.
Objetivos: Estudiar las características de los recién nacidos con de síndrome de Down en nuestro servicio entre 1982 y 2002.
Material y métodos: Se estudiaron retrospectivamente 75 casos diagnosticados de síndrome de Down mediante recogida de datos en las historias clínicas. Se consideró dicho diagnóstico con cariotipo compatible. Se investigó edad materna, edad gestacional, tipo de parto, sexo, presencia de cardiopatía, malformaciones digestivas, trastornos hematológicos, trastornos respiratorios, hipoglucemia, alteraciones traumatológicas, hipotiroidismo, patología oftalmológica.
Resultados: Incidencia de 1/800 RN vivos. Edad materna 33,7 ± 6 (DE), edad gestacional 38,7 ± 1 (DE); tipo de parto: eutócicos 64 %, cesárea 34 %, fórceps 3 %; varones 44 %, mujeres 66 %. Presencia de cardiopatía en el 36 % de los casos. De ellos el 40 % fueron ductus arteriosos persistentes. Malformaciones digestivas en el 14 % incluyendo atresia duodenal, malrotación intestinal, atresia colorrectal, páncreas anular, fístula traqueoesofágica, atresia biliar. Trastornos hematológicos 14,6 % de los cuales el 70 % fueron policitemias. Trastornos respiratorios 12 % de los cuales 40 % fueron distrés respiratorios adaptativos. Hipoglucemia en 6,5 %. Alteraciones traumatológicas 5,5 %, de ellas 75 % fueron luxación congénita de caderas. El 3 % presentaron hipotiroidismo y un 1,5 % tuvieron patología oftalmológica en forma de catarata neonatal.
Conclusiones: El síndrome de Down es una cromosomopatía que asocia pluripatología importante que debe ser descartada en la época neonatal. La incidencia de cardiopatía observada es inferior a la descrita en la literatura. También existen variaciones importantes en cuanto al tipo de cardiopatía. Ambos hallazgos se deben probablemente a la ausencia en nuestro hospital de cirugía cardíaca infantil y al hecho de derivar a otros centros los casos con sospecha prenatal de cardiopatía congénita quirúrgica. La incidencia de hipotiroidismo observada es superior a la de otros estudios previos.
P28110:35 h
SINDROME DE MARFAN NEONATAL
José Camilo López Pena, Gemma Ginovart Galiana, Carlos Salvador Goyanes Sotelo, Zoraida Rubio, M.ª José García Borau, Elisenda Moliner Calderón, Sonia Brio Sanagustín, Bibiana Pineda Prats, Almudena Sánchez Álvarez, Eduardo Tizano
Hospital de la Santa Creu i Sant Pau, Barcelona y Universidad Autónoma, Barcelona.
Introducción: El síndrome de Marfan (SM) es una enfermedad genética, cuya frecuencia estimada es de 1/10.000 nacidos. Herencia autosómica dominante en 70 % de los casos. El SM se debe a mutaciones en el gen FBN1, que codifica para la fibrilina, este gen se localiza en el brazo largo del cromosoma 15. La existencia de unas 500 mutaciones diferentes dificulta su diagnóstico molecular. Actualmente el diagnóstico sigue basándose en criterios clínicos.
Caso: Varón fruto de 2.ª gestación de 37+6 semanas. No antecedentes familiares de interés. Gestación bien controlada. No incidencias perinatales. Ecografías prenatales: agenesia arteria umbilical izquierda. Eje cardíaco desviado a la derecha. EF al nacimiento: Dolicocefalia, aracnodactilia, pectum excavatum, extremidades largas en relación al tronco. Hiperlaxitud articular de manos, pies, codos y rodillas. Soplo sistólico III/VI en foco mitral. Ecografía transfontanelar: normal. Homocisteína en plasma: normal. Estudio fondo de ojo: normal. Estudio genético SM: en curso. Ecocardiografía: válvula mitral redundante, con prolapso de ambas valvas en aurícula izquierda. Regurgitación mitral. Válvula aórtica bicúspide, con insuficiencia de grado ligero. Dilatación importante del anillo, raíz y aorta ascendente. Fracción de ejección VI del 72 %. Ante la ausencia de signos de insuficiencia cardíaca se realiza seguimiento ambulatorio. A los 2 meses de vida presenta diaforesis y dificultad respiratoria coincidiendo con las tomas. Radiografía de tórax: índice cardíaco-torácico 0,7. Ecocardiografía: fracción de ejección 69 %. Válvula mitral redundante con prolapso evidente de ambas valvas. Insuficiencia mitral severa. Dilatación importante de cavidades izquierdas. Válvula tricúspide redundante con prolapso de valvas y regurgitación severa. Válvula aórtica bicúspide con insuficiencia ligera. Se realiza restricción hídrica y se inicia tratamiento con furosemida y digoxina. Valorado por cirugía cardíaca (Hospital Vall d'Hebron), se realiza sustitución protésica de válvula mitral, a los 3 meses de vida. Posteriormente presenta insuficiencia cardíaca progresiva que no responde a tratamiento médico, es éxitus a los 4 meses.
Conclusión: La forma neonatal del SM es la forma menos frecuente y más grave. La afectación global y precoz de las válvulas cardíacas condiciona su mal pronóstico en los primeros meses de vida. En la actualidad el complejo diagnóstico molecular hace difícil el establecimiento del consejo genético.
P28210:40 h
ESTENOSIS HIPERTROFICA DE PILORO (EHP) EN RELACION AL TRATAMIENTO POR ERITROMICINA
Raquel Amo Rodríguez, Amo Jiménez, Miguel Fiol, Pedro Balliu Badía, Carmen Clavero
Hospital Son Dureta, Palma de Mallorca (Baleares).
Introducción: La asociación de EHP en relación al uso sistémico de eritromicina en el neonato fue descrito ya en los años 70. Se ha observado un aumento del riesgo de EHP en RN tratados con eritromicina en las 2 primeras semanas de vida. Se precisan nuevas investigaciones para aclarar el mecanismo exacto por el cual se produce este efecto.
Objetivos: Descripción de un caso clínico de asociación de Estenosis Hipertrófica de Píloro (EHP) en relación al uso de eritromicina.
Caso clínico: Recién nacido a término, de peso adecuado para la edad gestacional, que es trasladado desde otro hospital por distrés respiratorio y sospecha de aspiración meconial. Parto eutócico, con amniorrexis preparto, líquido amniótico teñido. Apgar: 4-6-8. Ph cordón: 7,13. Reanimación tipo III. Presenta dificultad respiratoria en aumento precisando intubación en el hospital emisor. Se mantiene intubado 4 días con necesidad de parámetros respiratorios elevados (SIMV, VAFO) y posteriormente ventilación no invasiva (N-IMV, N-CPAP) durante 6 días, persistiendo dificultad respiratoria. Presenta alteración de parámetros analíticos y clínica de infección respiratoria: hipoventilación, crepitantes bibasales, que no se acompaña de cambios radiológicos, por lo que se pauta tratamiento con cefotaxima y vancomicina. Como no existe mejoría clínica, se realiza fibrobroncoscopia (normal) y se obtiene cultivo del aspirado broncoalveolar y serologías de infección respiratoria (negativos). Ante la sospecha de infección por germen no sensible al tratamiento, se decide tratar empíricamente a los 16días de vida con eritromicina, (durante 9 días) con mejoría respiratoria progresiva. A los 5 días del tratamiento con eritromicina inicia cuadro de vómitos, se cambia a una leche con hidrolizado de proteínas sin mejoría, se realiza RAST a PLV que resulta negativo y en las ecografías de abdomen se objetiva EHP, que persiste a pesar de retirar la eritromicina por lo que se interviene a los 30 días de vida. El postoperatorio transcurre sin incidencias, con resolución del cuadro digestivo.
Conclusiones: 1) Valorar el riesgo-beneficio del uso de eritromicina sistémica en período neonatal. 2) Vigilar los posibles signos, síntomas de EHP cuando se decida tratamiento con eritromicina en este período, que facilite un diagnóstico precoz, evitando las complicaciones. 3) Informar a la familia sobre los posibles efectos secundarios.
P28310:45 h
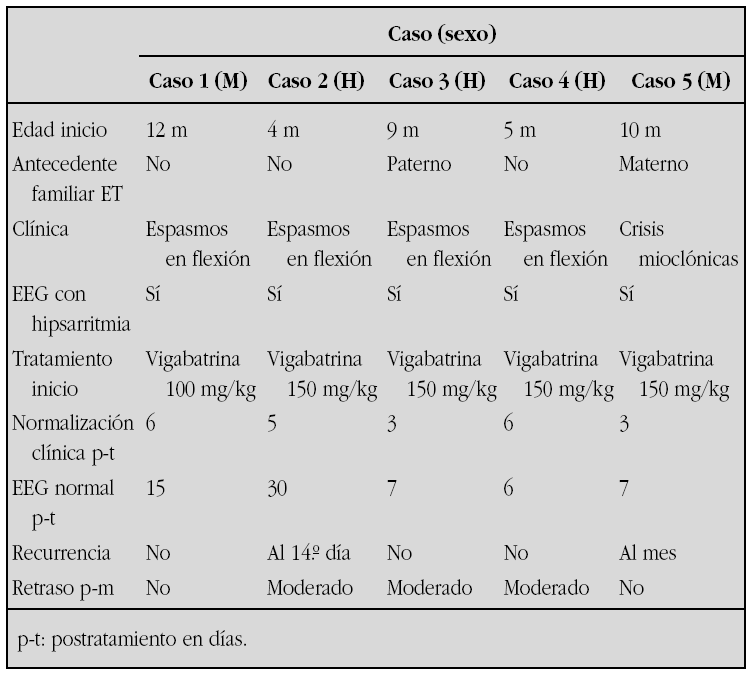
LETARGIA EN EL PRIMER DIA DE VIDA
Juan Mayordomo Colunga, Marta Costa Romero, Mónica García González, Elena Taborga Díaz, Laura Somalo Hernández, Rosa P. Arias Llorente, Aleida Ibáñez Fernández, José Blas López Sastre
Hospital Universitario Central de Asturias, Oviedo (Asturias).
Introducción: La hiperglicinemia no cetósica neonatal es un raro error innato del metabolismo, de herencia autosómica recesiva, cuya base fisiopatológica es un fallo en el sistema de degradación de la glicina (Gly), importante neurotransmisor a nivel de médula espinal y tallo cerebral actuando sobre interneuronas inhibidoras, y coagonista del Glutamato sobre receptores tipo NMDA a nivel del córtex cerebral. Presentamos un caso de un neonato con estupor y arreactividad a estímulos.
Caso clínico: Recién nacida a término que ingresa a las 20 h de vida por hipotonía, letargia, llanto escaso y débil, dificultad para la alimentación, ausencia de movimientos espontáneos, episodios de hipo, reflejo de Moro ausente y succión débil. Fenotipo normal, Apgar 9/10, somatometría RN: peso 2965 g (P25-50), talla 49 cm (P50), PC 33 cm (25-50). Chequeo infeccioso negativo, no acidosis, cuerpos reductores en orina negativos y amonio sérico normal. Aumento de glicina sérica (1905 μmol/l), urinaria (108975 μmol/g creatinina) y en LCR (217 μmol/l), con relación GlyLCR/Glysuero de 0,2. EEG iniciales compatibles con daño cerebral inespecífico; posteriormente patrón de paroxismo-supresión.
A las 48 h de vida apneas repetidas por lo que se conecta a ventilación mecánica modo SIMV hasta el día 14, en el que se extuba, permaneciendo con respiración espontánea a partir de entonces. Succión ausente con alimentación mediante sonda nasogástrica, no episodios de crisis cerebrales durante su ingreso en nuestro centro.
En la actualidad crisis de espasmo rebeldes a tratamiento con benzoato sódico, dextrometorfano, topiramato y vigabatrina, con marcada hipotonía y escasa movilidad espontánea.
Comentarios: El diagnóstico definitivo de esta entidad se establece midiendo el grado de actividad del sistema de degradación de la Gly en biopsia hepática, aunque es diagnóstico una ratio GlyLCR/Glysuero > 0,08. Inicialmente presentan un patrón de EEG de burst-supression, que evoluciona a un trazado de hipsarritmia típico o atípico a partir de los tres meses de edad.
No se conoce ningún tratamiento efectivo. El dextrometorfano, y en algún caso el benzoato sódico, son capaces de controlar parcialmente las crisis cerebrales, pero no evitan la progresión retraso psicomotor severísimo, con muy mal pronóstico vital.
P28410:50 h
QUILOTORAX NEONATAL SEVERO: ¿OCTREOTIDA COMO PRIMERA ELECCION?
M. Esther Guerrero Vega, Simón Pedro Lubián López, Manuel Mendoza Jiménez, Isabel Benavente Fernández, Estefanía Romero Castillo, Juan Mena Romero, Enrique Robles Caballos, Manuel Matías Vega
Hospital Universitario Puerta del Mar, Cádiz.
Introducción: El quilotórax congénito es una de las causas más frecuentes de hidrotórax neonatal, descartadas las causas inmunes, no inmunes e infecciosas. Puede ser causa de distrés respiratorio importante en el neonato. Su incidencia es baja: 1 por cada 2.000 ingresos en UCIN. La tasa de mortalidad oscila entre el 20-60 % dependiendo de la edad gestacional, del diagnóstico precoz, de la rapidez de instauración del tratamiento y de la duración y severidad del quilotórax.
Caso clínico: Recién nacido con diagnóstico intraútero de hidrotórax. Gestación controlada que cursa con hipertensión arterial materna tratada con nifedipino. Parto pretérmino a las 35 semanas de gestación. Peso: 3.300 g. Apgar 3 y 5, al primer y al quinto minuto de vida. Precisa reanimación tipo IV.
Exploración: Mala vitalidad. Palidez cutáneo mucosa. Mala perfusión periférica. Hipoventilación marcada en hemitórax derecho con murmullo vesicular conservado en el izquierdo. Latido cardíaco desplazado a la izquierda. Taquicárdico. PAM 28. Exámenes complementarios al ingreso normales salvo radiografía de tórax con derrame masivo en hemitórax derecho e inicio de este en base izquierda.
Se procede a la realización de toracocentesis confirmándose el diagnóstico de sospecha al obtener un líquido de lechoso, amarillento con análisis citoquímico compatible con quilotórax. Precisa colocación de drenajes en ambos hemitórax obteniéndose un volumen de 700 ml diarios durante la primera semana.
Tratamiento: 1) Toracocentesis con colocación de drenaje pleural en ambos hemitórax. 2) Soporte inotrópico con dopamina, dobutamina y noradrenalina. 3) Perfusión intravenosa de octreótida (3 mg/kg/h). 4) Antibioterapia intravenosa con imipenem y vancomicina. 5) Transfusiones de plasma fresco congelado. 6) Nutrición parenteral inicial y posteriormente enteral con fórmula láctea rica en triglicéridos de cadena media. 7) Inmunoglobulinas.
Evolución: A pesar de la inestabilidad hemodinámica y respiratoria presentada en los primeros días de vida, evoluciona favorablemente con descenso progresivo del volumen de quilo evacuado. Presentó hiperglucemia y fiebre como efectos secundarios del octreótida. Se extuba a los 18 días de vida y a los 21 se retira el último drenaje pleural. Conclusiones: Comprobamos la eficacia del octreótida en el tratamiento del quilotórax congénito con escasos efectos secundarios.
P28510:55 h
ESTUDIO SOBRE LA EFICACIA DEL TRATAMIENTO DEL DUCTUS ARTERIOSO PERSISTENTE CON UN SEGUNDO CICLO DE INDOMETACINA
M. Isabel Hernández Bernal, Javier Castro, Javier Cruz Bértolo
Hospital Universitario 12 de Octubre, Madrid.
Antecedentes y objetivos: Existen pocos trabajos en la literatura que analicen la eficacia de la utilización de un segundo ciclo de indometacina para el tratamiento del DAP tras el fracaso del primero. El objetivo de este trabajo es determinar la eficacia de ese segundo ciclo y determinar los posibles factores asociados a su éxito.
Métodos: Se estudiaron los recién nacidos pretérmino (RNPT) nacidos en el Hospital 12 de Octubre durante 1995-2003 menores de 32 semanas de EG (n = 829). 54 fueron tratados con un 2.º ciclo corto de indometacina. Se recogieron datos de sus historias clínicas: período prenatal (corticoides prenatales, corioamnionitis, pre-eclampsia), del RNPT (sexo, EG, peso al nacer) días de vida de administración de cada ciclo, procesos intercurrentes (EMH, surfactante, sepsis, fototerapia) y complicaciones posteriores (DBP, NEC, ROP, secuelas neurológicas o muerte). La variable principal fue el éxito o fracaso del 2.º ciclo. Se realizó análisis estadístico mediante de test de chi cuadrado (variables categóricas) y test t-Student (v. continuas).
Resultados: De los 829 RNPT menores de 32 semanas de EG 267 presentaron DAP (32 %). 238 fueron tratados con un primer ciclo: 138 respondieron (58 %). 54 de los no respondedores recibieron un 2.º ciclo con una eficacia de 33,3 % (18/54). La eficacia fue mayor en los RNPT mayores de 27 semanas de EG (p = 0,026). Los RNPT con un peso mayor de 1.000 g presentaron una tendencia a una mayor respuesta (p = 0,14). Entre los factores intercurrentes estudiados observamos que la presencia de sepsis peritratamiento se asociaba significativamente a una mayor frecuencia de fracaso del tratamiento (p = 0,005). No se encontraron diferencias significativas en la presencia de secuelas posteriores en relación al fracaso o éxito del ciclo. Se analizaron las opciones terapéuticas tras el fracaso del 2.º ciclo: la opción quirúrgica (21/36) fue la más utilizada (sin complicaciones en ningún niño), el uso de un 3.º ciclo fracasó en los 9 niños en los que se indicó. La opción conservadora funcionó en 2 niños que no presentaban alteraciones hemodinámicas.
Conclusiones: El tratamiento de DAP con un 2.º ciclo de indometacina presenta más posibilidades de éxito en los RNPT mayores de 27 semanas, así como en los niños que no presenta sepsis peritratamiento. Consideramos la opción quirúrgica como la más adecuada tras el fracaso del 2.º ciclo.
P28611:00 h
PERFORACION ESOFAGICA IATROGÉNICA
Emma Albiñana Vallés, Leonor García Maset, Blanca Alfaro Ponce, Amalia Ortín Pujante, Carlos Paredes Cencillo
Hospital Clínico Universitario, Valencia.
Aunque tradicionalmente se han considerado como inocuas, existen ciertas técnicas que no lo son tanto.
Recién nacido pretérmino (29 semanas), bajo peso extremo (910 g), remitida a nuestro servicio a los 16 días de vida por hidroneumotórax y sospecha de perforación esofágica vs fístula traqueoesofágica con mediastinitis.
En el hospital de origen objetivan derrame pleural derecho de características compatibles con la alimentación enteral, de aparición coincidente con el inicio de la misma. En la radiografía de tórax practicada se visualiza sonda nasogástrica ubicada en seno costofrénico posterior derecho, que se retira previo al traslado. Se realiza tránsito esófago-gástrico en el que se aprecia trayecto fistuloso de 1 cm de longitud y 1 mm de sección, que sale de la parte posterior de la primera porción de esófago. Se inicia tratamiento antibiótico, ante la sospecha de mediastinitis, con vancomicina, cefotaxima y metronidazol durante 15 días. Se suspende alimentación enteral durante 4 días y posteriormente se reinicia mediante sonda orogástrica colocada bajo control ecográfico. La evolución clínica es favorable y el tránsito esófago-gástrico de control es normal.
Ante cualquier procedimiento hay que extremar los cuidados para la realización del mismo, más aún si se trata de recién nacidos pretérmino, pues algo que parece tan inocuo y sencillo como la introducción de una sonda orogástrica puede provocar patología de riesgo para el paciente.
P28711:05 h
MORBILIDAD EN RECIÉN NACIDOS DE PARTOS MULTIPLES
Izaskun Miner Kanflanka, Nere Arostegi Kareaga, Uxue Astigarraga Irureta, Patricia Esparza Paz, Juncal Echevarría Lecuona, Luis Paisán Grisolia
Hospital Donostia, San Sebastián (Guipúzcoa).
Objetivos: Evaluar las patologías asociadas a los recién nacidos de partos múltiples (PM) ingresados y su distribución según edad gestacional (EG) y peso neonatal.
Material y métodos: Estudio observacional, por revisión de informes de alta del año 2004. Se analizaron, según EG y peso, las patologías más frecuentes: dificultad respiratoria, enterocolitis necrosante (ECN), infecciones, alteraciones ecográficas cerebrales. Se estudió la estancia media y la asociación con las distintas patologías, mediante los estadísticos x2 y ANOVA. Se aceptó p < 0,05 como significativa.
Resultados: Ingresaron 108 PM, 49,1 % varones y 50,9 % mujeres. La distribución de estas patologías según EG (en semanas) y peso (en gramos) fue la siguiente:

La ecografía, realizada en 41/108 casos (38 %), fue patológica en 9/41 (22 %); 5/9 (56 %) en EG ≤ 30 semanas, 4/9 (44 %) en 31-34 semanas y 0 en ≥ 35 semanas (p < 0,037). La estancia media, de 23,6 días (d), se prolongó en peso ≤ 1.000 g (56,3 d), dificultad respiratoria (40,6 d), uso de ventilación mecánica (62,2 d), surfactante (63,6 d), infecciones (38,2 d) y ECN (72,8 d) (p < 0,00001).
Conclusiones: La morbilidad de partos múltiples está relacionada con el bajo peso y la menor edad gestacional. DR, ECN e infecciones fueron las patologías más frecuentes. La estancia media fue mayor en aquellos con bajo peso, dificultad respiratoria, necesidad de ventilación mecánica o surfactante, infecciones o ECN.
P28811:10 h
DÉFICIT DE ORNITIN TRANSCARBAMILASA. A PROPOSITO DE UN CASO
Julia Gisbert Mestre, Elena de Frutos Moneo, Agustín Molina Merino, Luis Fernández Martín-Bilbatua, Francisco Javier Estañ Capell, Carlos Paredes Cencillo
Hospital Clínico Universitario, Valencia.
Introducción: Los trastornos del ciclo de la urea son un grupo de 5 enfermedades. La sintomatología cuando debutan en el período neonatal es predominantemente neurológica por el acúmulo de amonio, el cual hay que depurar con rapidez por el daño irreversible que produce.
Caso clínico: Primera gestación de una madre sana de 34 años con un embarazo controlado, de curso normal. No antecedentes obstétricos de interés. Parto a término, vaginal, precisando ventilación con presión positiva, recuperándose rápidamente; se calcula un Test de Apgar de 6/10, quedando con su madre en la maternidad. El peso al nacimiento es de 3.980 g. Tras un período inicial libre de 48 h, presenta hipotonía y rechazo alimentario ingresando ante la sospecha de sepsis iniciándose tratamiento con ampicilina y amicacina. Los análisis realizados no sugieren infección y el hemocultivo, cultivo de LCR y urinocultivo fueron estériles. La ecografía transfontanelar fue normal. En el equilibrio ácido-base presenta leve alcalosis respiratoria. El deterioro neurológico progresa requiriendo ventilación mecánica, trasladándose a la Unidad de Cuidados Intensivos (UCIN) del hospital de referencia. A su llegada, con 72 h de vida, presenta amonio en sangre de 1.031 μmol/l por lo que se realiza exanguinotransfusión y posteriormente diálisis peritoneal, y tratamiento médico con benzoato, fenilbutirato y L-carnitina. En sangre y orina, se objetiva ausencia de citrulina en suero y excreción urinaria aumentada de ácido orótico, compatible con déficit de ornitín transcarbamilasa. A pesar de las medidas de depuración no se pudo conseguir reducir el nivel de amonio en suero con una cifra máxima de 2.600 μmol/l. El niño falleció a los 12 días de vida. Se realizó estudio genético en el niño y en la madre, detectándose la mutación 905 A < G en el exón 9 del gen OTC, que cambia el aminoácido histidina en posición 302 por arginina, siendo la madre portadora heterocigota. Esta mutación se había descrito anteriormente y se asocia a fenotipo neonatal grave.
Conclusiones: La hiperamoniemia debe tenerse en cuenta en cualquier recién nacido con sintomatología neurológica. El diagnóstico precoz es importante para instaurar un tratamiento debido al daño irreversible que puede producir. Si el tratamiento médico no es eficaz hay que adoptar medidas de depuración extrarrenal entre las cuales la más efectiva parece ser la hemodiafiltración veno-venosa.
P28911:15 h
MIOCARDIOPATIA DILATADA NEONATAL POR AMITRIPTILINA
Pilar A. Crespo Suárez, Montserrat López Franco, Nathalie Carreira Sande, Alejandro Pérez Muñuzuri, Sabela Martínez Soto
Hospital Clínico Universitario, Santiago de Compostela (A Coruña).
Presentamos el caso de un neonato de 16hr de vida ingresado por policitemia. Madre y padre ex-ADVP y con hepatitis C crónica. Segundo embarazo de madre de 27 años que recibió durante la gestación metadona (90 mg/día), diazepam y lorazepam. Fumadora de 5 cigarrillos/día. PEV a las 36 semanas. Peso RN: 2.520 g. Apgar 9/10. Se realizó exanguinotransfusión parcial sin incidencias. Al segundo día de vida inició clínica compatible con síndrome de abstinencia neonatal por lo que se pautó tratamiento con fenobarbital. Al cuarto día de vida presenta taquicardia, mala coloración, cutis marmorata, hepatomegalia de un través y tercer ruido cardíaco. Hemograma, bioquímica y LCR normales. Ante sospecha de sepsis por manipulación en la canalización umbilical se instauró tratamiento con teicoplanina. Se realizó radiografía de tórax evidenciándose cardiomegalia y prominencia hiliar. En ecocardiograma: dilatación de aurícula y ventrículo izquierdos, insuficiencia leve de todas las válvulas, fracción de acortamiento 11 % y fracción de eyección 26 %. ECG: taquicardia sinusal. Tóxicos en orina: presencia de amitriptilina, metadona, nicotina, cafeína y metamizol. La madre reconoció haber tomado amitriptilina durante la gestación aunque sólo hasta el quinto mes, si bien los hallazgos toxicológicos no eran concordantes. Se estableció el diagnóstico de miocardiopatía dilatada por amitriptilina, potenciada por metadona. Se inició tratamiento con dobutamina a 10 μg/kg/min con buena respuesta pudiéndose retirar a los 4 días. Ecocardiograma de control: FE 79 %, FA 44 %, sin datos de insuficiencia valvular.
La amitriptilina es un antidepresivo tricíclico, con semivida de 10-50 h. Entre sus efectos adversos (1-9 %) destacan somnolencia, hipotensión ortostática, taquicardia, depresión miocárdica y prolongación de PR y QRS. Con el uso de antidepresivos tricíclicos periparto se han descrito problemas cardíacos, irritabilidad, distrés respiratorio, espasmos musculares, crisis convulsivas y retención urinaria. La metadona produce un aumento de los niveles plasmáticos (70-170 %) de antidepresivos tricíclicos, con potenciación de su efecto y toxicidad, por inhibición del metabolismo hepático.
Conclusión: La importancia de indagar sobre los fármacos ingeridos durante el embarazo y conocer las interacciones entre los mismos, ya que estas pueden provocar potenciación de sus efectos aumentando la toxicidad sobre el neonato.
P29011:20 h
ESTUDIO DE LA INCIDENCIA Y EVOLUCION SEROLOGICA DE CASOS DE RIESGO DE SIFILIS CONGÉNITA EN LOS ULTIMOS 8 AÑOS
Jesús Lucas García, Ramón Aguilera Olmos, Ariadna Alberola Pérez, Bárbara Gomila Sard, Ferrán Illana i Carbonell, Flavia Pronzato Cuello
Hospital General, Castellón.
Antecedentes y objetivos: El objetivo del estudio es conocer la progresión de la incidencia anual y la evolución serológica de los casos de riesgo de transmisión vertical de sífilis congénita (SC), ya que en general los casos de SC se diagnostican con los datos obtenidos al nacimiento, sin tener en cuenta la evolución serológica.
Métodos: Estudio descriptivo retrospectivo. Ámbito: Hospital General Castellón. Período: enero 1997-diciembre 2004. Población: Casos de riesgo de transmisión vertical de SC basándose en criterios del Centers Disease Control.
Resultados: Durante este período han nacido 19.315 RN, identificándose 20 casos de riesgo (0,1 %), de los cuales 3 (15 %) se diagnosticaron de neurosífilis por VDRL reactivo en LCR. La incidencia de casos de riesgo ha aumentado de 0,39 en 1997 a 3,25 en 2004. Señalamos que el 80 % de las madres son extranjeras. En la serología de los casos de neurosífilis, al nacimiento, tanto los anticuerpos treponémicos (AcT) como los anticuerpos no treponémicos (AcNT) fueron positivos y sólo uno de ellos presentó títulos de AcNT 4 veces superiores que los maternos, el único que presentó clínica. En dos de tres, tanto AcT como los AcNT se negativizaron entre los 6 y 12 meses, mientras que en el que presentó clínica, no se pudo obtener la evolución serológica. En el resto de casos de riesgo, en 4 ocasiones fueron positivos ambos Acs (anticuerpos), mientras que en los otros sólo los AcT fueron positivos, excepto uno. Ambos Acs se negativizaron antes de los 6 meses.
Conclusión: En nuestro medio, la incidencia de RN con riesgo de transmisión vertical de SC ha aumentado, destacando un alto porcentaje de neurosífilis (15 %) en relación con otras series. Entre las causas subrayamos el incremento de la inmigración en nuestra área geográfica. Según la bibliografía, en la mayoría de los RN con SC, tratados o no, los AcT persisten más de 18 meses (incluso de por vida), aunque puede existir un 10 % de personas infectadas, tratadas precozmente, en las que se pueden negativizar. En nuestro estudio destacamos dos casos de neurosífilis, en los que los títulos se negativizaron antes del año. El diagnóstico, con los datos obtenidos al nacimiento, es un diagnóstico de presunción por lo que creemos que es necesario un seguimiento de la evolución serológica de los casos de riesgo que nos permita confirmar la SC a posteriori.
P29111:25 h
RABDOMIOMA CARDIACO Y ESCLEROSIS TUBEROSA. A PROPOSITO DE UN CASO
Pilar M.ª Caro Aguilera, Celia Gómez Robles, Marta García Ramírez, Raquel Gil Gómez, Vicente Parra Márquez, Antonio Jurado Ortiz
Hospital Materno-Infantil Carlos Haya, Málaga.
Introducción: Los tumores cardíacos son infrecuentes en la infancia, si bien el rabdomioma cardíaco es el más común. Hasta el 50 % pueden asociarse a Esclerosis tuberosa, permitiendo, en ocasiones, el diagnóstico precoz de esta enfermedad.
Caso clínico: RNAT (39 + 2 s). PRN 2.806 g con diagnóstico prenatal de tumoración cardíaca. Exploración física: soplo sistólico III/IV en borde esternal izquierdo, pulsos presentes y simétricos, presión arterial normal. Analítica: normal. Ecocardiograma: pequeña tumoración en ápex de ventrículo izquierdo, otra mayor en valva posterior mitral que se extiende por el aparato subvalvular y músculo papilar. Una pequeña en valva anterior mitral y a nivel subaórtico sin obstrucción del flujo. La función valvular es normal. En el ventrículo derecho existen varias tumoraciones, dos de ellas en el tracto de salida sin comprometer el flujo a dicho nivel. Ecografía de cráneo: imágenes hiperecogénicas en zona subcortical profunda a nivel frontoparietal y otra de 17 mm a nivel temporal derecho de similares características. RM: nódulos pequeños subependimarios, con señal brillante en T1, (hamartomas) en paredes laterales de ambos ventrículos. Lesión hiperintensa en T1 en lóbulo frontal derecho que corresponde a un tuber. En ambos hemisferios se ven múltiples imágenes más pequeñas de características similares. Ecografía abdominal: normal. EEG: normal.
Conclusiones: La mayoría de los rabdomiomas se resuelven espontáneamente y no tienen repercusión hemodinámica, si bien orientan al diagnóstico de Esclerosis tuberosa. Los rabdomiomas cardíacos diagnosticados prenatalmente están relacionados con el posterior desarrollo de Esclerosis tuberosa. Ante la presencia de estas tumoraciones debería realizarse un estudio para descartar dicha enfermedad.
CARDIOLOGIA
Zona póster II (planta segunda del Auditorio)
P29210:15 h
SINDROME DE WPW EN LA INFANCIA: FORMAS DE PRESENTACION Y SEGUIMIENTO DE 25 CASOS
David Bartoli, Rosa M. Perich Durán, Silvia Teodoro Marín, Vitoria Aldecoa Bilbao, Gisela Viaplana Bartolí,
Corporació Sanitaria Parc Taulí, Sabadell (Barcelona).
Objetivo: Determinar la forma de presentación, diagnóstico, tratamiento y evolución de los casos de WPW diagnosticados en una Unidad de Cardiología Pediátrica de un hospital comarcal.
Material y métodos: Estudio retrospectivo y recogida de datos de un total de 25 pacientes menores de 15 años diagnosticados de WPW en los últimos 20 años en nuestro hospital.
Resultados: 25 pacientes: 12 niños y 13 niñas. La edad media al diagnóstico fue de 6,8 años. 2 pacientes eran hermanos. En 4 pacientes se halló cardiopatía estructural: 3 CIV y 1 Ebstein. 1 caso presentaba malformaciones extracardíacas asociadas. El diagnóstico se realizó mediante ECG en el 88 % de los pacientes y en el resto por hallazgo de vía anómala oculta en el estudio electrofisiológico. El 40 % de los casos se diagnosticó de manera casual por estudio electrocardiográfico de rutina y el 60 % debutó clínicamente con episodios de TPSV. La TPSV se autolimitó en el 53 %, precisó maniobras vagales en el 26 % y tratamiento farmacológico en el 20 % (1 caso con shock cardiogénico). Se practicó ablación de la vía anómala en el 80 % de los casos sintomáticos, siendo incompleta en el 25 %. 2 pacientes precisaron una segunda ablación y en 1 caso se hallaron vías anómalas múltiples. Tras la ablación, el 84 % se mantuvo asintomático y sin tratamiento.
Comentarios: 1) No hubo diferencias entre sexos. 2) La cardiopatía más frecuente asociada fue la CIV. 3) El 40 % de los casos se diagnosticó de forma casual manteniéndose asintomáticos durante todo su seguimiento. 4) El 12 % presentaba vías anómalas ocultas. 5) Las crisis de TPSV fueron autolimitadas o controladas con maniobras vagales en el 80 %. 6) El tratamiento con ablación fracasó en el 25 % de los casos. 7) El 84 % de los pacientes se mantuvo asintomático y sin tratamiento médico.
P29310:20 h
TETRALOGIA DE FALLOT CON COMUNICACION INTERVENTRICULAR RESTRICTIVA
Juana M. Espín López, Francisco J Castro García, Fuensanta Escudero Carceles, Miguel Navalón Pérez, José Manuel Guía Torrent
Hospital Universitario Virgen de la Arrixaca, Murcia.
Introducción: La asociación de tetralogía de Fallot (TF) y comunicación interventricular (CIV) restrictiva es rara, pudiendo ocasionar problemas en el diagnóstico y en el manejo clínico y empeorar el pronóstico de la cardiopatía. El objetivo de esta comunicación es describir los hallazgos clínicos y ecocardiográficos en una paciente con TF y CIV restrictiva.
Caso clínico: Recién nacida de 48 h de vida con cianosis apreciada desde el nacimiento, con empeoramiento progresivo. En la exploración física destacaba cianosis moderada, pulsos normales, discreta taquipnea, hepatomegalia y soplo pansistólico III/VI con epicentro en mesocardio. La radiografía de tórax reveló moderada cardiomegalia con flujo pulmonar disminuido. El electrocardiograma mostró ritmo sinusal a 128 latidos/min, eje de QRS a + 180° e hipertrofia ventricular derecha. En el ecocardiograma destacaba fosa oval permeable con flujo bidireccional, CIV subaórtica amplia con un mamelón de tejido de la valva septal tricuspídea protruyendo hacia la misma, ocluyéndola casi totalmente en sístole y ocasionando presiones suprasistémicas en ventrículo derecho y flujo turbulento con gradiente pico derecha-izquierda de 60 mmHg; discreto cabalgamiento aórtico, infundíbulo muy estrecho con mínimo paso, ductus permeable con flujo continuo aorta-pulmonar y ramas pulmonares de 4 mm de diámetro. La paciente precisó la administración de Prostaglandina E1 para mantener la permeabilidad ductal previamente a la corrección quirúrgica. Fue intervenida a los 12 días de vida mediante ampliación del tracto de salida de ventrículo derecho y posterior cierre de la CIV, falleciendo en el postoperatorio por disfunción ventricular derecha grave.
Discusión: La asociación de TF o atresia pulmonar y CIV restrictiva aumenta la mortalidad quirúrgica de la cardiopatía. Habitualmente se produce por tejido accesorio de la válvula tricúspide que ocluye parcialmente la CIV de forma similar al mecanismo de cierre espontáneo de los defectos septales ventriculares aislados. Este tejido puede ser identificado adecuadamente mediante ecocardiografía.
P29410:25 h
CORAZON DERECHO HIPOPLASICO: DIAGNOSTICO TRAS 11 AÑOS DE EVOLUCION
Rosa Garrido Uriarte, Carolina López Martínez, Sara Rupérez Peña, Patricia Company Maciá, Ana Peña Busto, Inés Esteban Díez, José M. Bazán Ocón, Jesús Blázquez Regidor
Complejo Hospitalario San Millán-San Pedro, Logroño (La Rioja).
Introducción: Un 8 a 10 por 1.000 de los recién nacidos vivos tienen una cardiopatía congénita (CC). La mitad aproximadamente, presentarán síntomas en el período neonatal siendo esta la causa de alrededor del 10 % de las muertes durante este período. Se estima que el 85 % de los niños nacidos con CC sobrevivirá hasta la vida adulta, gracias al avance de los procedimientos terapéuticos realizados en la infancia.
Caso clínico: Niña de 11 años remitida desde un Centro de Protección de Menores, para estudio de disnea de moderados esfuerzos y cianosis cutáneo-mucosa. Importante distocia social. No constan revisiones médicas ni vacunación. Exploración física: Buen estado general. Talla: P97. Peso: P50. Tensión arterial: 111/78 Frecuencia cardíaca: 70 lpm. Saturación de oxígeno: 80-84 %. Eupneica en reposo Cianosis marcada labial y de extremidades. Buena perfusión periférica con pulsos periféricos palpables y simétricos. Acropaquias en pies y manos. ACP: normal. Resto sin hallazgos. Pruebas complementarias: Hemograma: 7'23 mill/ml Hematíes, 59'5 Htco. Bioquímica: Glucosa, Urea, Creatinina, iones, perfil hepático, Alfa1 antitripsina, Mantoux, Test del sudor, Ceruloplasmina, Proteinograma, Hormonas tiroideas, Cortisol, Serología de hepatitis, Electroforesis de hemoglobina, Metahemoglobina en sangre: Sin alteraciones. Eritropoyetina: 48,9 mU/ml, Fondo de ojo: Normal. ECG: Ritmo sinusal; Eje 70°. Radiografía. de tórax: normal. TC cerebral: Normal. Ecocardiografía: Se observa ventrículo derecho hipoplásico y comunicación interauricular moderada. Cateterismo cardíaco: Ventrículo derecho hipoplásico con ausencia de la porción trabeculada, buena cámara de entrada, tricúspide normal e infundíbulo de buen calibre. Tronco y ramas pulmonares reducidas debido a que el flujo está también reducido. Cortocircuito derecha-izquierda importante y puro a través de la comunicación interauricular. Importante desaturación sistémica: VP: 100 %; AI: 90 %; AO: 92 % y repercusión manométrica: AP: 26/17/20; PCP: 17; VP: 19; VI: 96/13-17.
Conclusión: En el caso clínico se aporta una malformación no descrita en la literatura. La intervención propuesta es la técnica de Glenn bidireccional. La supervivencia al año de intervención es superior al 90 %, siendo la presión media de la arteria pulmonar (≥ 17 mm Hg) la variable más importante relacionada con la evolución.
P29510:30 h
HIPERTENSION PULMONAR PRIMARIA EN UN NIÑO DE TRES AÑOS
Javier Calzada Barrena, Aitor Ruano López, Montse Vázquez Ingelmo, Leire García Sarriugarte, Raquel Fernández Martínez, Anartz Fernández Prieto, Javier Ayala Curiel
Hospital de Basurto, Bilbao (Vizcaya).
Introducción: La hipertensión pulmonar primaria es una enfermedad de patogenia desconocida, con una incidencia de 1-2 casos por millón de habitantes y año. Se define por una presión media de la arteria pulmonar mayor de 25 mmHg en reposo o 30 mmHg con el ejercicio.
Caso clínico: Niño de 3 años que presenta episodios de dolor torácico, palidez y taquicardia, en relación con el ejercicio, desde hace 5 meses. Los episodios ceden espontáneamente en 4-5 min. No refiere otra sintomatología. Antecedentes: Embarazo gemelar, segundo gemelo. Ingreso en UCIN por prematuridad. Exploración: FC:140x'. FR:35x'. PA: 98/49. SpO2: 94 %. BEG. Buen color. ACP: Ritmo de galope. Latido hiperdinámico. Soplo sistólico I/VI, eyectivo, en punta. 2R fuerte. Pulsos palpables. Resto de la exploración negativa.
Pruebas complementarias: ECG: Signos de hipertrofia de cavidades derechas, con desviación del eje hacia la derecha. Radiografía de tórax: Cardiomegalia leve a expensas de cavidades derechas. Botón pulmonar prominente. Ecocardiografía: AD y VD muy dilatados. Tricúspide dilatada con IT severa de Vmáx 485-490 cm/s (gradiente VD-AD 95 mmHg). IP de Vmáx 300 cm/s (PDAP 35 mmHg). Cateterismo cardíaco: Hipertensión arterial pulmonar primaria. Test vasodilatador negativo. Evolución y tratamiento: Una vez establecido el diagnóstico y ante la no respuesta al test vasodilatador, se instaura tratamiento con prostaciclina inhalada. A pesar de ello, el paciente presenta crisis hipóxicas, falleciendo al mes del diagnóstico.
Discusión: Esta rara patología parece estar producida por un predominio de sustancias vasoconstrictoras en las arterias pulmonares, con lesión vascular, produciendo síntomas inespecíficos como disnea, dolor torácico y síncopes de esfuerzo. Se deben descartar otras causas de hipertensión pulmonar. La elección del tratamiento dependerá del grado de afectación y de la respuesta o no al test vasodilatador.
Conclusión:1) La hipertensión pulmonar primaria es una rara causa de dolor torácico en la infancia. 2) No existen tratamientos farmacológicos curativos en la actualidad, aunque la combinación de nuevos fármacos parece mejorar la supervivencia. 3) La principal causa de fallecimiento es el fallo cardíaco derecho. 4) La supervivencia actual es del 90 % a los 4 años con Epoprostenol.
P29610:35 h
DETECCION DE CARDIOPATIAS CONGÉNITAS EN POBLACION INMIGRANTE EN ATENCION PRIMARIA; UN PROBLEMA EN AUMENTO
Daniel Gómez Sánchez, Olga Peñalver Giner, Julia Gisbert Mestre, Ignacio Izquierdo Fos
Hospital de Gandía y Centro de Especialidades Francesc de Borja, Valencia.
Antecedentes y objetivos: Debido a la llegada de una población inmigrante infantil de edades muy variadas y con escasos controles médicos se están detectando cardiopatías congénitas hemodinámicamente significativas en estados evolucionados. Se muestran los casos detectados en un Hospital terciario con una población inmigrante pujante.
Métodos y resultados: Se recogen los pacientes remitidos desde los centros correspondientes a 2 áreas y que acudieron al los respectivos Centros de Salud a realizar un primer control, a regularizar su calendario vacunal o a solicitar un certificado de salud escolar.
Paciente 1: 9 años, Bulgaria. Talasemia major. Miocardiopatía dilatada. Insuficiencia cardíaca.
Paciente 2: 12 años, Bolivia. Comunicación interauricular o.s. de 25 mm con dilatación de cavidades derechas y presión elevada en ventrículo derecho. Disnea a moderados esfuerzos.
Paciente 3: 9 años, Ecuador. Comunicación interauricular o.s. de 19 mm con dilatación de cavidades derechas y presión elevada en ventrículo derecho.
Paciente 4: 11 años, procedente de Bulgaria. Ductus arterioso permeable de 6 mm Ao-pu con dilatación e hipertrofia de cavidades izquierdas. Asintomático.
Paciente 5: 11 años. Problema social. Diagnóstico: Canal auriculoventricular transicional con ostium primum amplio y dilatación de cavidades en dextrocardia. Asintomático.
Paciente 6: 9 años. Senegal. Comunicación interventricular infundibular. Insuficiencia aórtica moderada por prolapso valva septal. Dilatación cavidades izquierdas. Disnea a esfuerzos moderados.
Paciente 7: 4 años. Inglaterra. Ductus arterioso permeable de 5mm Ao-Pu. Dilatación cavidades izquierdas. Asintomática.
Conclusiones: La mayoría de las cardiopatías congénitas hemodinámicamente significativas se diagnostican y/o tratan en los primeros meses de vida. Con el aumento de la población inmigrante infantil ha aumentado el número de cardiopatías en estados evolucionados que requieren en ocasiones tratamientos quirúrgicos inmediatos, recordándonos la importancia de los controles en los CAP.
P29710:40 h
RELACION DEL BNP CON EL GRADO LA INSUFICIENCIA CARDIACA
M.ª del Mar Santos Sebastián, Andrés Alcaraz Romero, Jesús Cecilio López-Menchero Oliva, Carlos Romero Román, Patricia Aparicio García, Gema Arriola Pereda, Raúl Roberto Borrego Domínguez, Teresa Álvarez Martín
Hospital General Universitario Gregorio Marañón, Madrid.
Objetivo: Describir los valores de BNP en niños sin problemas cardiológicos y en niños con diferentes cardiopatías, y estudiar las posibles diferencias entre aquellos con y sin clínica de IC.
Métodos: Estudio prospectivo, descriptivo y observacional. Los valores plasmáticos de BNP se determinaron en 122 niños de diferentes edades sin cardiopatía (1 mes a 18 años), y en 61 niños con diferentes cardiopatías atendidos en consulta, planta de cardiología o UCI entre abril y noviembre de 2004. Los niños con cardiopatías se clasificaron en: cardiopatía congénita (cc) sin IC, cc con IC (cortocircuito izquierda-derecha y/o obstrucción izquierda) y miocardiopatías (mc). El grado de IC se valoró con sistemas de puntuación clínica. Las concentraciones de BNP se han determinado mediante electroinmunoquimioluminiscencia (Advia Centaur, Bayer®). Los datos se presentan como mediana (P25-P75), y se utiliza el test de Mann-Whitney para las comparaciones intergrupo y correlación de Spearman.
Resultados: Los valores de BNP en los controles fueron 13,9 ± 10,0 pg/ml, y no hubo diferencias en relación con la edad o sexo. Se incluyeron 61 niños de edades comprendidas entre los 2 meses y los 17 años (media de 51,7 meses): 28 con cc sin IC, 18 con cc e IC y 15 con mc. Los valores de BNP obtenidos en los diferentes grupos se presentan en la siguiente tabla (pg/ml), como mediana (P25-P75):

Conclusiones: Los valores de BNP están elevados en los niños con IC, en mayor grado en los niños con mc. Los niños con cc sin IC muestran valores de BNP normales. El BNP es un marcador de IC en niños.
P29810:45 h
NIVELES DE HOMOCISTEINA EN NIÑOS DE 11-12 AÑOS (ESTUDIO RIVAS-VACIAMADRID)
Enrique Villalobos Pinto, M. Teresa Morales San José, M.ª José Peláez García de Salazar, Marciano Sánchez Bayle Hospital del Niño Jesús, Madrid y Centro de Salud La Paz, Rivas-Vaciamadrid (Madrid).
Introducción: La homocisteína es un factor de riesgo cardiovascular identificado recientemente. Existen pocos estudios sobre homocisteína en niños españoles.
Objetivos: Determinar niveles de homocisteína en población de 11-12 años de Rivas-Vaciamadrid.
Sujetos y métodos: Muestra de 195 niños (104 niños y 65 niñas) de la edad reseñada. Ajuste a una distribución normal.
Resultados: La media de los niveles de homocisteína fue de 6, 44 (DE = 2, 10). No hubo diferencias significativas según el género o antecedentes familiares de factores de riesgo cardiovascular. Sí con la apoproteína A (r = 0, 188, p < 0, 01).


Conclusión: Los niños estudiados presentan valores adecuados de homocisteína. El porcentaje de niños con valores considerados de riesgo fue bajo.
P29910:50 h
CARDIOPATIAS CONGÉNITAS EN LA PROVINCIA DE BADAJOZ Y SUS AREAS DE SALUD: INCIDENCIA Y ANALISIS CLINICO
Isabel Arias López, Emilia M. Martínez Tallo, Francisco Campo Sampedro, Juan José Cardesa García
Hospital Universitario Infanta Cristina, Badajoz, Hospital Materno-Infantil, Badajoz y Universidad de Extremadura, Badajoz.
Antecedentes y objetivos: Cada vez es mayor la importancia clínica y social de las cardiopatías congénitas (CC), por la mayor supervivencia y los esfuerzos para mejorar su calidad de vida, lo que exige nuevos enfoques asistenciales. Objetivos: conocer la incidencia de las CC en la provincia de Badajoz y en sus distintas Áreas de Salud (AdS) y analizar sus características clínicas fundamentales.
Métodos: Estudio retrospectivo sobre las historias clínicas de todos los pacientes nuevos en la Unidad de Cardiología Pediátrica (Centro de Referencia Provincial) en el año 1997. Se ha valorado el debut clínico, edad, tiempo de consulta, relación o asociación con otras patologías malformativas y genéticas. Se revisa el seguimiento a lo largo de los cinco años siguientes para identificar tendencias evolutivas.
Resultados: De los 742 pacientes que acuden por primera vez sólo 116, el 15,6 %, tenían una patología cardíaca. Las tasas de CC/1.000 RN vivos oscilan entre 24 en el AdS de Badajoz y 12 en el AdS de Don Benito. Las diferencias se deben a la derivación directa a Centros de Referencia Nacional, y a que en el AdS de Badajoz se contabilizaron todas las CC, incluidas CIV muscular pequeña, CIA tipo OS pequeñas, válvula aórtica bicúspide y prolapso de válvula mitral leve y arritmias congénitas. Excluidas estas, la tasa se sitúa en 16/1.000 RN vivos, similar a las de las AdS de Mérida y Llerena-Zafra, y las de otros estudios. La patología más frecuente fue la CIV (5/1.000 RN) y la CIA tipo ostium secundum. No encontramos diferencias significativas en los tipos de CC que se dan en las cuatro AdS. No existen diferencias de sexo. La distribución del debut clínico es homogénea a lo largo de los trimestres y meses de 1997. El 70 % debutan en el primer año de vida; y el 50 % en el primer mes. Al nacimiento, el 40 % estaban asintomáticos, se apreció soplo en un 26 %, y un 7 % manifestaban cianosis. El principal motivo de consulta es el soplo cardíaco: 64 %. El 91 % de los casos se diagnosticaron en la primera visita. Existía algún factor de riesgo para CC en el 18 %. Un 14 % tenían alguna enfermedad genética tipada. En un 25 % se asocian otras malformaciones. Un 67 % tenían una sola CC, 12 % tenían dos y 11 % tres o más. Se practicó ecocardiografía al 98 %. Requirieron alguna prueba extraordinaria el 25 % y se realizó cirugía en el 25 %. El 80 % de los CC no se dan de alta durante los 5 años de seguimiento.
P30010:55 h
ESCLEROSIS TUBEROSA DE DIAGNOSTICO PRENATAL
Emilia Urrutia Maldonado, M. del Mar Rodríguez Vázquez del Rey, Susana Roldán Aparicio, M.ª Matilde Vidal Alarcón, Sebastián Manzanares, Mercedes Casado Rodríguez, Encarnación Montes Pecete, José Miguel Pérez de la Cruz
Hospital Universitario Virgen de las Nieves, Granada.
La esclerosis tuberosa es un síndrome neurocutáneo caracterizado por el crecimiento de hamartomas en distintos órganos. La herencia es autosómica dominante, aunque muchos de los casos son esporádicos. Los rabdomiomas cardíacos son lesiones frecuentemente asociadas con la esclerosis tuberosa, y desempeñan un papel importante en el diagnóstico de esta enfermedad. Sin embargo, la identificación prenatal de los tumores intraútero es poco común. Presentamos el caso de un recién nacido con diagnóstico prenatal de probable esclerosis tuberosa.
Caso clínico: Primigesta sin antecedentes, remitida para estudio al detectarse en la ecografía realizada a las 36 semanas de gestación varias formaciones tumorales intracardicas. Se realiza eco-Doppler fetal, observándose dos grandes tumores en ventrículo izquierdo, uno de los cuales está situado en el tracto de salida del ventrículo izquierdo dando lugar a estenosis subaórtica moderada. Ante la sospecha de esclerosis tuberosa se realiza RM que detecta malformación frontoparietal izquierda. El niño nace bien a las 41 semanas mediante cesárea y a la exploración presenta un soplo sistólico eyectivo II/VI en foco aórtico. El eco-Doppler a las 24 h de vida detecta la presencia de ocho tumores intracardíacos, dos de ellos de gran tamaño, uno de los cuales determina una estenosis aórtica moderada. La RM cerebral posnatal muestra hallazgos compatibles con síndrome neurocutáneo tipo esclerosis tuberosa. A los 14 días de vida el niño se encuentra asintomático.
Comentarios: 1) La presencia de arritmias fetales y/o detección de tumores intracardíacos mediante ultrasonografía prenatal, debe hacer sospechar el diagnóstico de esclerosis tuberosa antes del nacimiento. 2) Aunque los rabdomiomas son histológicamente benignos y se describe su tendencia a la regresión espontánea, su tamaño y localización pueden condicionar un comportamiento clínico desfavorables. Su diagnóstico prenatal permite una primera aproximación al pronóstico y a la actitud terapéutica.
P30111:00 h
CASO LETAL DE ENFERMEDAD DE KAWASAKI CON ANEURISMA CORONARIO GIGANTE EN NIÑO DE 2 AÑOS
Raquel Carceller Beltrán, Rebeca Sarrat Torres, Elena Javierre Miranda, Gemma Terrer Manrique, Cristina Fernández Espuelas, M. Dolores García de la Calzada, Joaquín de Felipe Villaverde, José Salazar Mena
Hospital Materno-Infantil Miguel Servet, Zaragoza.
Introducción: La Enfermedad de Kawasaki es una vasculitis aguda, en ocasiones difícil de diagnosticar por no cumplir siempre los criterios clínicos que la definen (casos atípicos). Si no se le aplica tratamiento de forma precoz, un 25 % de los casos presentan como complicación aneurismas coronarios, que pueden llegar a comprometer la vida del niño, sobre todo si superan los 8 mm (aneurismas gigantes).
Caso clínico: Varón de 2 años con cuadro de fiebre elevada de 14 días sin foco, con molestias articulares ocasionales y discreto eritema faríngeo y conjuntival bilateral. No adenopatías, edemas ni rash cutáneo. No mejoría de la fiebre a pesar de tiempo transcurrido y tratamiento con Amoxicilina-Clavulánico. Analítica: VSG 120, PCR 173 mg/l y trombocitosis de hasta 640.000/μl. Cultivos, serologías, Mantoux y estudios de inmunidad normales. Se sospecha Enfermedad de Kawasaki atípica y se pauta tratamiento con inmunoglobulina (Ig) i.v. y AAS a 100 mg/k/d, mejorando los síntomas en 2 días tras su inicio. En ECO-Cardiografía de control a las 3 semanas se aprecia una dilatación aneurismática en la bifurcación de la coronaria izquierda, con aumento de tamaño hasta los 10 mm. Cinco meses después el niño falleció por un infarto de miocardio como complicación del aneurisma coronario gigante.
Comentario: Destacamos la gran importancia de sospechar una Enfermedad de Kawasaki (más complicado en los casos atípicos) para poder así establecer un tratamiento precoz (antes del 10.º día de fiebre) con Ig i.v. (a 2 g/k en dosis única según últimas revisiones). Esta medida es nuestra única arma para prevenir la formación de aneurismas coronarios, reduciéndose el riesgo de un 25 % a un 5 %.
P30211:05 h
ABORDAJE COMBINADO EN LA EXTRACCION DE CATÉTERES INTRAVASCULARES RETENIDOS
Alberto Parente Hernández, Óscar Sánchez París, Agustín Cañizo López, Ana Laín Fernández, Julio Cerdá Berrocal, Luis Zunzunegui Martínez, Juan José Vázquez Estévez
Hospital Materno-Infantil Gregorio Marañón, Madrid.
Objetivo: Los cuerpos extraños intravasculares secundarios a la extracción incompleta de los catéteres intravasculares de larga duración es una complicación poco frecuente y de difícil solución. El uso exclusivo de un abordaje quirúrgico abierto no es siempre efectivo, además de ser laborioso y tener una alta morbilidad asociada.
Materiales y métodos: Presentamos 2 pacientes con restos de catéter tipo Port-A-Cath en el territorio de la vena cava superior, seccionados durante su extracción inicial.
Resultados: Se realizó en ambos el uso combinado de la cirugía abierta en el punto de entrada del catéter con un abordaje hemodinámico intravascular. Esta asociación nos permitió la localización y visualización del resto de catéter, además de impedir su migración durante la manipulación mediante la fijación hemodinámica intravascular. El abordaje quirúrgico de la puerta de entrada del catéter se realizó para liberar las adherencias con los tejidos que impedían su extracción. Con técnica de cateterismo y el uso de un lazo-guía fijamos la zona proximal del catéter para extraerlo por vía femoral (caso 1) o bien para su localización y extracción por el abordaje abierto cervical (caso 2).
Conclusión: Este abordaje combinado nos permite la localización y el control visual directo durante las maniobras de extracción, permitiendo la resolución del problema por ambas técnicas (cirugía y hemodinámica), además de reducir la morbilidad del proceso.
P30311:10 h
MIOCARDITIS FULMINANTE PORMYCOPLASMA PNEUMONIAE:A PROPOSITO DE UN CASO
Marta Vera Estrada, Jéssica Ortiz, Geòrgia Sarquella Brugada, Yolanda Fernández, Iolanda Jordán García, Fredy Prada Martínez, Jordi Pou Fernández
Hospital Sant Joan de Deu, Barcelona.
Introducción y objetivo: La miocarditis es una entidad poco frecuente en pediatría y su etiología es desconocida en la mayoría de los casos. La presentación clínica es variable y presenta un amplio espectro de gravedad. Se presenta el caso de una paciente con miocarditis fulminante que requirió trasplante cardíaco.
Caso clínico: Chica de 15 años, sin antecedentes de interés, que acudió a urgencias por vómitos y dolor en hipocondrio derecho de 9 días de evolución. No otra sintomatología. No consumo de fármacos o tóxicos. Exploración física: aceptable estado general con palidez cutánea. Auscultación cardiorrespiratoria normal. Dolor a la palpación a nivel de epigastrio e hipocondrio derecho sin signos de irritación peritoneal, hepatomegalia de 5 cm. Analítica sanguínea: AST 320 U/l, ALT 260 U/l, elevación discreta de bilirrubina total y conjugada y tiempo de protrombina 60 %, resto de bioquímica normal. Ingresó para estudio con sospecha de hepatopatía aguda. A las pocas horas de ingreso inició disnea con taquipnea, crepitantes pulmonares bilaterales y taquicardia con soplo sistólico. Radiografía de tórax: cardiomegalia con infiltrado intersticial bilateral. Se trasladó a UCIP donde se inició tratamiento farmacológico de soporte. Se realizó ecocardiografía que mostró grave dilatación del ventrículo izquierdo con contractilidad ventricular muy deprimida compatible con miocardiopatía dilatada. Ante la sospecha de miocarditis se inició tratamiento con gammaglobulina intravenosa y se solicitaron serologías según protocolo. La paciente presentó un rápido empeoramiento clínico con inestabilidad hemodinámica a pesar del tratamiento por lo que se trasladó a centro de referencia para tratamiento de rescate. Se colocó balón de contrapulsación aórtico sin mejoría, requiriendo asistencia biventricular en espera de trasplante cardíaco. La biopsia endocárdica confirmó el diagnóstico de miocarditis aguda. Las serologías realizadas confirmaron la infección aguda por Mycoplasma pneumoniae. A los 7 días del traslado se realizó trasplante cardíaco con evolución favorable.
Comentarios: La asociación de Mycoplasma pneumoniae con miocarditis y pericarditis es extremadamente rara. La presentación clínica más común es la insuficiencia cardíaca aguda. El principal tratamiento es de soporte, presentando un tercio de los casos curación total, otro tercio curación con algún grado de disfunción miocárdica y el resto evolucionan a muerte o trasplante cardíaco.
P30411:15 h
PERICARDITIS PURULENTA: ¿CUAL ES LA MEJOR ESTRATEGIA TERAPÉUTICA?
Carme Farrán Balcells, Sonia Cañadas Plazón, Pedro Domínguez Sampedro, M.ª Queralt Ferrer Menduiña, Dimpna Albert Brotóns, Luis Miró, Jordi Roqueta Mas, Concepción Figueras Nadal, Fernando Paredes Carmona, Manuel Quintana
Hospital Materno-Infantil Vall D'Hebron, Barcelona.
Introducción: La pericarditis purulenta (PC) es una enfermedad grave para la que no existe una única estrategia terapéutica universalmente aceptada.
Casos clínicos.Caso 1 (1998). Niño de 20 meses. Fiebre de 4 días, en tratamiento con eritromicina, y signos de fallo cardíaco. Presenta leucocitosis y PCR de 25 mg/dl. Ecocardiografía: derrame pericárdico. Pericardiocentesis: 125 mL de exudado purulento; Gram sin gérmenes; cultivo negativo. Se interviene: drenaje, apertura de ventana pericárdica y colocación de catéteres pleuropericárdicos para irrigación con povidona yodada 0,5 % (3 días), y 4 dosis de urocinasa (1.ª 10.000 U, 2.ª 15.000 U, 3.ª y 4.ª 20.000 U; cada 6 h). Tratamiento E.V. con cefotaxima (se substituye por meropenem por infección nosocomial por Pseudomonas aeruginosa) y vancomicina (3 y 1 semana, respectivamente). Cultivos negativos. Estudio inmunológico y función tiroidea normales. Alta a las 3 semanas. A los 5 años, la exploración y la ecocardiografía son normales.
Caso 2 (2004). Niño de 2 años, no inmunizado frente a neumococo, en el que se detecta roce pericárdico. Fiebre de 5 días, tratado con amoxicilina-clavulánico. Presenta leucocitosis y PCR de 32,4 mg/dl. Ecocardiografía: derrame pericárdico. Pericardiocentesis: líquido purulento; Gram: cocos grampositivos (¿neumococo?); cultivo negativo. Se interviene: limpieza pericárdica y colocación de catéteres para irrigación con povidona yodada 0,5 % (4 días), y 9 dosis de urocinasa (10.000 U; cada 8 h). Ante evolución tórpida (progresión a constricción) se practica pericardiectomía subtotal. Tratamiento E.V. con cefotaxima y cloxacilina (1 semana), y después, por mala evolución, con piperacilina-tazobactam y vancomicina (10 días), seguidas de amoxicilina-clavulánico (1 semana). Cultivos negativos. Estudio inmunológico normal. Se detecta hipotiroidismo subclínico transitorio. Alta a las 3 semanas. A los 2 meses la exploración es normal mostrando la ecocardiografía un mínimo patrón restrictivo.
Conclusiones: Parece indiscutible que la PC debe tratarse precozmente con antibioterapia E.V y drenaje pericárdico. Queda por demostrar la efectividad de otras opciones terapéuticas y su adecuada secuencia de aplicación. A falta de estudios, un primer paso debe ser el compromiso individual por seguir estrategias terapéuticas homogéneas.
P30511:20 h
RETRASO EN EL DIAGNOSTICO DE CARDIOPATIA CONGÉNITA ENTRE LA POBLACION INMIGRANTE
Juana M. Espín López, Fuensanta Escudero Carceles, Miguel Navalón Pérez, Francisca Teruel Carrillo, Francisco J Castro García, José Manuel Guía Torrent
Hospital Universitario Virgen de la Arrixaca, Murcia.
Objetivos: Valorar la influencia de los inmigrantes en la natalidad y comparar la edad de diagnóstico en las distintas cardiopatías, entre los nacidos en España y los nacidos en otros países.
Métodos: Recogemos todos los nacidos vivos en nuestro hospital y el porcentaje de hijos de inmigrantes, entre enero de 2000 y diciembre de 2004. También revisamos todas las cardiopatías congénitas en niños de hasta 11 años de edad, estudiadas en nuestra sección en ese mismo período, agrupando las distintas cardiopatías según criterios clínico-hemodinámicos, en: grupo I, cortocircuitos izquierda derecha, grupo II, obstrucciones al tracto de salida derecho e izquierdo sin cianosis y grupo III, cardiopatías cianógenas. Posteriormente, comparamos la edad de diagnóstico de cada una de ellas entre los nativos e inmigrantes nacidos aquí y los inmigrantes nacidos en sus países.
Resultados: El número de nacidos vivos ha aumentado, en esos 5 años de 6.780 a 7.395 (8,3 %) y en ese aumento ha influido el número de inmigrantes que ha pasado de 271 a 1.366 (del 4 % al 18,5 %). El total de cardiopatías estudiadas en ese tiempo ha sido de 759, de las que el 65 % pertenecen al grupo I, 20 % al grupo IIy 15 % al III. El 23 % de los niños con cardiopatía son hijos de padres inmigrantes. La edad de diagnóstico en niños menores de 3 meses (nacidos en España) es similar en los nativos y los de padres inmigrantes, sin embargo hay diferencia significativa entre los nativos y los inmigrantes no nacidos en España (mediana 18 vs 45 días, p = 0,003). En cuanto a los grupos hemodinámicos, aunque hay un retraso en el diagnóstico en los tres grupos, entre los niños inmigrantes, este alcanza significación estadística sólo en los grupos I y III (p = 0,004 y 0,003 respectivamente).
Conclusiones: 1) La inmigración está contribuyendo al incremento de la natalidad en nuestra Comunidad Autónoma. 2) Las cardiopatías congénitas en hijos de inmigrantes representan ya casi la cuarta parte del total. 3) Hay un retraso significativo en la edad de diagnóstico entre los nativos y los hijos de inmigrantes no nacidos en España, hecho que no ocurre cuando se trata de hijos de inmigrantes ya nacidos en España.
P30611:25 h
SINDROME DE CORAZON IZQUIERDO HIPOPLASICO: VARIACIONES TEMPORALES DE LA PREVALENCIA EN UNA POBLACION DEFINIDA GEOGRAFICAMENTE
Francisco Rodríguez Argente del Castillo, Sergio Muñoz Sánchez, Alejandro de Goicoechea Vera, Esther Ocete Hita, Natalia Cutillas Abellán, Ángeles Ruiz Extremera
Hospital Clínico Universitario San Cecilio, Granada.
El síndrome de corazón izquierdo hipoplásico es un defecto cardíaco congénito letal en niños que plantea dilemas y controversias tanto en su manejo como en el planteamiento ético-profesional.
Introducción: Documentar la prevalencia e incidencia de las cardiopatías congénitas proporciona una información muy útil para la práctica pediátrica, la planificación de la asistencia y la investigación etiológica. Nuestro propósito es analizar las variaciones temporales en la incidencia del síndrome del corazón izquierdo hipoplásico ocurridas en los últimos cinco años en una población bien definida.
Métodos: Utilizamos datos de los registros de la Unidad de Cuidados Intensivos Pediátricos y de la Unidad de Cardiología Infantil del H.C.U. Se identificaron todos aquellos recién nacidos ingresados con el diagnóstico de Síndrome de Corazón Izquierdo hipoplásico durante los años de 1999 a 2004.
Resultados: De 1999 a 2004, el registro recogió 14 casos de hipoplasia del ventrículo izquierdo entre 15.000 niños, con una prevalencia de 0,93 por cada 1.000 recién nacidos. La prevalencia aumentó a 1,3 por cada 1.000 recién nacidos desde 2003 a 2004. La prevalencia e incidencia variaron en función de la geografía.
Conclusiones: La prevalencia del síndrome de corazón izquierdo hipoplásico está aumentando en el área estudiada. Así como algunos de los nuevos casos se pueden explicar por la mejora en la comunicación de casos y el diagnóstico, otros se deben a cambios en la distribución de factores de riesgo en la población. Las bases de estos cambios no es totalmente conocida. Ulteriores estudios serán necesarios para determinar la naturaleza y distribución de estos factores.

