

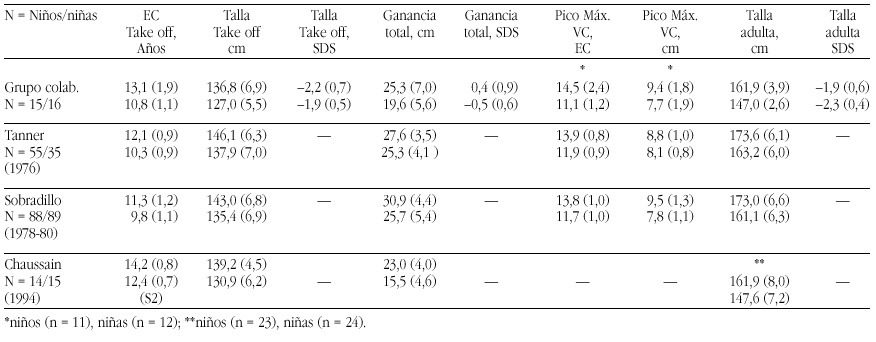
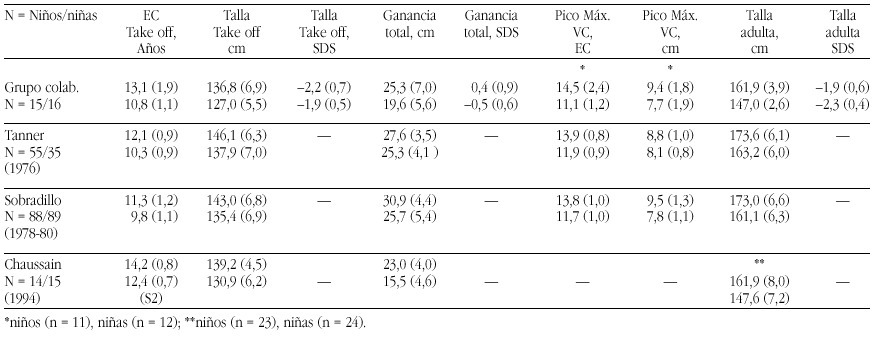
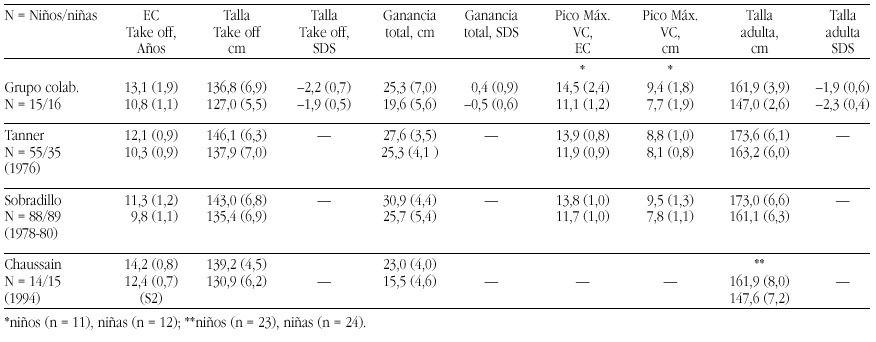
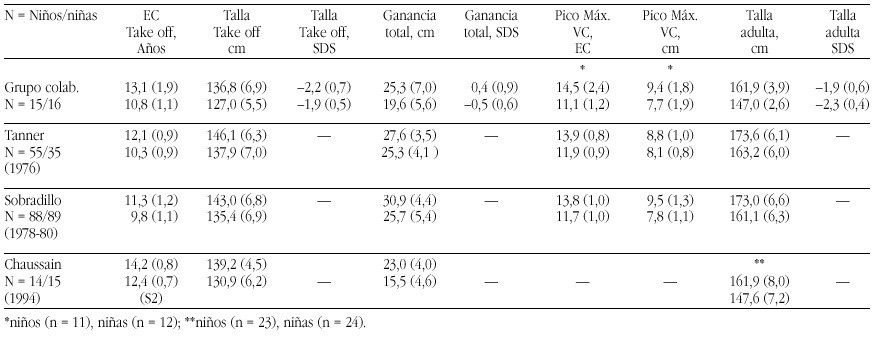
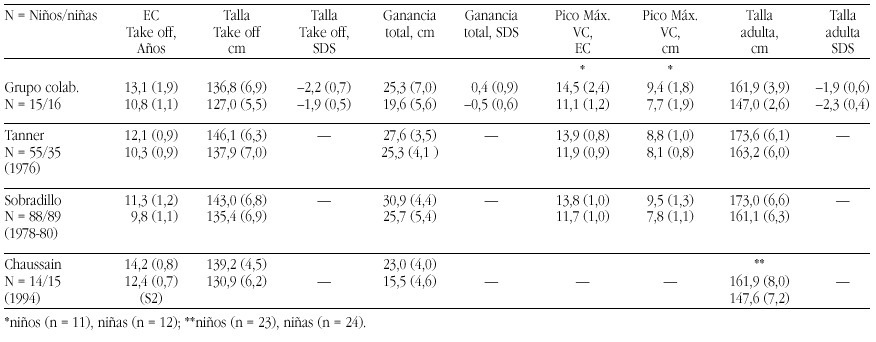
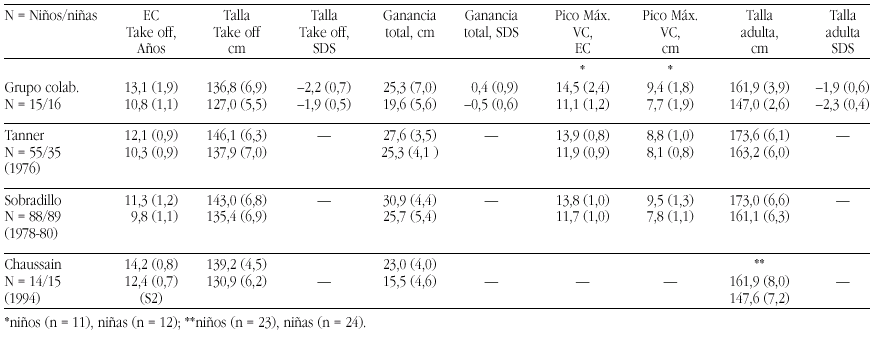
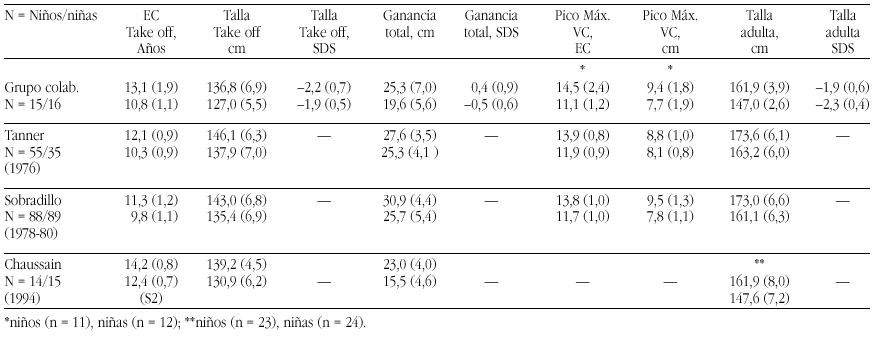
El hipotiroidismo congénito (HC) es la patología endocrinológica congénita más frecuente, con una prevalencia variable según zonas geográficas, entre 1:2500 y 1:4000 recién nacidos. En la actualidad, los programas de screening del HC detectan la mayoría de casos y permiten un tratamiento precoz de la enfermedad, evitando las graves secuelas neurológicas y motoras del hipotiroidismo en los primeros años de la vida. Genéricamente, el HC puede clasificarse en permanente y transitorio.
La mayoría de casos de HC permanente (80-85%) se debe a trastornos embriológicos de la glándula (disgenesia) que pueden llevar tanto a la ausencia total de la misma (agenesia) como a su localización anormal a lo largo del trayecto migratorio que ésta recorre en el cuello (ectopia). En el 15-20% de ocasiones existe, sin embargo, una glándula tiroidea de tamaño normal o agrandado (bocio) de localización correcta. Este grupo etiológico corresponde a los defectos congénitos en la síntesis de hormonas tiroideas o dishormonogénesis tiroidea. La base genética de disgenesias y de dishormonogénesis tiroideas empieza a conocerse en su complejidad a través de la localización de defectos moleculares en pacientes con los diversos fenotipos tiroideos, pero aún el porcentaje de casos de HC que en la actualidad somos capaces de filiar a nivel genético es reducido, reflejando el conocimiento todavía parcial que se posee respecto de los procesos embriológicos y de hormonosíntesis en el tiroides.
La incidencia de HC transitorio es alta en Europa y muy infrecuente en Norteamérica, Japón y Australia. Su etiología se ha relacionado con el paso transplacentario de anticuerpos bloqueantes del tiroides o de medicación antitiroidea de la madre al feto o bien con deficiencias endémicas de yodo o intoxicación por productos yodados (antisépticos o contrastres para procedimientos radiológicos) en el recién nacido. Sin embargo, cuando se investiga la etiología del HC transitorio en series de distintos países, invariablemente se encuentra un porcentaje de casos, denominados "idiopáticos", cuya causa permanece sin identificar. Hasta el momento no se conocen causas genéticas relacionadas con alteraciones transitorias en la síntesis o secreción de hormonas tiroideas.
En 1997 en el laboratorio de Endocrinología Pediátrica de la Universidad de Amsterdam (Prof. De Vijlder, Dra. Ris-Stalpers) iniciamos un proyecto que persigue la identificación de nuevos genes específicos de tiroides y la conexión de posibles defectos en estos genes con casos de HC cuya base genética es aún desconocida. La estrategia de clonaje escogida se basó en una técnica de reciente implantación en Biología Molecular que permite conocer el perfil de expresión completo y cuantitativo de todos los genes que se expresan en un tejido: el Análisis en Serie de la Expresión Génica o SAGE (por Serial Analisis of Gene Expression). A continuación se revisan los fundamentos moleculares de la función tiroidea y junto con los hallazgos alcanzados hasta el momento, tanto en la aplicación de la técnica SAGE a nuestros objetivos como en la identificación de nuevos genes tiroideos, sus defectos moleculares y sus correspondientes correlatos clínicos en pacientes con HC.
Fundamentos moleculares de la función tiroidea
Virtualmente todos los genes que codifican proteínas tiroideas, ya sean enzimáticas o de carácter estructural, que participan en la síntesis hormonal del tiroides, pueden ser origen de defectos hereditarios que originen hipotiroidismo (figura 1)1. El conocimiento cada vez más preciso de estos procesos de síntesis y el desarrollo de las técnicas de clonación molecular están conduciendo a un conocimiento contínuo de nuevas proteínas específicas implicadas en la hormonogénesis tiroidea. Al papel esencial de la tiroglobulina (Tg) y la tiroperoxidasa (TPO) como soportes estructural y enzimático de la síntesis hormonal en el tiroides, se van sumando nuevas moléculas como el receptor de TSH (R-TSH) o la proteína Gsa acoplada a este receptor, identificadas ahora como causantes de otras dishormonogénesis de presentación clínica bien diferenciada. La existencia, previamente intuida, de nuevas proteínas tiroideas, como la de un transportador activo de yodo en la membrana basal del tireocito, fue confirmada a través de la clonación del gen denominado "Simportador de Sodio y Yodo" o NIS (por "Na+/I- Symporter"). Posteriormente, se ha identificado un nuevo transportador de yodo (Pendrina, codificado por el gen PDS) esta vez localizado en la membrana apical, en relación con la que puede constituir la más frecuente de las dishormonogénesis tiroideas, el Síndrome de Pendred. La clonación de los tres genes encargados de codificar las desyodasas tiroideas (DIO1, DIO2 y DIO3), y en especial la de la desyodasa tipo 1 por su contribución a la formación intratiroidea de T3, completaba el número de genes cuya actividad participa directamente en el proceso de síntesis o secreción hormonal en el tiroides. Recientemente, la identificación de 2 oxidasas tiroideas (ThOX1 y ThOX2), encargadas de la producción de peróxido de hidrógeno (H2O2) en la luz folicular, engrosa la lista de genes conocidos que muestran una expresión específica o restringida al tiroides. En los apartados siguientes se detalla el proceso de identificación de las nuevas oxidasas tiroideas y los estudios sobre sus implicaciones clínicas.

Figura 1. Representación esquemática del proceso de biosíntesis y secreción de hormonas tiroideas en el tireocito junto a las proteínas implicadas en la hormonogénesis. Tg, tiroglobulina; TPO, tiroperoxidasa; R-TSH, receptor de tirotropina; Gsa, subunidad (de la proteína G estimuladora; NIS, transportador basal de sodio y yodo; Pendrina: transportador apical de cloro y yodo; S.G.H2O2: sistema generador de peróxido de hidrógeno. Esquema modificado de Moreno, 19991.
Un apartado especial lo forman los factores de transcripción específicos de tiroides, conocidos como TTF-1, TTF-2 y Pax-8, algunos de los cuales se han relacionado recientemente tanto con defectos dishormonogénicos como con alteraciones disgenéticas de la glándula tiroidea. Estas proteínas nucleares no participan directamente en los procesos de síntesis hormonal del tireocito, pero son responsables del mantenimiento del fenotipo tiroideo a través del estímulo de la transcripción de genes fundamentales para el mismo, como los de Tg, TPO, R-TSH o NIS. Por último, existen en el tiroides actividades bioquímicas bien conocidas, como la "dehalogenacion"y reciclaje del yodo intracelular tras la lisis de la Tg, que aún no han sido caracterizadas desde el punto de vista molecular. La alteración funcional de los genes que codifican estas proteínas tiroideas, si bien conduce a hipotiroidismo como expresión clínica común, origina también algunos rasgos fenotípicos característicos de cada dishormonogénesis que permiten su diferenciación clínica o bioquímica (tabla 2).
La herencia de las dishomonogénesis es de carácter mendeliano y, con alguna excepción, sigue un patrón autosómico recesivo. El hipotiroidismo presente en la osteopatía de Albright, un defecto en el gen de la proteína Gsa que produce una falta de respuesta intracelular a la TSH, es el único tipo de dishormonogénesis que se hereda de forma dominante, aunque su penetrancia es muy variable.
El carácter recesivo de la mayoría de dishormonogénesis nos hace presumir que la existencia de un único alelo activo de estos genes es suficiente para mantener una función tiroidea normal. Sin embargo, la haploinsuficiencia (actividad de uno sólo de los alelos) de algunos genes tiroideos podría originar fenotipos tiroideos menores, leves o incompletos cuando se sumasen otras circunstancias limitantes de la hormonogénesis, como la carencia de yodo. Una mejor caracterización, tanto clínica como molecular, de estos defectos parciales de la función tiroidea contribuiría de forma importante a clarificar la etiología de los tipos de hipotiroidismo aún catalogados de idiopáticos.
El generador tiroideo de H2O2
La incorporación del yodo a la molécula de tiroglobulina (Tg) es un paso clave en la hormonognesis tiroidea. En la actualidad se conocen 4 elementos necesarios en la reacción de organificación del yodo: la pendrina, que transporta el yodo a la luz folicular a través de la membrana apical; la TPO, enzima catalizadora de la reacción; la Tg, aceptor final del yodo, y un sistema de oxidasas capaz de generar peróxido de hidrogeno (figura 2)2. El peróxido de hidrogeno (H2O2) es un substrato imprescindible para la actividad enzimática de la TPO, participando no sólo en la oxidación previa del yodo sino también en el posterior acoplamiento entre yodotirosinas para formar las hormonas tiroideas. En este sentido, la falta de H2O2 constituye per se un factor limitante de la organificación y, consecuentemente, de la síntesis de hormonas tiroideas.

Figura 2. Representación del sistema de membrana generador de peróxido de hidrógeno (H2O2). Mediante oxidación de NADPH hasta NADP en el lado citoplasmático de la membrana apical se generan electrones que son transportados a través de la membrana para formar H2O2 en el lado opuesto. La TPO cataliza la yodación y el acoplamiento de la Tg en presencia de H2O2 como agente oxidante en el lado folicular de la membrana. Esquema tomado de De la Vieja et al, 19972.
La existencia de un sistema generador de H2O2 específico del tiroides y localizado en la membrana apical del tirocito fue demostrada en los años 80 a través de la localización de peróxido de hidrógeno en el folículo tiroideo3,4 y la posterior purificación parcial de la correspondiente actividad enzimática5. Diversos estudios bioquímicos sobre el generador tiroideo de H2O2 identificaron que su actividad era dependiente de Calcio y que utilizaba NADPH como donador de electrones6. Más tarde se comprobó que la producción de H2O2 es susceptible de estimulación por TSH, lo cual sugería que a los genes implicados en este sistema de oxidasas (por entonces aún no identificados) tendrían expresión específica en tiroides y que la(s) oxidasa(s) tiroidea(s) debía(n) considerarse nuevos marcadores de diferenciación tiroidea7.
Utilizando una estrategia de clonaje basada en la purificación de la actividad enzimática, Dupuy et al identificaron recientemente un fragmento de cDNA que codificaba una oxidasa tiroidea de 1210 aminoácidos y 138 kDa de peso molecular que denominaron p138Tox8. Con posterioridad, y mediante screening de una librería de células tiroideas en cultivo, De Dekens et al clonaron 2 cDNAs que codificaban 2 proteínas oxidasas de 1551 y 1548 aminoácidos (aproximadamente 180 kDa) que denominaron respectivamente ThOX1 y ThOX29. Ambas proteínas estaban localizadas en el cromosoma 15q15.3 y se acumulan en la membrana apical de la célula tiroidea. La secuencia de p138Tox resultaba ser el fragmento carboxiterminal de la proteína ThOX2. Las proteínas ThOX tienen una estructura primaria que incluye 7 dominios transmembrana, 3 lugares de unión para NADPH (Nicotinamide Adenin Dinucleotide Phosphate, forma reducida) y 1 para FAD (Flavin Adenin Dinucleotido), 5 posibles lugares de glucosilación y, de manera similar a ciertas oxidasas de las plantas, 2 lugares de interacción con el calcio conocidos como EF-Hands (figura 3). Los EF-Hands, presentes en otras familias de proteínas, controlan la actividad funcional de las mismas, induciendo cambios conformacionales que parecen necesarios para el inicio de su actividad.

Figura 3. Estructura de las proteínas ThOX. Este modelo de topología transmembrana y la presencia de los diversos dominios funcionales se basa en predicciones que han de ser corroboradas experimentalmente. NADPH: motivo de unión (a modo de "bolsillo") para el NADPH; FAD: motivo de unión a FAD; EF-Hand: motivos de unión al calcio; I-VI: dominios transmenbrana. Inside: lado citoplasmático de la membrana apical; Outside: lado folicular de la membrana apical. Esquema tomado de De Dekens et al, 20009.
Los estudios funcionales sobre estas proteínas se encuentran en fase muy inicial, así como lo que conocemos sobre la fisiología global del generador tiroideo de H2O2 (componentes, interacciones, regulación y control de la producción de peróxido de hidrógeno). Sin embargo, y por analogía con otros sistemas de oxidasas y estudios enzimáticos10, es muy probable que existan otros componentes adicionales a las proteínas ThOX1 y 2, flavoproteínas o estructuras reguladoras de la producción de H2O2 (un agente es per se nocivo para la célula) que en el futuro completen nuestra visión sobre el sistema tiroideo de oxidasas.
Implicaciones clínicas del generador tiroideo de H2O2
Tras la identificación de estas 2 proteínas oxidasas tiroideas y desde el punto de vista clínico, cabía preguntarse qué tipo de patología podría estar relacionada con posibles defectos de producción de H2O2 en el tiroides. Mientras el exceso de producción de peróxido de hidrógeno y otras "especies activas de oxígeno" (ROS) se ha relacionado con procesos mitogénicos y cáncer11, el hipotiroidismo congénito con defecto de organificación del yodo podría ser la expresión clínica más plausible. Basándonos en la dualidad de este sistema de oxidasas tiroideo e hipotetizando que la falta de actividad de una de las 2 proteínas ThOX podría ser parcialmente compensada por la actividad de la otra, comprobamos la hipótesis de que ciertos defectos parciales de organificación del yodo pudiesen ser debidos a mutaciones en los genes ThOX.
Para ello realizamos una selección retrospectiva de pacientes con HC que presentaban defectos parciales de organificación del yodo (detectados con descarga i.v. de perclorato) cuya causa molecular fuese desconocida. Se excluyeron posibles alteraciones en los 3 elementos restantes de la hormonogénesis, aplicando 3 criterios de exclusión: cualquier dato bioquímico o funcional que sugiriese defectos de TPO (como descargas con perclorato > 90%), sordera o alteraciones auditivas familiares (como signo de posible Síndrome de Pendred) y datos bioquímicos o clínicos que sugiriesen un posible defecto en la síntesis de Tg. Once pacientes con estas características fueron identificados retrospectivamente en la base de datos del programa holandés de screening de HC desde el año 1981.
Nuestros datos de expresión génica en una librería SAGE de tiroides (ver apartado correspondiente) indicaban que ThOX2 tiene un nivel de expresión superior al de ThOX1 (datos no publicados). Por este motivo decidimos iniciar el screening de mutaciones del gen ThOX2. La organización genómica del gen fue determinada a través de 2 clones genómicos presentes en la base de datos GenBank: el gen estaba compuesto por 33 exones codificantes que fueron amplificados del DNA de sangre periférica de los pacientes. Utilizando como método de screening de mutaciones la cromatografía líquida (DHPLC)12 y la secuenciación directa de los productos de PCR con un patrón cromatográfico alterado, identificamos 2 mutaciones inactivantes (stop codon) en 2 pacientes con HC y DOY parcial13. Las mutaciones se presentan en heterocigosis y consisten en codones de terminación prematuros que codifican proteínas truncadas en un 50% de su extensión, excluyendo todos los motivos funcionales de la proteína THOX2: 6 de los siete dominios transmembrana, los tres sitios de unión al NADPH, el de unión a FAD y los dos de unión al Calcio (EF-Hands). La segregación familiar de estas mutaciones sugiere un patrón de herencia autosómico dominante.
El fenotipo clínico que presentaban estos dos pacientes era el mismo: un hipotiroidismo congénito leve y de carácter transitorio. El test de perclorato, realizado en la tercera semana de vida, indicaba descargas de entre el 40 y el 60% (figura 4). Todas las causas conocidas de HC transitorio estaban ausentes en estos pacientes, incluyendo la ausencia de intoxicación por yodo, comprobada a través de yodurias normales en el período neonatal. Tras el inicio de tratamiento sustitutivo con levotiroxina a dosis estándar, los 2 pacientes requirieron dosis cada vez menores de T4. A la edad de tres años (dosis de T4: 1-2 mg/kg/día) fueron deprivados de tratamiento, siendo capaces de mantener el eutiroidismo.

Figura 4. Test de descarga con perclorato en 2 pacientes con hipotiroidismo congénito transitorio y mutaciones en el gen THOX2. Tras la captación de I123 por el tiroides, la administración intravenosa de perclorato (NaCl O4-) produce la descarga del 40-60% del isótopo fuera del tiroides. Estos resultados indican la existencia de un defecto parcial en la organificacion del yodo (DOYP).
El defecto parcial en la síntesis de hormonas tiroideas de estos pacientes puede deberse a haploinsuficiencia (junto con el mutado, existe un alelo normal de ThOX2) o un efecto dominante negativo (por interacción inapropiada de la proteína mutada con otros componentes del sistema de oxidasas). Es posible que, al tratarse de defectos incompletos de la hormonogénesis, la expresión bioquímica de estos hipotiroidismos congénitos "borderline" solo sea detectable cuando los requerimientos de hormonas tiroideas son máximos, esto es, durante los iniciales meses de la vida. Estos hallazgos constituyen la primera evidencia de que determinados casos de HC transitorio tienen una causa genética y que se deben a defectos parciales de la producción de H2O2 en el tiroides.
Nuevos genes específicos de la fisiología tiroidea
La identificación de nuevas proteínas implicadas en el desarrollo embriológico y los procesos de hormonosíntesis tiroidea en los últimos años ha posibilitado un conocimiento más profundo e integrado de la fisiopatología tiroidea. Sin embargo, estos conocimientos distan mucho de ser completos. En el laboratorio de Endocrinología Pediátrica de la Universidad de Amsterdam iniciamos un proyecto cuyo objetivo principal era el clonaje de nuevos cDNAs con un patrón de expresión específico de tiroides o, al menos, restringido al tiroides y a otros pocos tejidos adicionales. El fundamento es que todos los genes específicos de tiroides que hasta el momento conocemos muestran este tipo de patrón de expresión y que, las proteínas que respectivamente codifican han demostrado a posteriori ser relevantes en diversos aspectos de la fisiología tiroidea. Asimismo (y apoyando la finalidad clínica de nuestro proyecto) todos estos genes se han encontrado mutados o delecionados en pacientes con diversos tipos de hipotiroidismo, la mayoría de ellos de expresión clínica congénita.
Nuestra estrategia de clonaje está basada en el Análisis en Serie de la Expresión Génica o SAGE. Tras la construcción de una genoteca SAGE de tejido tiroideo normal y el posterior desarrollo de un método computarizado para seleccionar los cDNAs con expresión preferente en el tiroides, hemos identificado un grupo de genes cuya caracterización genómica (y función de las proteínas que respectivamente codifican) es objeto de estudio en la actualidad.
La técnica SAGE y el tiroides
El SAGE es una técnica descrita en 1995 por Velculescu et al. que permite el análisis completo y cuantitativo de todos los genes expresados en un tejido determinado14,15. Cada molécula de RNA presente en el tejido (o cultivo celular) es representada por un secuencia muy corta (10 pares de bases) que se denomina "cola SAGE" (por SAGE-tag) y que normalmente se localiza en la parte final (3') de la molécula de RNA. La abundancia de estas "colas" cortas de secuencia se cuantifica a través de un programa software especialmente diseñado para la técnica.
La figura 5 representa esquemáticamente los principales pasos a seguir en la construcción y análisis de una genoteca SAGE. De manera resumida, tras la extracción del RNA total del tejido objeto de estudio y la purificación de su RNA mensajero, se obtienen las "colas SAGE" con el concurso de 2 enzimas de restricción (NlaIII como "anchoring enzyme" y NotI como "tagging enzyme"). Posteriormente se procede a la concatenación de estas colas cortas de secuencia y a su subclonaje en vectores apropiados. Finalmente se procede a la secuenciación en serie de estos clones y a la cuantificación, y ordenación en función a su abundancia, de todas estas mini-secuencias a través de un programa software. El uso de la base de datos conocida como Genbank, donde se almacenan las secuencias de todos los genes identificados hasta el momento, permite conocer la presencia (y, en su caso, la abundancia) de los genes en el tejido que se estudia. Cuando las colas SAGE no encuentran su correspondencia (o "match") en la mencionada base de datos, se las denomina colas "No-match" y se asume que forman parte de genes presentes en el tejido pero que aún no han sido identificados.

Figura 5. Gráfico esquemático de la técnica SAGE (Serial Analysis of Gene Expression).
Con objeto de analizar el perfil completo de expresión génica en la glándula tiroidea, construimos una genoteca SAGE de una muestra de tejido tiroideo normal16. Se secuenciaron un total de 10,994 de estas "colas", que representaban un total de 6.099 genes presentes en el tiroides. Cerca de un tercio de ellos (1.839) eran genes ya conocidos y aproximadamente dos tercios (4.260) correspondían a genes aún por identificar. Dentro de los genes conocidos, el gen de Tg es el más abundante en el tiroides, con un porcentaje de expresión cercano al 2%. El gen de TPO muestra una expresión 10 veces menor (0,24%). La abundancia relativa de otros genes específicos de tiroides en nuestra genoteca SAGE figura en la tabla 2. Estos datos, por un lado, nos han permitido conocer de manera cuantitativa el perfil completo de expresión génica en el tiroides y, con respecto al objetivo de nuestro proyecto, posibilitan la identificación de nuevos cDNAs y proteínas especificos de la fisiología tiroidea.


Identificación de nuevos genes específicos de tiroides
Con objeto de identificar nuevos genes tiroideos, concentramos nuestra atención en las denominadas colas "No-match" o secuencias cortas que representan genes aún por caracterizar. Una primera dificultad a salvar era el número extremadamente elevado de "colas" SAGE (4.260) potencialmente interesantes. La segunda, que, de las más de 4.000 colas ausentes en el GenBank, una amplia mayoría habrían lógicamente de corresponder a genes expresados ampliamente en muchos tipos celulares (los denominados "housekeeping genes") y tan sólo una pequeña proporción representarían genes específicos de tiroides.
Para posibilitar el estudio en profundidad de un número adecuado de colas SAGE procedimos a desarrollar un método de selección de las "colas SAGE" con expresión preferencial en tiroides (Moreno et al, aceptado para publicación en Genomics). Este método, basado en la comparación de los niveles de expresión de cada una de las colas en 10 tejidos humanos diferentes, utiliza un algoritmo matemático que hemos denominado TPE (por Tissue Prefferential Expression). Para comprobar la validez del método, analizamos las primeras 80 colas "No-match" de nuestra genoteca asignando un valor TPE a cada una de ellas. Seleccionando las colas con un TPE más alto (TPE>7) como aquellas con mayor probabilidad de corresponder a genes específicos de tiroides, comprobamos experimentalmente (tanto por northern blot como por RT-PCR) que efectivamente estos cDNAs tenían una expresión preferencial en tiroides. Finalmente procedimos al screening de una genoteca de cDNA de tiroides para identificar las secuencias completas de estos cDNAs. Tres colas "No-match" correspondientes a los números 56 (NM56), 41 (NM41) y más recientemente la 159 (NM159) sirvieron para identificar 3 nuevos cDNAs17 cuya organización genómica y posible homología con otros genes procedimos a analizar:
Cola "No-Match no. 56
El clon correspondiente a la cola NM56 contenía la secuencia parcial (3 kb) de un cDNA con secuencias de unión para NADPH y FAD. Además, mostraba homologías con oxidasas presentes en el ratón y el cerdo y con algunas oxidasas humanas. Siguiendo una estrategia de clonaje diferente (purificación proteica), una secuencia más larga pero aún incompleta de este mismo cDNA fue identificada por Dupuy et al. y denominada g138tox (Thyroid oxidase de 138 kDa)8 y más adelante por De Dekens et al9, denominándola ThOX2. El que uno de nuestros clones positivos correspondiese a componentes del sistema generador de H2O2 del tiroides constituía una comprobación práctica de la validez de nuestra estrategia de clonaje. El screening de mutaciones en el gen ThOX2 iniciado a continuación en pacientes con defectos de organificación parcial de yodo (DOYP) ha posibilitado la identificación de mutaciones en pacientes con HC transitorio descrita en el apartado anterior.
Cola "No-Match" no. 41
Usando la segunda cola seleccionada, clonamos un gen de expresión restringida al tiroides. Tiene un RNA mensajero de 1,3 Kb y ocupa 20 kb de DNA genómico dividido en 8 exones y 7 intrones. Se halla localizado en el brazo largo del cromosoma 16. Sus niveles de expresión, determinados por northern blot, son elevados en tiroides y presentes a nivel bajo en células endocrinas del pulmón y el estómago. Este gen codifica una proteína de 40,1 kDa que se ha traducido in vitro en un sistema de lisado de reticulocitos. Diversos programas de computador predicen la posible localización de esta proteína en la membrana extracelular o bien su posible secreción al medio extracelular, la existencia de sitios de fosforilación y su capacidad de dimerización a través de puentes disulfuro. La homología de la proteína NM41 con otras proteínas (Rhodopsina, Peripherina) que funcionan como moléculas de adhesión intercelular (ICAMs), nos lleva a explorar la posibilidad de que esta nueva proteína pueda desempeñar funciones similares en el tiroides. Muchas de estas ICAMs son moléculas intermediarias y efectoras de procesos de migración celular, posibilitando la interacción con otras moléculas localizadas en superficies celulares adyacentes. La función de esta proteína es actualmente objeto de estudio en nuestro laboratorio. Trabajamos con la hipótesis de una posible implicación de NM41 en migración tiroidea o bien en el mantenimiento de la organización folicular del tejido tiroideo. La comprobación estas hipótesis nos llevaría a considerar a NM41 como un gen candidato en alteraciones disgenéticas tiroideas (especialmente ectopias) en pacientes con hipotiroidismo congénito.
Cola "No-Match" 159
Recientemente, una tercera cola SAGE nos ha llevado a la identificación de un cDNA, también de expresión específica en tiroides, cuya organización genomica se encuentra en estudio. Se halla localizado en el brazo corto del cromosoma 6 y la secuencia parcial del cDNA del que disponemos encuentra similitudes con genes homólogos en ratón, C. elegans y Drosophila Melanogaster, indicando una conservación evolutiva importante. A nivel protéico existen similitudes con diversas oxidasas que utilizan NADH como donador de electrones. La posibilidad de que esta proteína forme parte del complejo sistema generador de peróxido de hidrógeno en el tiroides también se investiga en la actualidad. En este sentido, pacientes con defectos parciales de organificación del yodo en los que no se encontraron mutaciones en ThOX2 ni en ThOX1 serían candidatos evidentes para screening de mutaciones en este gen.
Conclusiones
El hipotiroidismo congénito es la expresión clínica común de un grupo de alteraciones de naturaleza tanto genética como ambiental. Los defectos moleculares identificados hasta el momento en genes tiroideos se relacionaban exclusivamente con casos de HC permanente. Sin embargo, existen evidencias de que determinados casos de HC transitorio pueden deberse también a alteraciones genéticas que inducen defectos leves y parciales de la hormonosíntesis tiroidea.
La aplicación de las nuevas técnicas de biología molecular a la identificación de nuevos genes específicos de tiroides ha de contribuir en el futuro al conocimiento más detallado de los procesos celulares relevantes en fisiología tiroidea y al esclarecimiento etiológico de los casos de hipofunción tiroidea que aún clasificamos como idiopáticos.
Agradecimientos
Este trabajo ha sido financiado en parte con la Beca de Investigación de la Sociedad Europea de Endocrinología Pediátrica (ESPE), financiado por Novo/Nordisk AS y con el Premio de Investigación de la Sociedad Española de Endocrinología Pediátrica (SEEP), financiado por Lilly.

