

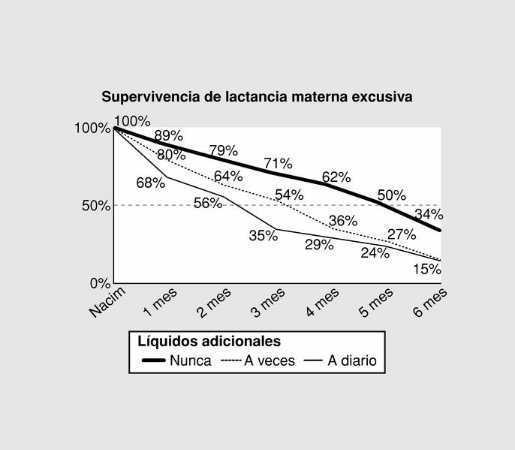
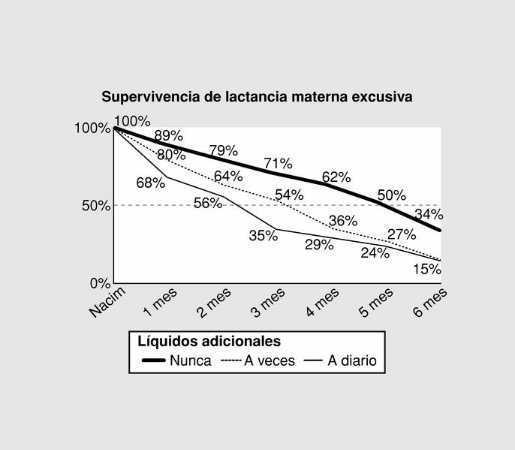
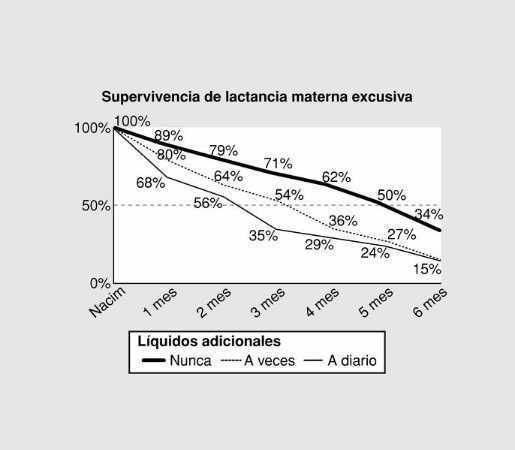
Jueves, 31 de mayo (15,30 - 16,30 h)
Endocrinología
Concordancia entre DR y DQ en diabéticos tipo 1
P. Giralt Muiña, M. Urranz Ardanaz, L. Santillana Ferrer, E. Galiano Fernández, M.A. Márquez de la Plata Legaz e I. Condado Sánchez de Rojas
Hospital Nuestra Señora de Alarcos, Ciudad Real.
Antecedentes: Se ha demostrado que la diabetes tipo 1 es una enfermedad poligénica; sin embargo desde hace varios años se ha considerado la región HLA de clase II como la más importante en la producción de ésta enfermedad. Se ha visto que la mayoría de los diabéticos tipo 1 son DR3 o DR4. Posteriormente se observó que el 95% de los DR4 que tenían un alelo DQB 0302 eran diabéticos mientras que los DR4 DQB 0301 no lo eran, por lo que se ha sugerido que es más importante este grupo de moléculas.
Los DQ tienen 2 cadenas: alfa y beta. Los alelos de la cadena beta 0302 y los de la alfa 0301, se han ligado al DR4. Los alelos de la cadena beta 0201 y los de la alfa 0501, se consideran ligados al DR3. En el presente trabajo observamos que la correlación no es tan exacta.
Métodos: Se realiza el estudio genético DQ a 48 pacientes diabéticos tipo 1 que previamente tenían efectuado un estudio DR.
El tipaje HLA-DR se realiza por técnicas serológicas. Microlinfotoxicidad con enriquecimientos de linfocitos B. No se valoraron los subtipos. Los alelos HLA-DQ se determinaron por reacción en cadena de la polimerasa, con amplificación específica de alelo (PCR-SSP). Se utilizaron cebadores específicos para los genes DQA1 y DQB1 (Protans, Ger). Los productos amplificados se separaron por electroforesis en gel de agarosa al 2% y se asignaron las amplificaciones específicas del alelo.
Resultados: Solamente el 58% de los DQ corresponden con el DR esperado. De un total de 48 pacientes estudiados en 28 se encontró una relación entre DR y DQ. En 10 pacientes DQA1*0501,0301/DQB1*0201,0302 sólo 5 eran DR3-DR4. De 19 pacientes DQA1*0501/DQB1*0201 12 eran del grupo esperado DR3. En 14 pacientes DQA1*0301/DQB1*0302 7 eran DR4 y en otros 5 pacientes diabéticos tipo 1 que presentaban otros DQ sólo 1 era DR4 y los otros 4 eran de DR catalogados como no diabetógenos.
Conclusiones: Los estudios genéticos efectuados en diabéticos tipo 1 por biología molecular (DQ) y los realizados por serología (DR), no concuerdan en casi la mitad de los casos si no se analizan los subtipos DR.
¿Es el Polimorfismo del Gen del Enzima de Conversión de la Angiotensina un marcador de Nefropatía Diabética?
B. Orive Olóndriz, A. Rodríguez Estévez y S. Salazar Urcelay
Hospital Txagorritxu, Vitoria.
La diabetes mellitus insulin dependiente, tiene una alta morbilidad cardiovascular y su pronóstico vital está ligado a la existencia de Nefropatía Diabética, probablemente determinada por anomalías en la hemodinámica renal y por una predisposición genética.
Objetivo: Reportar un caso de Nefropatía Diabética de aparición precoz, a pesar de un buen control metabólico.El paciente tiene un Polimorfismo Inserción/Delección del Gen de Enzima de Conversión de Angiotensina, correspondiente a un Genotipo DD, considerado un factor de riesgo para la Nefropatía Diabética y su evolución a la Insuficiencia Renal Crónica.
Varón que tiene ahora 20 años, previamente sano, con antecedentes familiares de diabetes no ID, que hace el debut de su diabetes con cetoacidosis cuando tenía 5 años (Sept-1985). Bien controlado, con cifras de Hb A1C medias de 7,35%. 5 años más tarde (Sept 1990) presenta un Síndrome Nefrótico completo, con proteinuria de 90 mg/m2/h; hipoalbuminemia de 1,9 gr/l e hipercolesterolemia de 387 mg/dl. Su filtrado glomerular era de 120 ml/m/1,73 y la tensión arterial normal. La biopsia renal mostró lesiones de glomeruloesclerosis segmentaria y focal con aumento dela matriz mesangial y engrosamiento de la pared de las arteriolas aferentes, compatibles con Nefropatía Diabética. El nivel plasmático de ECA fue de 57,7 U/L (en suero, por técnica cinética) y su genotipo para el gen de ECA, fue homocigoto DD. Se inició precozmente tratamiento con inhibidores de ECA e hipolipemiantes y en la actualidad, 10 años después del inicio de la nefropatía, tiene un filtrado de 140 ml /m/1,73, T.A normal; proteinuria de 4 gr/día con albuminemia en el límite bajo de la normalidad. No tiene retinopatía y sí un hipotiroidismo subclínico, posiblemente agravado por la intensa pérdida de proteínas por orina.
Conclusiones: La aparición de Nefropatía en niños diabéticos, en estadíos precoces de la enfermedad y con adecuado control metabólico puede estar condicionado genéticamente por el Polimorfismo del gen de ECA. El adecuado manejo de la hiperlipidemia y el tratamiento antiproteinúrico con Inhibidores de ECA, enlentecen el deterioro de la función renal.
Torsión de hidátides testiculares y edad prepuberal
C. Moreno Hurtado, A. Blesa, C. Lineros, R. Núñez Núñez, L. Rodríguez, I. Santiago Álvarez, R. Cabrera García y E. Blesa Sánchez
Hospital Universitario Infanta Cristina, Badajoz.
Introducción: La torsión de hidátides testiculares constituye una entidad clínica de importancia relativa, en cuanto que su patogenicidad parece limitada pero su diagnóstico se presta a confusión con otras causas más preocupantes de escroto agudo, como torsión testicular y orquiepididimitis. Su llamativa prevalencia en la edad prepuberal y la posible influencia de los cambios hormonales propios de ella, junto al incremento de incidencia en los últimos años, nos han animado a realizar el presente trabajo.
Pacientes y métodos: Se realiza un estudio retrospectivo, Grupo I, en el que se valora en todos los pacientes intervenidos en nuestro Centro entre Enero de 1984 y Diciembre de 1998, la edad, el año en que fueron intervenidos, y el diagnóstico diferencial planteado por el cuadro de dolor escrotal agudo que presentaban.
Se realiza otro estudio prospectivo, Grupo II, de marzo de 1999 a marzo de 2000, en el que se valoran distintas variables clínicas relacionadas con la torsión, junto a una estimación del estado nutricional de los pacientes (Índice de Masa Corporal), el estadio de maduración (según Tanner) del vello pubiano y el pene, así como el volumen testicular.
Resultados: La edad de los 61 pacientes del Grupo I fue de 9,37 ± 2,58 años. La incidencia de torsión de hidátide durante los últimos 5 años del estudio casi triplicó la de los 10 primeros años. El diagnóstico preoperatorio de torsión de hidátide se hizo en 33 de los 61, siendo de torsión testicular en 6 y de escroto agudo inespecífico en 22.
Los 21 pacientes del Grupo II, de 9,83 + 3,03 años, presentaron un Índice de Masa Corporal de 19,58 +3,77. El 90% presentaron un desarrollo peneano y del vello pubiano en estadio I o II. El volumen testicular fue de 2,16+1,66 ml.
Conclusiones: El estudio retrospectivo confirma el llamativo incremento en la frecuencia de torsión de hidátides testiculares en los últimos años, así como su incidencia predominante en niños que van a iniciar o han iniciado recientemente el desarrollo puberal, lo que confirma el estadio de desarrollo sexual estimado en el estudio prospectivo, que, además, nos muestra que estos pacientes tienen un Índice de Masa Corporal próximo al percentil 90. Parece por lo tanto probable la existencia de un efecto hormonal, relacionable con el estadio prepuberal, como desencadenante del cuadro patológico, aunque no explique su incremento durante los últimos años.
Diabetes Mellitus tipo 2 en Pediatría: A propósito de un caso
J. Galán de Dios, M. Cabrera Lafuente, E. Patricia Torres Begara, I. González-Casado y R. Gracia-Bouthelier
Hospital Universitario La Paz, Madrid.
Introducción: La Diabetes Mellitus (DM) tipo 2 representa el 90% del total de las diabetes mellitus, y aunque clásicamente se describe por encima de los 30 años, va a constituir el 5% de los debuts diabéticos en menores de 20 años. En algunas series se observa un aumento en la incidencia en jóvenes, en probable relación a cambios en los hábitos alimenticios y a la mayor prevalencia de obesidad.
Caso clínico: Niña de 11 años y 9 meses remitida por su pediatra por un cuadro de candidiasis vulvovaginal, en el curso del cual se objetivó glucosuria e hiperglucemia (glucemia capilar en ayunas de 310 mg/dl). Desde 15 días antes la paciente refería enuresis 2ª, poliuria y polidipsia. No presentó pérdida de peso en los meses previos. Importante obesidad con un IMC de 33,2 de larga evolución. AP: Peso elevado al nacimiento. Estudiada a la edad de 3 años por obesidad y talla alta, con normalidad en el estudio endocrinológico. AF: Madre obesa, con hipercolesterolemia. Tía paterna DM tipo 2 diagnosticada a los 30 años. Exploración física: BEG. Obesidad generalizada (peso 89 kg, talla 163, IMC:33,5). Lesiones de acantosis nígricans en cuello. Candidiasis del área vaginal. Resto normal. Tanner III. Al ingreso: glucemia capilar de 329 mg/dl., glucosuria++ y cetonuria++, sin acidosis. Exploraciones complementarias: HbA1C: 12,9%. Péptido C: 4,3 ng/ml. Colesterol 301 mg/dl; HDL: 41 mg/dl. Función tiroidea normal. Estudio de autoinmunidad para DM tipo 1 negativo. Evolución: Ante sospecha de debut DM tipo 1 (hiperglucemia, cetosis y debut brusco), se inicia insulinoterapia a dosis inferiores a las habituales ( 0,4 U/kg/día) por el elevado peso de la paciente, y control de la ingesta de hidratos de carbono según el método de raciones. Lográndose un excelente control metabólico desde los primeros días. Se observó un descenso progresivo de las necesidades de insulina, a la vez que mejoraba el IMC. Por lo que, ante la sospecha de DM tipo 2, se suspende la insulinoterapia al 5º mes de evolución.
Comentario: En la DM tipo 2 existe una resistencia periférica a la insulina con un déficit relativo en la secreción de la misma. A diferencia de la tipo 1, no se evidencia destrucción autoinmune de las células ß, ni asociación clara a un HLA concreto, aunque sí hay una clara predisposición genética aún no definida, suele tener un comienzo más larvado y se asocia menos frecuentemente a cetosis (sólo un 14% de los debuts en jóvenes). Son factores de riesgo la obesidad, el sedentarismo, la hiperlipemia y la edad avanzada. El diagnóstico diferencial con DM tipo 1 en fase de luna de miel se basa en la asociación a obesidad y acantosis nigricans, y la negatividad del estudio de autoinmunidad. Nuestro caso ilustra cómo la DM tipo 2 no es exclusiva de la edad adulta, y que en algunas ocasiones la forma de debut es atípica: cetosis y clínica de corta evolución; debiéndose prestar especial atención a los debuts diabéticos asociados a obesidad.
Valores de referencia hormonales de función corticosuprarrenal en recién nacidos sanos
J.A. López Medina, A. García Suárez, L. Fidel Moltó Ripoll, M.P. Pinel Julián, F. Perán Mesa, R. López-Jurado Romero de la Cruz, C. Fuentes Gutiérrez y M. de Felipe Jiménez-Casquet
Hospital Universitario Virgen de las Nieves, Granada.
Antecedentes: En las determinaciones hormonales las fluctuaciones fisiológicas, junto con el error analítico, son suficientes para dificultar la interpretación de las mismas. Problemas adicionales plantean los recién nacidos para los que existen escasas referencias de valores normales, dificultando el diagnóstico de patologías de una extrema urgencia, por ejemplo la hiperplasia adrenal congénita con pérdida salina. El objetivo de este estudio ha sido estimar los valores de referencia de las concentraciones séricas/plasmáticas basales de hormona corticotropa (ACTH), cortisol, 11-desoxicortisol, 17 hidroxiprogesterona (17-OHP), la actividad de renina plasmática (ARP), aldosterona, -4-androstendiona (4A), testosterona libre y sulfato de dehidroepiandrosterona (DHA-S) en niños recién nacidos sanos granadinos, mediante los inmunoanálisis utilizados habitualmente en este medio.
Métodos: La población fue de 30 recién nacidos sanos (15 niños y 15 niñas), con una vida media de 4,68 horas de vida (2,4-6,9 h), edad gestacional media de 39,7 semanas (38-42 s), y peso medio de 3380 g (2.700-4.070 g). Se ha determinado ACTH mediante IRMA (Nichols Inst. Diag., CA 92675) y ARP mediante RIA (immunotech 177 13276 France); el cortisol y sus principales precursores (DHA-S, 17-OHP y 11 desoxicortisol) mediante métodos RIA (Immunotech 177 13276, France), así mismo han sido medidos por RIA los niveles de Testosterona libre (DPC CA 90045), 4A (INCSTAR Co, MN 55082-0285 USA) y Aldosterona (immunotech 177 13276, France). Los rangos de normalidad de las poblaciones estudiadas han sido obtenidos, tras comprobar la normalidad y aleatoriedad de los datos obtenidos a partir de dicha población, mediante los test d'Agostino y de Hart. Se han establecido dichos rangos con un intervalo de confianza del 95 %, representando los mismos al 90 % de la población. La existencia de diferencias entre los resultados obtenidos en los grupos de sexo sometidos a estudio se han analizado mediante un test de análisis de varianza (ANOVA), habiéndose aceptado la significación de las diferencias entre los mismos para una p < 0,05.
Resultados:
Cortisol (ng/mL)Niños Niñas Ambos: (=)(=)30-118
ACTH (pg/mL)--6-108
DHA-S (ng/mL)--1566-4618
17-OHP (ng/mL)--5-40
11-desoxicortisol (ng/mL)--23-68
ARP (ng/mL/h)--11-55
Aldosterona (pg/mL)--360-1700
Testosterona libre (pg/mL)2,0-15,61,4-3,2-
4A (ng/mL)--0,35-9,32
Las únicas diferencias significativas entre sexos corresponden a los niveles de Testosterona libre (P < 0,05), no existiendo diferencia significativa para el resto de las hormonas estudiadas
Conclusiones: No se constatan diferencias de concentración en función del sexo para ACTH, cortisol, DHA-S, 17-OHP, 11-desoxicortisol, ARP, aldosterona y 4A. Los niveles de testosterona libre presentan diferencias significativas entre sexos. El establecimiento de valores de referencia adecuados y la utilización de relaciones entre las tasas de precursores y de metabolitos permite la adecuada detección de alteraciones hormonales en el eje suprarrenal. Podemos concluir que la utilización de una metodología analítica multicomponente puede ayudar al correcto diagnóstico de la mayoría de las alteraciones metabólicas neonatales ligadas a esteroides.
Fibrinolisis alterada en el niño obeso hiperinsulinémico
M. Valle Jiménez, R. Martos Estepa, F.J. Ruz Rodríguez, F. Gascón Luna, F. Bermudo Guitarte y R. Cañete Estrada
Hospital Comarcal Valle de los Pedroches, Pozoblanco, Centro de Salud de Pozoblanco, Pozoblanco.
Introducción: El PAI-1, el mayor inhibidor fisiológico de la fibrinolisis endógena, se encuentra aumentado en el sujeto obeso, pudiendo ser el responsable del deterioro progresivo de la actividad fibrinolítica que se produce con el incremento de peso. Existen evidencias clínicas y experimentales que lo relacionan con el desarrollo de aterotrombosis. Nosotros hemos descrito en niños obesos prepúberes concentraciones significativamente elevadas de PAI-1, t-PA y fibrinógeno, respecto de niños no obesos. Tomando como valor de referencia la concentración basal de insulina de éstos niños no obesos, en este trabajo separaremos el grupo de niños obesos en normo e hiperinsulinémicos, y estudiaremos en ellos algunos aspectos de la hemostasia.
Pacientes y métodos: Estudiamos 17 niños obesos (IMC superior al p90 para su edad y sexo) hiperinsulinémicos (insulina basal superior a la media más dos SD de la insulina basal en niños no obesos) y 44 obesos normoinsulinémicos. Todos de edades emprendidas entre seis y nueve años. Determinamos la concentración banal de insulina (enzimoinmunoanálisis de micropartículas), glucosa (hexoquinasa), t-PA, PAI-1 (enzimoinmunoanálisis), y fibrinógeno (ensayo funcional usando trombina).
Resultados y conclusiones:
Obesos Obesos Normoinsulinémicos Hiperinsulinémicos
(n = 44)(n = 17)p
Edad (años)7,62 ± 1,147,89 ± 0,240,884
IMC (kg/m2)21,88 ± 0,3323,54 ± 0,310,005
Índice cintura/cadera0,837 ± 0,0070,867 ± 0,010,023
Glucosa (mg/dl)85,46 ± 0,8988,06 ± 1,120,108
Insulina (mU/ml)6,33 ± 0,3613,04 ± 0,79 < 0,001
t-PA (ng/ml)3,76 ± 0,244,11 ± 0,260,407
PAI-1 (ng/ml)34,5 ± 3,2447,5 ± 4,100,029
Fibrinógeno (g/l)2,86 ± 0,092,87 ± 0,140,953
Se representa la media ± el error estándar de la media.
No encontramos diferencias entre los dos subgrupos de obesos en la concentración de fibrinógeno, a diferencia de lo que ocurre entre obesos y no obesos. No obstante, los niños obesos hiperinsulinémicos pueden constituir (dentro de los niños obesos) una subpoblación de mayor riesgo aterogénico, por presentar niveles superiores de PAI-1, parámetro relacionado con un deterioro de la actividad fibrinolítica.
Lipodistrofia parcial congénita. Presentación de un caso
L. Ortiz González, E. Navarro Hinojosa, A. Vilela, J. Parra, J.F. Mesa, M. Portillo, I. Saez y J.M. Arroyo Fernández
Hospital General de Mérida, Mérida.
Introducción: La lipodistrofia parcial congénita es un trastorno infrecuente caracterizado por una pérdida gradual y simétrica del tejido adiposo subcutáneo de la cara, con afectación variable del tórax, abdomen, caderas y brazos, respetando las extremidades inferiores. Predomina en el sexo femenino (4:1). Comienza generalmente a partir de los 5 años y siempre en la cara, que puede adoptar un aspecto cadavérico, con un establecimiento completo del cuadro al llegar a la pubertad. En algunos casos tiene lugar una hipertrofia simultánea de la grasa subcutánea de la mitad inferior del cuerpo con una notable desproporción con respecto a la mitad superior del mismo. En otros casos puede existir una hemilipodistrofia que afecta a la mitad de la cara o del cuerpo. En algunos pacientes se han observado trastornos renales, alteraciones del metabolismo hidrocarbonado y lipídico, hipocomplementemia y síntomas neurológicos centrales. Es una entidad de causa desconocida y en la actualidad no se dispone de tratamiento eficaz.
Presentamos un caso de una niña afecta de una lipodistrofia parcial congénita sin otras alteraciones endocrinas, renales o neurológicas acompañantes en la actualidad.
Caso clínico: Niña de 6 años de edad que consulta por pérdida progresiva de grasa facial de un año de evolución. Antecedentes familiares: sin interés clínico. Antecedentes personales: embarazo controlado sin patologías. Parto eutócico. Peso RN: 3.500 g. Período neonatal sin incidencias. Curvas de peso y talla desde el nacimiento mantenidos en el P25. Desarrollo psicomotor normal. Escolarización normal y rendimiento académico satisfactorio. Exploración física: excelente estado general. Buena coloración de piel y mucosas. Atrofia de grasa cutánea en cara con respeto del resto de la economía corporal. Ausencia de hipertricosis y acantosis. No adenopatías ni edemas. ACR normal. Abdomen blando, depresible, no doloroso a la palpación, no masas ni organomegalias. Genitales externos normales de mujer. Exploración neurológica normal. ORL normal. Aparato locomotor normal. Exámenes complementarios: Hemograma normal. VSG (1ª hora): 21 mm. Glucosa, urea, creatinina, sodio, potasio, calcio, fósforo, proteinograma, perfil hepático y lipídico: normales. Hormonas tiroideas, FSH, LH, PRL, PTH, cortisol, estradiol, DHEAS: normales. GH basal y tras ejercicio:1,6 y 6,3 mcg/L respectivamente (normales). Somatomedina C: 106 UI/L (normal). Insulina, Péptido C y GH basales y tras SOG dentro de límites normales. Edad ósea correspondiente a la edad cronológica. Rx. cráneo y ecografía abdominal normales. Cariotipo: 46 XX, normal.
La evolución general de la niña es favorable sin que se hayan manifestado en la actualidad signos o síntomas relacionados con los posibles trastornos descritos asociados a esta entidad.
Comentarios: La lipodistrofia parcial congénita es una enfermedad con un buen pronóstico general sobre todo si no existen otras alteraciones orgánicas acompañantes a las manifestaciones faciales. Desde el punto de vista estético, especial atención requiere la repecursión psicólogica y social de la enfermedad. En este sentido, dado el carácter progresivo del proceso y su máxima expresión en la adolescencia, conviene considerar el apoyo de un equipo multidisciplinar.
Hipogonadismo Hipogonadotrófico Secundario a Malformación Hipofisaria en una niña con Trisomía 9p
C. Mata Fernández, C. Azcona San Julián, V. Alzina de Aguilar y J. Narbona García
Clínica Universitaria de Navarra, Pamplona.
El hipogonadismo hipogonadotrófico puede ser ocasionado por una alteración del eje hipotálamo-hipofisario de etiología muy diversa, que da lugar a una secreción insuficiente de gonadotrofinas. Presentamos el caso de una niña con hipogonadismo hipogonadotrófico secundario a malformación hipofisaria, asociado a otras manifestaciones clínicas, cuyo cariotipo muestra una trisomía 9p.
Recién nacida a término, fruto de un embarazo con retraso moderado del crecimiento desde el séptimo mes de embarazo. El peso de recién nacida es de 2.710 g (p3-10), la longitud de 48 cm (p10-25) y el perímetro craneal de 31,5 cm ( < p3). Al nacimiento se aprecian rasgos dismórficos: microcefalia, facies tosca, nariz prominente, orejas de implantación baja, clinodactilia del quinto dedo bilateral, hiperlaxitud articular y tendencia al equinovaro. El cariotipo realizado en sangre periférica muestra cromosomopatía tipificada como trisomía 9p, heredada de su madre, cuyo cariotipo es: 46XX,inv(9),t(9;12)(q13;q24).
Su desarrollo psicomotor a lo largo del seguimiento realizado es limítrofe, y se diagnóstica de una hipoacusia neurosensorial bilateral con pérdida en frecuencias conversacionales de 35-40 dB, así como de una disfasia mixta (agnosia verbal auditiva y apraxia bucal). La paciente es referida a nuestra consulta de Endocrinología Pediátrica a los 13,5 años de edad. Su desarrollo estaturoponderal muestra un descenso en la talla del percentil 50 al percentil 3, desde los 6 hasta los 13,5 años. La edad ósea muestra dos años de retraso respecto a la edad cronológica, así como desmineralización ósea difusa. La paciente no muestra signos de desarrollo puberal y presenta un enlentecimiento de la velocidad de crecimento (p3), con una relación segmento superior/inferior de 1, por lo que se determinan gonadotropinas basales, estradiol, IGF-I e IGFBP-3, detectándose niveles casi inapreciables de gonadotropinas y estradiol. Los niveles de IGF-I se encuentran en -1DS y los de IGFBP-3 en (M/+1DS) en relación a su edad ósea. Se realiza un test de LHRH que muestra una respuesta plana. La secreción de GH tras estímulo con Clonidina es normal. La resonancia magnética cerebral muestra una silla turca de aspecto vacío con una adenohipófisis hipoplásica, sin identificarse la neurohipófisis, y una anomalía de la fosa posterior compatible con síndrome de Dandy-Walker. En la ecografía pélvica se observa una estructura tubular correspondiente a un útero rudimentario, sin identificarse anejos. La densitometría ósea realizada muestra osteopenia (DEXA lumbar -2,12 DS). Se inicia tratamiento sustitutivo con estrógenos.
El hipogonadismo hipogonadotrófico (HH) es la segunda causa de retraso puberal. Puede ser de origen congénito o adquirido, y dentro de las causas congénitas deberse a un déficit aislado de gonadotrofinas o asociarse un síndrome polimalformativo. En un 5-10% de pacientes se han identificado mutaciones en distintos genes responsables de la producción de gonadotropinas. El HH de origen autosómico recesivo puede ser causado por mutaciones en el gen del receptor de las gonadotropinas, de la leptina y de su receptor. Deficiencias aisladas de FSH y LH pueden deberse a mutaciones en la subunidad beta. Existen también deficiencias combinadas de hormomas hipofisarias originadas por mutaciones en el gen PROP-1 y HESX, ambas incluyen HH. Los signos clínicos asociados al hipogonadismo pueden representar pistas para localizar el gen causante, sin embargo, en nuestro caso las diversas manifestaciones clínicas asociadas no podemos encuadrarlas dentro de ningún síndrome ni podemos asegurar que sean debidas a la anomalía cromosómica que presenta.
Valoración de la mineralización ósea mediante osteosonografía digital en niños y adolescentes con antecedente de parto pretérmino
C. Mata Fernández, D. Jiménez Villarreal, C. Azcona San Julián, J. Gimeno Ballester, E. Burghard, M. Portas y V. Alzina de Aguilar
Clínica Universitaria de Navarra, Pamplona.
Los principales factores implicados en la adquisición de una adecuada masa ósea son los nutricionales (principalmente la ingesta de calcio y vitamina D), hormonales y genéticos. La osteosonografía (medición de la mineralización ósea mediante ultrasonidos) mide la velocidad de trasmisión del ultrasonido en la metáfisis distal de la falange proximal de los cuatro dedos de la mano no dominante. Se elige este lugar por poseer un recambio óseo de características similares al tejido óseo corporal, y ser muy sensible a las variaciones en el metabolismo mineral. Este es un método nuevo que posee un coeficiente de variación muy bajo (CV = 0,7%) confiriendo gran precisión y fiabilidad a la medida.
El objetivo de este trabajo es valorar la mineralización ósea en niños con antecedente de parto pretérmino mediante osteosonografía digital así como medir la ingesta diaria de calcio, vitamina D y el ejercicio físico realizado por estos niños.
La mineralización ósea se ha valorado mediante osteosonografía digital (equipo DBM Sonic-1200), en la metáfisis distal de las falanges proximales del 2º al 5º dedos de la mano no dominante, obteniendo la velocidad del sonido a través del hueso amplitud dependiente (Ad-SOS, m/s) y utilizando como valores de referencia los realizados por Gimeno et al (1999).
Se recogen al nacimiento, la edad gestacional , el peso y la longitud, factores de riesgo de osteopenia (retraso de crecimiento intrauterino y tratamiento con corticoides en el período neonatal). Se determina la edad en el momento de la medición de la masa ósea, el peso, la talla, y el índice de masa corporal (IMC). Así mismo, se realizó una encuesta nutricional para el cálculo de la ingesta diaria de calcio y del grado de ejercicio físico. El grado de actividad física se determinó según la escala modificada de Kröger y colaboradores: Clase I: Actividad física sólo en el colegio. Clase II: Realización de 3 a 5 horas de deporte semanal extraescolar. Clase III: Realización de más de 5 horas de deporte semanal extraescolar.
Los resultados se expresan en media (intervalo de confianza). Se estudian 43 niños (11 niños y 32 niñas) de edad media 8,7 años (IC 95%: 7,4; 10,0) cuya edad gestacional fue de 32,2 semanas (IC 95%: 31,2; 33,1), la longitud de recién nacido de 41,7 cm (p25) (IC 95%: 40,0; 43,4) y el peso de recién nacido de 1798,2g (p50) (IC 95%: 1628,5; 1968,0). Patología en el período neonatal: 8 niños recibieron tratamiento con corticoides, 7 niños presentaron enfermedad de membrana hialina, 3 niños están afectos de encefalopatía neonatal, y 3 niños mostraron retraso del crecimiento intrauterino. Datos antropométricos en el momento de la determinación de la mineralización ósea: Talla-SDS: 0,5 (IC 95%: 0,1; 1,0); peso-SDS: 0,5 (IC 95%: 0,2; 0,9); IMC-SDS: 0,4 (IC 95%: -0,1; 0,7). La Ad-SOS-SDS: -0,2 (-0,7; 0,3). La ingesta diaria media de calcio y de vitamina D es de 1012 mg (IC 95%: 907; 1118) y 10,5 mcg (IC 95%: -0,5; 21,4), esta últiam inferior (p < 0,001) a los requerimientos recomendados (RDA, 1989). La media del ejercicio físico realizado es de 2 horas semanales. Los valores de mineralización ósea (Ad-SOS, m/s) se correlacionan de forma positiva con la edad (r = 0,80; p < 0,001), con la talla (r = 0,74; p < 0,001) y con el peso (r = 0,67; p < 0,001). Se establece una correlación negativa entre la edad y las horas de ejercicio físico (p < 0,001). El número de horas de ejercicio físico es menor cuanto mayor es la edad del niño (r = 0,76; p < 0,05).
La mineralización ósea valorada mediante osteosonografía está por debajo de la media para la edad en niños con antecedente de parto pretérmino. La ingesta de vitamina D en este grupo de niños es inferior a la cantidad diaria recomendada (RDA, 1989). Sin embargo la ingesta diaria de calcio es adecuada. No se observa una relación estadísticamente significativa entre la mineralización ósea y el ejercicio físico.
Hipoparatiroidismo idiopático en la infancia: a propósito de un caso
E.P. Torres Begara, I. González Casado, A.M. Jiménez Martín, M. Cabrera Lafuente, R. Márquez Moreno, M.ªM. Hawkins Solís y R. Gracia-Bouthelier
Hospital Universitario La Paz, Madrid.
Introducción: El hipoparatiroidismo es una patología que se produce como consecuencia de alteraciones en la síntesis o la secreción de la PTH, o en la acción periférica de ésta en los órganos diana ( pseudohipoparatiroidismo). La clínica es variada y depende sobre todo de la hipocalcemia, que se manifiesta fundamentalmente como tetania. A continuación describimos un caso de hipoparatiroidismo idiopático en un varón de 13 años de edad.
Caso clínico: Niño de 13 años que comienza de forma brusca con parestesias en miembros y calambres musculares, sin otra sintomatología acompañante. A la exploración presenta hipertonía generalizada, hiperreflexia, fasciculaciones y signos de Trousseau y de Chvostek positivos. Exploraciones complementarias: Hemograma normal; bioquímica sanguínea e ionograma: Ca 6 mg/dl, P 8,2 mg/dl, Ca iónico 0,6 mmol/l, Mg 1,7 mg/dl, resto normal; orina: Calciuria 0,23 mg/kg/día, Fosfaturia 28 mg/kg/día; metabolismo del hierro: normal; PTHi en plasma: 16 pg/ml( N: 20-60); TSH 0,93 uUI/ml, T4 l 0,74 ng/dl; Cortisol urinario: 20 mcg/24 h; Vitamina D:13 ng/ml; Acs antitiroideos y antiparatiroideos negativos; Ecografía de tiroides y paratiroides normal; Rx abdomen simple y serie ósea normales; Cariotipo 46 XY,CATCH 22 negativo. Se detectó mutación en el exón 2 del gen de la mioadenilato desaminasa (en heterocigosis). Se inició tratamiento con gluconato cálcico al 10% intravenoso lento a dosis de 9 mg de calcio/kg de peso, con monitorización en cuidados intensivos, pasándose a las 48 horas a calcio vía oral. Las cifras de Ca y P se normalizaron completamente a los 9 días. Se instauró tratamiento domiciliario con vitamina D (calcitriol), aporte oral de calcio y restricción de alimentos ricos en fósforo.
Conclusiones: A pesar de no ser frecuente en la edad infantil, ante una hipocalcemia debe descartarse siempre un hipoparatiroidismo. La etiología del hipoparatiroidismo es múltiple: en el período neonatal suele deberse a agenesia o aplasia de las paratiroides, mientras que más tardíamente suelen ser de causa autoinmune o idiopática. No hay descritas en la bibliografía relaciones entre el hipoparatiroidismo y el gen de la mioadenilato desaminasa, cuyo déficit ocasiona miopatía.
Diabetes tipo 1 en la edad pediátrica: características al debut
M.R. Jiménez Liria, E. García García, B. Chamizo Moreno, M.M. Sánchez Gutiérrez, F. Lendínez Molinos, J.L. Gómez Llorente, J. Aguirre Rodríguez, J. Momblán de Cabo, M.ªJ. Belmonte Martín y R. Rodríguez López
Hospital Torrecárdenas, Almería.
Objetivos: Describir las características al debut de los niños menores de 14 años con diabetes tipo 1. Comparar la situación clínica al diagnóstico en tres grupos de edad y dos períodos de tiempo consecutivos.
Material y métodos: Estudio retrospectivo de las historias clínicas de los niños menores de 14 años diagnosticados de diabetes tipo 1 en nuestro servicio, en la última década. Se recogieron variables epidemiológicas (sexo, edad al debut, mes de nacimiento y mes de debut) y clínicas (tiempo desde inicio de síntomas de hiperglucemia y presencia de cetoacidosis). Se agruparon los pacientes en tres grupos de edad al debut y en dos períodos de tiempo de cinco años, anterior y posterior al 1995.
Resultados: Se incluyeron 70 pacientes (39 varones), de 4,9 años de edad media (Rango 1-11,9 años). El 39% nacieron en los meses de verano, y el 34% debutaron en los meses de invierno. El tiempo medio desde el inicio de los síntomas de hiperglucemia fue de 1,2 meses y el 37% presentaban cetoacidosis.
Las variables en los tres grupos de edad fueron las siguientes:
< 5 años5-10 años> 11 años
Tiempo inicio síntomas (meses)1,570,430,60
Cetoacidosis13/377/192/7
En el período anterior a 1995 presentaron cetoacidosis 7/22 y en el posterior 11/34.
Conclusiones: Destaca el prolongado tiempo desde el inicio de los síntomas de hiperglucemia, hasta el diagnóstico de diabetes, sobre todo en el grupo de edad de menores de 5 años. Un 37 % debutan en situación de cetoacidosis, sin diferencias en los tres grupos de edad ni en los dos períodos de tiempo.
Infectología
Estudio de diferentes aspectos de la mortalidad pediátrica debida a procesos infecciosos en nuestro departamento durante quince años (1985-1999)
J.L. Ruibal Francisco, B. de Miguel Lavisfer, M.T. Lacasa Pérez, O. Pérez Rodríguez y C. Martínez Hernández
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: Las enfermedades infecciosas representan la tercera causa de muerte pediátrica en nuestro departamento. Suponen el 11% del total de éxitus. En este trabajo se han investigado diferentes aspectos sobre la mortalidad de los niños debida a dichos procesos en el período de tiempo comprendido entre los años 1985-1999.
Material y métodos: Se han analizado de manera retrospectiva, las historias clínicas de todos los pacientes pediátricos fallecidos en relación con procesos infecciosos con edades comprendidas entre el mes de vida y los 18 años de edad, en el período de tiempo transcurrido entre el 1 de enero de 1985 y el treinta y uno de diciembre de 1999. Se han estudiado el número total de pacientes fallecidos, la etiología de sus fallecimientos, la causa inmediata de muerte que condicionó su muerte, y su distribución por sexo y grupos de edad. Los datos se han expresado como estadística básica (media, desviación típica, máximo y mínimo). Asimismo en datos absolutos y porcentajes sobre el total de los analizados.
Resultados: Los pacientes fallecidos por patología infecciosa han sido 14 de los que 9 eran varones (64%) y 5 mujeres (36%). La edad media a la que murieron fue de 4,6 ± 5,1 (15,5, 0,15) años. El 64% (9) fallecieron entre los 0 y 5 años de edad, el 21% (3) entre los 5 y 10, el 7% (1) entre los 10 y los 15, y también el 7% (1) con más de 15. Dichos procesos se produjeron en tres pacientes inmunodeficientes (uno con una anemia aplásica, otro con un SIDA, y otro con una inmunodeficiencia combinada severa), y en 9 inmunocompetentes. La etiología de estos procesos fue por sepsis y/o meningitis meningocócicas en 6 (44%), en 2 casos(14%) por bronquiolitis, y en uno respectivamente (7%), por una gastroenteritis aguda que originó una deshidratación hipernatrémica, por una epiglotitis, por una sepsis por pneumocistis carinii, por una encefalitis herpética, por una septicemia por pseudomonas aureoginosa, y por una sepsis por el virus herpes tipo I. La causa inmediata del fallecimiento de estos pacientes fue en 7 casos (50%) por muerte cerebral, por fracaso multisistémico en 4 (29%) y en 3 (21%) por insuficiencia respiratoria.
Conclusiones: Las enfermedades infecciosas suponen una importante causa de mortalidad en niños menores de 5 años El 64% fallecen por debajo de esa edad. Existe una preponderancia a favor de los varones. La mayor parte de ellos se deben a sepsis y/o meningitis meningocócicas y lo hacen por muerte cerebral.
Validez del test inmunocromatográfico Binax para detección del antígeno de neumococo en orina, en niños con neumonía lobar
S. Guillén Martín, C. Ruano Fajardo, L. Molina, E. Salto, M.J. Martín Puerto, E. Giangaspro Corradi y J.T. Ramos Amador
Hospital 12 de Octubre , Madrid.
Introducción: La enfermedad neumocócica tiene gran importancia en la infancia, siendo la primera causa de neumonía bacteriana en menores de 5 años. Existe una gran dificultad para el diagnóstico etiológico de las neumonías, debido a la baja rentabilidad de los hemocultivos y de las técnicas clásicas de detección de antígenos, junto a la inespecificidad de las pruebas complementarias.
Material y métodos: El período de estudio comprende de febrero a noviembre del 2000. Los criterios de inclusión eran niños con neumonía lobar a quienes se realizó la técnica inmunocromatográfica Binax en orina no concentrada en las primeras 72 horas tras el diagnóstico de neumonía. Se consideraron sujetos control a 30 niños en el preoperatorio de cirugía, con una mediana de edad de 50 meses (Rango: 1-165 meses).
Resultados: En total, se identificaron 64 pacientes con diagnóstico de neumonía lobar con una mediana de edad de 24 meses, en un rango entre 5 y 167 meses. En 8 hubo derrame pleural. Del total, 46 tenían más de 15.000 leucocitos, en 28 de ellos el Binax fue positivo. En estos pacientes, la mediana de la proteína C reactiva fue de 12 mg/dl (Rango: 1,9-12 mg/dl). Esto demuestra una sensibilidad del 61%, con un intervalo de confianza del 95%, cuyos límites son 46% y 74%. Si consideramos el conjunto de neumonías, la técnica Binax fue positiva en 34 niños, esto supone una sensibilidad del 55%, con un intervalo de confianza del 95%, cuyos límites son 42% y 67%. De los 30 controles, la prueba fue positiva en 3 (10%), una especificidad del 90% con un intervalo de confianza del 95%, cuyos límites son 75% y 97%.
Conclusiones: 1) La técnica inmunocromatográfica de detección de antígeno de neumococo en orina, es útil en el diagnóstico etiológico de neumonías lobares. 2) Son necesarios más estudios, con mayor número de pacientes, para definir la validez de la prueba.
Epidedimitis por E. coli en un lactante de 5 meses
F. Pagone, G. Castillejo, J.S. Escribano, O. Salvadó, R. Jiménez, J.M. Barroso, J. Manuel y J. Montero
Hospital Universitario Sant Joan de Reus, Reus.
Introducción: La epididimitis es un hallazgo inusual antes de la pubertad y su identificación es importante debido a que los síntomas recuerdan a los de la torsión testicular. Presentamos un caso de epididimitis por E. coli en un lactante de 5 meses, sin malformaciones congénitas del tracto genito-urinario asociadas.
Caso Clínico: Lactante varón de 5 meses que consulta por inflamación testicular derecha e irritabilidad. E.F.: T axilar: 38,2 ºC. Irritable. Testículo izquierdo normal. Testículo derecho aumentado de tamaño, de consistencia dura, con eritema, calor y dolor a la palpación.
Hemograma: Hcto: 36,4%; Hb: 11,6 gr/L; Leucocitos: 22.700/mm3; S:69%; L:5,8%;
PCR: 38 mg/L. Sedimento orina: normal.
Ecografía testicular: Testículo derecho aumentado de tamaño de ecogenicidad heterogénea, con áreas hipoecóicas y flujo prominente. Aumento de flujo en parénquima testicular, epidídimo y cubiertas. Testículo izquierdo normal.
Urocultivo: E. coli.
Ecografía renal: normal. CUMS: normal.
Inicia tratamiento con ampicilina y gentamicina endovenosos, presentando remisión completa de la fiebre y disminución importante de los signos inflamatorios locales en las primeras 24 horas. Alta hospitalaria a los 9 días.
Comentarios: La inflamación aguda del epidídimo es una infección retrógada ascendente que, partiendo de la uretra y tras pasar por el deferente, llega al epidídimo. Los síntomas y los hallazgos físicos pueden ser similares a los de la torsión testicular: eritema escrotal, edema y dolor. Los datos de laboratorio como fiebre, leucocitosis, piuria y bacteriuria sugieren el diagnóstico de epididimitis. En niños en edad prepuberal, Enterobacterias y Pseudomonas representan la causa más común de epididimitis. Con frecuencia la infección está relacionada con anomalías estructurales predisponentes del tracto genito-urinario.
Una vez que se confirma un epididimitis en edad prepuberal, debe realizarse una ecografía renal a fin de descartar malformaciones congénitas asociadas. Hallazgos positivos como anormalidades ureterales o de vasos deferentes se han reportado en aproximadamente una cuarta parte de los casos.
Tuberculosis pulmonar y ganglionar generalizada por Mycobacterium africanum
M.L. Murga Sierra, M.A. García Herrero, A.M. Carrasco Torres, C. Torrijos Román, S. Vicente Mañanes, M.A. Ramírez Escobar
y E. García de Frías
Hospital Universitario Príncipe de Asturias, Alcalá de Henares.
El Mycobacterium africanum (MA) es el agente causal del 60-80% de los casos de tuberculosis en África central y en menor proporción en algunas zonas de Asia. En Europa su aislamiento es raro, aunque cada vez se describen mas casos debido a los movimientos migratorios.
Caso clínico: Niña de 8 años, de raza negra, procedente de Guinea Ecuatorial, que presenta en los últimos 8 meses fiebre hasta 39° y masa axilar derecha, con anorexia y pérdida ponderal. En sus antecedentes destacan varios episodios de paludismo tratados periódicamente, fiebre tifoidea hace 4 años y dos transfusiones sanguíneas; varios familiares han padecido tuberculosis. En la exploración al ingreso presentaba fiebre de 38° y gran deterioro del estado nutricional; destacaba una tumoración axilar de 4 cm, sin signos inflamatorios locales y con exudación amarillenta en zona de punción (prueba diagnóstica en su país de origen). Su auscultación pulmonar y el examen neurológico fueron normales. Su abdomen estaba distendido y con hepatomegalia de 20 mm. La analítica sanguínea detectó elevación de la proteína C reactiva (107 mg/dL), anemia (7,4 g/dL), hipoalbuminemia (2,84 g/dL) e hipergammaglobulinemia policlonal (4,24 g/dL). Su Mantoux fue positivo. La serología VIH fue negativa. La gota gruesa fue negativa para parásitos en sangre y se detectaron varios parásitos intestinales. En la Rx torax presentaba ensanchamiento mediastínico y adenopatías hiliares. El TC torácico detectó un infiltrado en lóbulo superior derecho y múltiples adenopatías axilares y supraclaviculares derechas junto a mediastínicas e hiliares bilaterales. El TC abdominal demostró hepatoesplenomegalia homogénea y múltiples adenopatías mesentéricas y en hilio esplénico. El estudio anatomo-patológico de la lesión axilar fue compatible con linfadenitis granulomatosa necrotizante, con tinción de auramina positiva. Las baciloscopias de jugos gástricos fueron negativas pero el cultivo confirmó el crecimiento de MA. La paciente fue tratada con Isoniazida, Rifampicina y Pirazinamida con desaparición de la fiebre a las 48 horas y disminución progresiva de las adenopatías axilares.
Conclusiones: 1) Destacamos la excepcionalidad del caso tanto por su severidad clínica como por las escasas infecciones por MA comunicadas en nuestro país. 2) Es importante epidemiológicamente la identificación de la especie porque permite detectar el tipo de brote y aplicar medidas de control.
Brote epidémico de parotiditis en una población de Madrid
R. Rodríguez Barrientos, M.G. Tamayo López, J.M. Pérez Arévalo, F. de la Vega Jiménez, M. López Tomás y G. Bettas Ferrero
Centro de Salud Rosa de Luxemburgo, San Sebastián de los Reyes, Centro Epidemiológico del Área 5 de la Comunidad de Madrid, Madrid.
Introducción: Presentamos un brote epidémico de parotiditis, que afecto a dos colegios de San Sebastián de los Reyes (Madrid), entre noviembre y diciembre del año 2000. Un total de 20 niños presentaron clínica compatible (tumefacción de una o ambas parótidas, acompañado de fiebre o febrícula) a pesar de estar correctamente vacunados.
Objetivos: Describir la presentación de un brote de parotiditis aguda en una población de niños ya vacunados, planteándonos su aparición con una posible menor inmunogenicidad de la vacuna con la cepa Rubini (Triviraten, Bema). Relacionar el brote epidémico de nuestra población con el aumento del número de casos ocurrido dentro del territorio de la Comunidad de Madrid, donde se utilizó l esta vacuna desde noviembre de 1996 hasta mayo junio de 1999.
Descripción: En noviembre y diciembre del año 2000 se detectaron en dos colegios de la localidad madrileña de San Sebastián de los Reyes niños que presentaron criterios clínicos de parotiditis. El número de casos fue de 20 en edades comprendidas entre 2 y 10 años. Se informo de ello a la Consejería de Sanidad de la Comunidad de Madrid, cuya actitud fue vacunar con la cepa Jeryll-Lynn (Priorix o MSD) a todos los contactos susceptibles de los casos, que no hubiesen recibido 2 dosis de vacuna con la cepa Jeryll-Lynn. La evolución en todos los casos fue favorable, no presentando ninguna complicación. Se comprobó que se habían producido más casos en el Área 5 (zona norte de la Comunidad de Madrid con una población de 600.000 habitantes, y una población pediátrica cercana a los 85.000 niños). El número de casos de parotiditis había aumentado en los últimos tres años, 1998 con 7 casos, 1999 con 18 casos y año 2000 con 120 casos. Podemos hablar por lo tanto de una situación generalizada de progresivo aumento del número de brotes. La revisión bibliográfica sobre la vacuna Rubini indica su baja capacidad inmunógena con respecto a la otra cepa que se utiliza habitualmente, cepa Jeryll-Lynn.
Conclusiones: La vacunación con la cepa Rubini (Triviraten, Berna), no es lo suficientemente inmunógena, permitiendo la aparición de brotes epidémicos. Ante un brote, la actitud correcta es vacunar con la cepa Jeryll-Lynn a todos los contactos susceptibles que no tuviesen dos dosis de la cepa Jeryll-Lynn (MSD o Priorix).
Intensa rabdomiolisis en el contexto de un síndrome del shock tóxico asociado a herida cutánea
A. Díaz Moro, L. Castañón López, J. Mallo Castaño, J.M. Marugán de Miguelsanz, J.P. Martínez Badás, C.N. Villafañe y H. González Aparcio
Hospital de León, León.
Introducción: El síndrome del shock tóxico (SST) es una enfermedad multisistémica aguda y grave, rara en la edad pediátrica, asociada a infección por Staphylococcus aureus y otros gérmenes, y mediada por toxinas. El diagnóstico es exclusivamente clínico, y se caracteriza por fiebre, hiperemia de mucosas y eritrodermia descamativa, que rápidamente progresa a hipotensión, shock, compromiso multiorgánico e incluso la muerte. La afectación muscular forma parte de las manifestaciones sistémicas de la enfermedad y se expresa desde el punto de vista bioquímico por una elevación variable en los niveles de los enzimas musculares, fundamentalmente de la creatinkinasa (CK).
Caso clínico: Niña de 3 años de edad, que 24 horas antes de su ingreso comienza bruscamente con fiebre, vómitos, diarrea profusa, oliguria y postración progresiva. Traumatismo leve en región frontal cuatro días antes.
Exploración física: Tª 39ºC, FC 150 lat/min, TA 75/35 mmHg. Afectación grave del estado general, mucosas secas, exantema generalizado escarlatiniforme, mala perfusión periférica. Herida inciso-contusa exudativa en región frontal izquierda. Orofaringe congestiva. No meningismo ni focalidad neurológica, alterna letargia con fases de agitación y lenguaje incoherente. Glasgow 12. Resto de la exploración normal.
Exploraciones complementarias: Leucocitos 10.600/mm3 (26N, 4L, 57C, 11 Metamielocitos, 2 Mielocitos), plaquetas 31.000/mm3, VSG 21, PCR 338. Afectación renal prerrenal: Urea 153 mg/dl, Cr 2,5 mg/dl, Na 124 mEq/l y EFNa 0,17%. Hipocalcemia (Ca total 6,9 mg/dl y Ca iónico 2,7 mg/dl), hipofosforemia (P 2,6 mg/dl) e hipoproteinemia (proteínas totales 4,4 gr/dl y albúmina 2,7 gr/dl). Afectación hepática: ALT/AST 250/550 U/L. Acidosis metabólica: pH 7,28, bicarbonato 14 mmol/L, EB -12 mmol/L. Coagulopatía: T.Cef 55 seg, T.P 34%, fibrinógeno normal. Amonio en plasma normal. Orina: hematuria +++ por tira reactiva, con sedimento normal. TAC craneal y LCR normales. Cultivo del exudado de la herida: S. aureus. Hemocultivos, cultivo LCR, frotis faríngeo, exudado nasal para virus respiratorios, coprocultivo y estudio virológico en heces negativos. ASLO negativo. Serología Sarampión, Rubéola, Echovirus, Coxackie B, Rickettsia y Leptospira negativa.
Evolución: tratamiento con expansión de la volemia, corrección de las alteraciones hidroelectrolíticas, y antibioterapia iv. A las 24 horas, presenta dolor intenso en masas musculares, sobre todo de extremidades inferiores, contractura, edema, signos inflamatorios locales e impotencia funcional; CK 21.800 U/L, LDH 2.369 U/L, y aldolasa normal. Al 8º día, se observa descamación furfurácea de predominio en manos y pies. La evolución posterior fue favorable desde el punto de vista clínico y analítico, con descenso progresivo de los niveles de CK y normalización paulatina del resto de las anomalías hematológicas y bioquímicas.
Comentario: El SST está desencadenado por la producción y liberación masiva de exotoxinas a partir de un foco séptico, en general evidente (heridas quirúrgicas, neumonías, sinusitis, empiemas, fascitis, osteomielitis, abscesos), aunque a veces puede ser insignificante o incluso pasar inadvertido.
Dentro del espectro clínico de la enfermedad, la afectación muscular es a veces la manifestación más relevante, dando lugar a cuadros de miositis o rabdomiolisis, que pueden llegar a ser muy intensos.
El diagnóstico diferencial se plantea con todas aquellas entidades que cursen con síndrome febril y exantema, fundamentalmente con la enfermedad de Kawasaki, la sepsis bacteriana, la fiebre de las Montañas Rocosas, el síndrome de la piel escaldada estafilocócica, el síndrome de Stevens-Johnson y la escarlatina.
El tratamiento consiste en la reposición del espacio intravascular, soporte inotrópico y respiratorio y corrección de las alteraciones específicas. Es importante el tratamiento adecuado del foco séptico con antibióticos y desbridamiento quirúrgico si es preciso.
El hecho de que una infección local aparentemente intrascendente, pueda ser responsable de un cuadro clínico potencialmente mortal, con fracaso circulatorio y compromiso multiorgánico, justifica la importancia de un rápido reconocimiento clínico y de un tratamiento precoz y agresivo.
Osteomielitis por Salmonella grupo B en un niño previamente sano
A.M. Carrasco Torres, P. Sevilla Ramos, J.A. Gómez Carrasco, P. González Santiago, A. Lassaletta Atienza y E. García de Frías
Hospital Universitario Príncipe de Asturias, Alcalá de Henares.
Introducción: La causa más frecuente de ostomielitis aguda hematógena es el Staphylococcus Aureus. La osteomielitis por Salmonella es infrecuente en pacientes sin patología de base (drepanocitosis, hemoglobinopatías). Presentamos el caso de un niño previamente sano con osteomielitis del tarso por Salmonella, con el antecedente de gastroenteritis aguda el mes anterior.
Caso clínico: Niño de 3 años y 5 meses que consulta en urgencias por claudicación en la marcha de la extremidad inferior derecha, sin traumatismo previo. Se realiza estudio radiológico, sin alteraciones, siendo tratado con vendaje compresivo. A los 5 días se retira el vendaje sin observar mejoría y observando inicio de fiebre, por lo que se decide el ingreso.
Exploración física: Tª37, 7ºC. Buen estado general. Buena coloración de piel y mucosas. Resto de la exploración sistemática normal. Tumefacción dolorosa rojo-violacea y caliente en zona media del dorso del pie derecho.
Exploraciones complementarias: Hemograma:15900 leucocitos (N50,9%, L40%) serie roja y plaquetaria normales. Extensión periférica sin datos patológicos. Inmunoglobulinas, C3 y C4 normales. PCR: 9,6 mg/l. ASLO: 87U. Factor reumatoide, ANA, serología de VIH y Brucella : negativos. Hemocultivo y Mantoux: negativos. Coprocultivo: positivo para Salmonella grupo B sensible a ampicilina y TMP-SMX .Estudio de hemoglobinopatías: normal. Rx del pie derecho en los primeros 7 días: normal. Control a los 12 días: imagen lítica en primera cuña pie derecho. RM pie derecho: Imágenes sugestivas de infección osteoarticular (1ª, 2ª cuña y primer metatarsiano). Gammagrafía ósea con Tecnecio-99: Hipercaptación en tarso derecho, no en otros lugares. Cultivo del material biopsiado: positivo para Salmonella grupo B.
Evolución: Recibe tratamiento con Cefotaxima intravenosa ; con evolución clínica y radiológica favorable. Tiempo total de tratamiento antibiótico 13 semanas. Seguimiento ambulatorio y alta con ausencia de signos y síntomas al mes de su ingreso.
Comentario: Queremos destacar la rareza de la osteomielitis hematógena en pacientes sin patología de base; en nuestro caso con origen gastrointestinal, como portador. Conviene dejar abierta la posibilidad de que en un futuro pueda describirse algún factor de origen inmune que explique estos excepcionales casos.
Botulismo Infantil. Presentación de un caso
J.A. López Torres, A.M. Peinado Garrido, R. Ávila Villegas, F.J. Garrido Torrecillas, M. Cobo Peralta y M. Villa Hernández Gómez
Hospital Universitario San Cecilio, Granada.
Introducción: El botulismo infantil se observa en lactantes de menos de 6 meses de edad. El mecanismo de producción se atribuye a la colonización del intestino por esporas de Clostridium botulinum, que generan las toxinas. La transmisión de la enfermedad no se produce de persona a persona y el origen de las esporas es incierto, postulándose el mismo en la miel de abeja, y en jarabe de maíz, no usado corrientemente en nuestro medio. El período de incubación en el caso del botulismo infantil es desconocido. Presentamos un caso de botulismo infantil.
Caso clínico: Lactante mujer de 4 meses de edad, con antecedentes personales y familiares sin interés. Se alimenta con lactancia materna y tiene puesta la 1ª dosis de DTP+Polio. Consulta en nuestro servicio de urgencias porque hace dos días que le encuentra dificultad para abrir los párpados, seguido de irritabilidad y decaimiento progresivo, de tal modo que en el momento de la consulta afirma la madre que no puede succionar. Refiere también que hace seis días que no tiene deposiciones, habiendo tenido previamente un hábito intestinal de 1 deposición cada 2 ó 3 días. La madre no había manipulado miel ni alimentos en conserva. A la exploración presenta un Glasgow 15/15, con llanto débil y quejumbroso, las pupilas en posición media con escasa respuesta a la luz, quedando los ojos entreabiertos. En respuesta a la tracción la cabeza cuelga pasivamente hacia atrás. Hipotonía con postura en libro abierto. Los movimientos oculares extrínsecos son normales. Sigue con la mirada los objetos y el rostro. Los miembros cuelgan con escasa respuesta de flexión. Los reflejos de estiramiento rotuliano son normales. Presenta, también, rinorrea mucosa. Se realizaron pruebas complementarias, resultando el sistemático de sangre y orina normal, los cultivos centrales de sangre, orina, heces y LCR fueron negativos así como las serologías séricas y de LCR. Se realizó una TAC craneal, donde se observa una leve atrofia cortical frontal. EEG con actividad de base adecuada a su edad con algún elemento lento difuso. Se hicieron determinaciones especiales de hormonas tiroideas, amonio y lactato, tóxicos en sangre y orina que fueron negativos. El fondo de ojo fue normal. Dado el cuadro clínico tan típico y característico, tal y como refiere la literatura médica, se solicitó una determinación de toxina botulínica y esporas de Clostridium en heces que fueron positivos, lo que cerró el diagnóstico.
Discusión: Deseamos resaltar que dentro del diagnóstico diferencial de la hipotonía en la lactancia ("bebé blando"), que comprende un amplio diagnóstico diferencial que incluye patología infecciosa, neuropatías, patología medular y miopatías, no se deje de evaluar esta enfermedad como posible diagnóstico, dentro de las consideradas enfermedades de la placa motora.
Fiebre tifoidea de presentación atípica
J.M. Barroso, X. Allúe Martínez, J.S. Escribano, C. Bardají, O. Salvadó, R. Jiménez y F. Pagone
Hospital Universitario Sant Joan de Reus, Reus, Hospital Universitario Juan XXIII, Tarragona.
Introducción: La fiebre tifoidea es una enfermedad sistémica de transmisión fecal-oral con una tasa de incidencia de 1,41 casos por cien mil habitantes. Existe menos del 1% de mortalidad y como complicaciones destacan: hemorragia (15-20%), perforación intestinal (1-10%), neumonías por aspiración o por sobreinfección, meningitis, espondilitis, condritis costales, colecistitis.
Caso clínico: Paciente varón de origen magrebí de 14 años que presenta cefalea, artromialgias generalizadas, ictericia conjuntival y dolor abdominal, con vómitos biliosos y anorexia de 4 días de evolución. En tratamiento con ibuprofeno y amoxicilina/clavulánico dos días antes del ingreso.
Exploración física: T.A.:105/70 mmHg. Regular estado general, somnoliento. Ictericia conjuntival, auscultación cardiorrespiratoria normal, abdomen blando, depresible, doloroso a la palpación; se palpa hepatomegalia y polo de bazo. No adenopatías, no petequias.
Exploraciones complementarias: Analítica sanguínea: urea: 20,5 mmol/L, creatinina: 228 mcmol/L, AST: 6,11 µkat/L, ALT: 2,56 µkat/L; GGT: 2,31 µkat/L, amonio: 43 mmol/L, LDH: 31,60 µkat/L, bilirrubina total: 110 mmol/L, Quick: 130%, albúmina: 29 gr/L. Analítica de orina: cuerpos cetónicos negativos, proteínas: +, urea: 315 mmol/L, creatinina: 5998 µmol/L, Na: 10 mEq/L, FENa: 0,2, bilirrubina: ++, urobilinógeno: ++. Ecografía abdominal: hepatomegalia moderada. Serologías a virus hepatitis A, B y C, CMV, VEB: negativas. Rx tórax: normal.
Presenta cuadro de hepatitis con insuficiencia renal prerrenal que se corrige a las 48 horas tras administración de líquidos. A los dos días de hospitalización inicia fiebre de 39,5ºC , persistiendo estado somnoliento , con distensión del abdomen por lo que se repite la ecografía abdominal donde se observa líquido intraperitoneal libre y engrosamiento de las paredes de la vesícula biliar, con barro biliar y edema importante de ambos riñones. Evoluciona tórpidamente, presentando inestabilidad hemodinámica y peritonitis aguda, por lo que se interviene de urgencias, constatándose peritonitis por perforación de vesícula biliar. Se realiza colecistectomía simple, con evolución postoperatoria adecuada, mejoría clínica y desaparición de la fiebre y del cuadro de colestasis. Tras recibir el resultado del hemocultivo positivo para Salmonella thyphi, se etiqueta el cuadro como fiebre tifoidea con perforación de vesícula biliar y se completa tratamiento antibiótico hasta 14 días.
Comentario: En este paciente la fiebre tifoidea se presenta como una hepatitis, con colestasis e insuficiencia renal prerrenal. Sigue una evolución tórpida y se complica con perforación biliar. Las causas más frecuentes de hepatitis son los virus (virus hepatitis B, virus Epstein-Barr, citomegalovirus), pero también entran en el diagnóstico diferencial la brucelosis, el paludismo y la fiebre tifoidea entre otros, así como hepatitis tóxicas y linfoma. Existe una forma rara de fiebre tifoidea llamada ataxoadinámica, que es una forma grave de curso hipertóxico, postración, fiebre elevada, diarrea, deshidratación y colapso que puede ser mortal.
Complicaciones cutáneas de la varicela en niños inmunocompetentes
N. Hernández González, M.J. García Miguel, F. Baquero Artigao, M.I. de José Gómez, J. Galán de Dios, C. Borque Andrés y F. del Castillo Martín
Hospital Universitario La Paz, Madrid.
Objetivos: Describir las complicaciones dermatológicas de la varicela en niños sanos que requirieron ingreso hospitalario. Estudiar la frecuencia, tipo de lesión, etiología, localización, tratamiento y evolución de dichas complicaciones.
Material y métodos: Revisión de las historias clínicas de los niños menores de 14 años hospitalizados por complicaciones cutáneas de la varicela durante un período de 14 años (1987-2000). La obtención de muestras para cultivo bacteriológico se realizó obteniendo material purulento directamente de las lesiones abiertas o mediante frotis del lecho de la lesión una vez retirada la costra. El análisis estadístico se realizó mediante el programa informático SPSS 9.0.
Resultados: Durante el período de estudio ingresaron 323 niños sanos diagnosticados de varicela. Las complicaciones cutáneas fueron las más frecuentes, encontrándose en 110 niños (34 %). La edad media fue de 3,5 ± 2,8 años (rango 2 meses-13 años). El 52 % eran varones. Se diagnosticaron un total de 133 infecciones cutáneas: 71 impétigos (53 %), 41 celulitis (31 %), 11 exantemas escarlatiniformes (8 %), 8 abscesos (6 %) y 2 linfangitis (2 %). Las localizaciones más frecuentes fueron: generalizadas en los impétigos, cabeza-cuello en las celulitis y tronco en los abscesos. Se cultivaron 64 muestras de exudado purulento, de las cuales 48 (75 %) fueron positivas. Las bacterias aisladas fueron: S. pyogenes 22 (46 %), S. aureus 16 (33 %), S. pyogenes + S. aureus 8 (17 %) y S. viridans 2 (4 %). Se realizaron 58 hemocultivos siendo positivos 9 (15,5 %): S. pyogenes en 7 casos y S. aureus en 2 casos. La media de días de hospitalización fue de 7,1 ± 4,8 (rango 1-38). Todos los casos evolucionaron hacia la curación sin secuelas.
Conclusiones: Las sobreinfecciones cutáneas representan la primera causa de hospitalización por varicela en niños sanos. El impétigo es la complicación más frecuente, seguido de la celulitis. La bacteria más frecuentemente aislada es S. pyogenes.
Pericarditis y Encefalitis por Virus Ebstein-Barr
A. Pericas Bosch, T. Pérez Porcuna, R. Porcel Rubio, N. Cortés Álvarez, C. Figueras Nadal, M. Boronat, S. Cañadas Palazón,
M. Pujol Jover, J.M. Bertran Sangés y A. Fitó Costas
Hospital Materno-Infantil Vall d'Hebrón, Barcelona.
Introducción: La infección por virus de Epstein-Barr (VEB) suele ser con frecuencia subclínica durante la infancia y a pesar de que su curso es habitualmente benigno y autolimitado, en ocasiones puede presentar complicaciones importantes.
Objetivos: Pretendemos llamar la atención sobre las complicaciones cardíacas y neurológicas de la infección por VEB, presentando dos casos que en el último año ingresaron en nuestra Unidad.
Material y métodos: El primer caso es una niña de 11 años que debuta con un cuadro de insuficiencia cardíaca sin otra clínica previa que febrícula y odinofagia. El ecocardiograma demuestra la existencia de un derrame pericárdico que requiere pericardiocentesis y monitorización en la Unidad de Cuidados Intensivos Pediátricos durante 12 días. Tras un estudio etiológico exhaustivo, el único hallazgo es la presencia de IgM antiVCA de VEB.
El segundo caso es una niña de 14 años que ingresa por fiebre, meningismo, afasia de expresión y debilidad muscular de extremidades inferiores que evoluciona hacia un cuadro de obnubilación con un Glasgow de 13. El estudio de LCR presenta discreta pleocitosis y proteinorraquia. La RMN cerebral demuestra lesiones corticales hiperintensas plurifocales de predominio frontal y temporal. Se detecta también como único hallazgo la presencia de IgM antiVCA de VEB.
Conclusiones: Las complicaciones cardiológicas y neurológicas en la infección por VEB son infrecuentes y en ocasiones pueden constituir la primera o única manifestación de la enfermedad, dificultando su diagnóstico.
Tratamiento de corta duración de la leishmaniasis visceral en pacientes pediátricos inmunocompetentes con anfotericina B liposómica
C. Figueras Nadal, M.J. García Miguel, F. Asensi Botet, R. Velasco Bernardo, A. Canals Baeza, Ll. Ciria Calavia, C. Fortuny Guasch, L. Santos Serrano, R. Cullell Hernández y J. de la Cruz Moreno
Hospital Materno-Infantil Vall d'Hebrón, Barcelona, Hospital Universitario La Paz, Madrid, Hospital Universitario La Fe, Valencia.
Antecedentes: La leishmaniasis visceral (LV) por Leishmania infantum continúa siendo endémica en el sur de Europa , especialmente en el área mediterránea . Los compuestos antimoniales pentavalentes han sido hasta la actualidad el tratamiento de elección, pero en los último años se han registrado fracasos terapéuticos y también importantes efectos adversos que han hecho reconsiderar dicho régimen.
Davidson y col. publicaron el primer uso clínico de anfotericina B liposómica (AnL) para el tratamiento de la leishmaniasis en 1991, y en 1996 establecieron la eficacia de un tratamiento de corta duración.
Objetivo: Siguiendo la pauta propuesta por Davidson, este trabajo pretende evaluar la eficacia y seguridad de AnL a una dosis máxima de 4 mg/kg los días 1-2-3-4- 5 y 10 para el tratamiento de LV infantil.
Métodos: Desde enero de 1997 hasta septiembre de 2000, se realizó un estudio prospectivo multicéntrico con la participación de 13 hospitales pediátricos, tratándose a 32 niños inmunocompetentes, de edad comprendida entre 7 y 84 meses (media 26,03 meses) que no habían recibido tratamiento previo. Se administró AnL a una dosis de 4 mg/kg los días 1-2-3-4-5 y 10 en 28 pacientes, y a una dosis de 3 mg/kg en otros 4. Todos los pacientes tenían síntomas de LV clínicamente activa. Se realizó control analítico los días 1-4-10 y 21 y los meses 3-6 y 12, realizándose un aspirado de medula ósea (AMO) y/o test serológico (IFA o ELISA) los días 1 y 21.
El día 21 se realizó un seguimiento clínico-analítico de todos los pacientes, a los 3 meses de 24, y a los 6 meses y al año de 19 y de 16 pacientes, respectivamente.
El día 1 se observaron parásitos en el aspirado de médula ósea en 27 niños (87,1%), y en los casos en los que no se observaron (n = 4) o en los que no se pudo realizar AMO (n = 1) los anticuerpos frente a leishmania fueron positivos.
Resultados: Todos los niños (100%) tuvieron una excelente respuesta clínica y el AMO a los 21 días fue normal en los 24 pacientes (100%) en que se realizó. En los otros 8 niños la eficacia del tratamiento fue evaluada por la respuesta clínica. Durante el período de seguimiento, no se observó ninguna recidiva (0%), ni ningún acontecimiento adverso (0%).
Conclusiones: El tratamiento de LV infantil con una dosis total de 24 mg/kg de AnL es en nuestro estudio altamente eficaz y seguro. Además, destacamos la ventaja de reducir la estancia hospitalaria a únicamente cinco días, logrando una mejoría notable en la calidad de vida de los niños y sus familiares e incluso una reducción en los costes generados por una larga hospitalización.
Microabscesos esplénicos como presentación poco frecuente de Salmonelosis
E. Mora González, A. Pérez Guerrero, S. García Calatayud, A. Pérez Puente, V. Madrigal Díez, M. Otero Fernández, C. Baza Vilariño,
E. Pérez Belmonte, M. Pacheco Cumani y M.A. Andrés Fraile
Hospital Universitario Marqués de Valdecilla, Santander.
La aparición en un niño de fiebre alta y percusión renal positiva sugieren el diagnóstico de sospecha de pielonefritis aguda, que requiere confirmación mediante urocultivo y debiera acompañarse de la realización de una ecografía abdominal.
Presentamos el caso de una niña de 10 años con dos episodios de fiebre alta, en el segundo de los cuales se evidencia además percusión renal izquierda positiva y dolor en fosa ilíaca izquierda. La orina muestra nitritos y proteínas positivos y bacteriuria. En sangre destaca neutrofilia del 73%, y una proteína C reactiva de 10,7 mg/dL.
Tras recoger urocultivo y hemocultivo, y ante la sospecha clínica y analítica de pielonefritis izquierda, se inicia tratamiento empírico con ceftriaxona endovenosa. La ecografía abdominal objetiva normalidad en ambos riñones, pero se observan múltiples lesiones hipoecoicas de menos de 1 cm. de diámetro en bazo y otra semejante en hígado, compatibles con microabscesos. Ante este diagnóstico ecográfico y con objeto de aumentar la cobertura antibiótica para anaerobios, se añade clindamicina endovenosa al tratamiento.
En el estudio microbiológico, destaca coprocultivo negativo y aglutininas positivas para el antígeno 0 de Salmonella typhi (dilución 1/160) y negativas para el antígeno H. El control de aglutininas a los 10 días: antígeno 0 positivo (dilución:1/320), antígeno H positivo (dilución:1/640).
La desaparición completa de los síntomas a los 3 días de iniciado el tratamiento y el diagnóstico serológico de salmonelosis permitieron completar el tratamiento por vía oral con amoxicilina-clavulánico.
A los 2 meses de completado el tratamiento, la niña permanece asintomática y la ecografía abdominal de control muestra la desaparición de la lesión hipoecoica hepática pero la persistencia de las lesiones esplénicas.
Los abscesos esplénicos suelen aparecer de forma aislada y en adultos inmunosuprimidos o con leucemias. Su hallazgo en un niño inmunocompetente debe hacer pensar en otras posibilidades diagnósticas especialmente de origen infeccioso como Salmonella, Brucella, Estafilococias, Mycobacterias, Bartonella e infecciones fúngicas. Aunque el diagnóstico etiológico de certeza de cualquier absceso debe incluir su punción y aspiración, en nuestro caso la presencia de múltiples lesiones pequeñas no permitió esta prueba.
Llamamos la atención sobre este caso por dos razones; una, su rara frecuencia en la edad infantil, y otra, para destacar el papel de la ecografía abdominal en el diagnóstico diferencial de las pielonefritis agudas.
Artritis monoarticular como única manifestación de Brucelosis en un niño de 5 años
J.J. Borraz Torca, L. Arruza Gómez, E. Aleo Luján, A. Retuerta Oliva, V. San Antonio Arce, B. Agúndez Reigosa, G. Villar Villar y B. Sánchez de la Vara
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: La Brucellosis es una zoonosis que afecta raramente a niños, generalmente en un contexto epidemiológico claro. Presentamos el caso de un niño de 5 años con artritis de rodilla y leve síndrome constitucional como única manifestación de una infección por dos especies distintas de Brucella (B. Abortus y B. Mellitensis).
Caso Clínico: Varón de 5 años de edad sin antecedentes personales y familiares de interés que acude a Urgencias por presentar inflamación, dolor e impotencia funcional en la rodilla derecha, así como febrícula y malestar general de 36 horas de evolución. En la exploración física se objetiva únicamente tumefacción, aumento de temperatura y dolor junto con contractura en flexión de la rodilla derecha, con hepatomegalia de 2-3 cm, esplenomegalia de 4 cm y febrícula (Tª 37,5ºC); siendo el resto de la exploración normal. En la anamnesis se encuentra que hacía unos 2 meses, en verano, el niño había viajado varias veces en la furgoneta de un familiar que se dedicaba al transporte de ganado. P.C.: sistemático de sangre: 8320 leucocitos ( 32 N, 55 L, 7 M), resto normal. VSG 15. PCR 1.0. Bioquímica: GOT 237, GPT 345, LDH 238. Anticuerpos antinucleares, anti músculo liso, anti célula parietal negativos. Factor reumatoideo, complemento, ASLO normales. Serología: Brucella abortus 1/640, Brucella mellitensis 1/640; Salmonella, CMV, VEB, toxoplasmosis, sífilis negativos. Mantoux negativo. Hemocultivos de larga incubación negativos. Radiografía de rodilla: discreto aumento de partes blandas. RMN de rodilla : derrame articular, compatible con artritis transitoria moderada. Se instaura tratamiento con antinflamatorios (ibuprofeno) inicialmente y una vez establecido el diagnóstico con trimetroprim/sulfametoxazol y rifampicina vía oral, siendo la evolución clínica satisfactoria y normalizándose la serología.
Conclusiones: La Brucellosis es una enfermedad poco frecuente en la infancia y que puede tener múltiples formas de presentación: desde cuadros prácticamente asintomáticas a formas multisistémicas de gravedad, pasando por presentaciones que afectan a un único órgano o sistema y que pueden hacer muy difícil el diagnóstico.
Descripción de una epidemia de meningitis por enterovirus. Estudio multicéntrico
G. Ares Mateos, C. Calvo Rey, M. Bueno Campaña, M.J. García Miguel, R. Velasco Bernardo, S. Caserío Carbonero y M.L. Navarro
Hospital Severo Ochoa, Leganés, Fundación Hospital Alcorcón, Alcorcón, Hospital Universitario La Paz, Madrid.
Objetivos: Describir el brote epidémico de meningitis virales por Enterovirus en niños vivida en los meses invernales del año 2000 en diversos hospitales de Madrid y Toledo (H. La Paz, H. Severo Ochoa, H. Fundación Alcorcón, H. 12 Octubre, H. Gregorio Marañon, H. Virgen de la Salud).
Pacientes y métodos: Se recogieron retrospectivamente las características clínicas y analíticas de las meningitis virales confirmadas etiológicamente mediante PCR en líquido cefalorraquídeo o mediante seroconversión a Picornavirus (sin otros procesos infecciosos concomitantes). Se calculó en todos los casos la puntuación en la escala de Boyer
Resultados: Se identificaron 90 casos de meningitis viral en niños con edades desde un mes hasta catorce años (63 niños y 27 niñas). El período de ingreso fue de Enero a Junio de 2000 (máxima incidencia en Marzo). En 70 casos se identificó virus en LCR por PCR y en 20 casos el diagnóstico se obtuvo por seroconversión a picornavirus (pacientes procedentes de Toledo). Las manifestaciones clínicas incluyeron fiebre (38,2 ± 0,68 ºC) salvo en uno de los pacientes. Un paciente presentó púrpura petequial leve y otro obnubilación. En cuanto a las características del LCR, el contaje celular fue de 225 ± 285 con un máximo de 1200 células y un porcentaje de PMN de 51,7 ± 30 % (en el 46,6% de los pacientes > 60%). Las proteínas fueron 41,8 ± 21,7 mg/dl y la glucosa 60,9 ± 10,1 mg/dl. En el hemograma se objetivó una leucocitosis media de 14505 ± 10899, con unos neutrófilos de 10060 ± 4317. La puntuación media en la escala de Boyer fue de 1,06 ± 0,87. En 6 pacientes la puntuación fue de 3.
Conclusiones: La epidemia de meningitis viral por Enterovirus ha mostrado una gran variabilidad clínica y analítica como lo demuestran la dispersión de los datos analizados, destacando la importante leucocitosis y neutrofilia. Sin embargo, la puntación en la escala de Boyer ha sido generalmente baja permitiendo un adecuado enfoque pronóstico y terapéutico de estos pacientes.
Abceso intraorbitario, complicación de una celulitis orbitaria
S. Martínez González, E. Morteruel Arizcuren, I. Pocheville Guruzeta, J.M. Arana Herrerías y M. Chouza Viturro
Hospital de Cruces, Baracaldo.
La celulitis periorbitaria es una patología relativamente frecuente en la edad pediátrica, aunque sus complicaciones (abscesos subperiosticos u orbitarios, afectación intracraneal, trombosis del seno cavernoso, afectación del globo ocular), son infrecuentes aunque potencialmente graves.
Se realizó una revisión de los casos pediátricos con diagnóstico de celulitis orbitaria ingresados en nuestro hospital en un período de 10 años (1990-2000). De ellos únicamente se diagnosticaron 3 casos que presentaron como complicación un absceso intraorbitario. Presentamos uno de estos casos.
Niño de 2 años y 5 meses hospitalizado por celulitis periorbitaria en tratamiento con antibioterapia i.v., que tras mejoría inicial comienza con síntomas de afectación intraorbitaria. Se realizó valoración oftalmológica y realización de un TAC de órbita, que objetivó la presencia de un absceso traorbitario de localización en cuadrante supero interno. Ante el hallazgo, se realizó un drenaje quirúrgico de la colección purulenta. El niño evolucionó sin secuelas durante el seguimiento posterior.
A pesar de la inusual aparición de complicaciones en las celulitis orbitarias, consideramos necesario en todos los episodios que requieran hospitalización además de la antibioterapia i.v., la realización de una exploración ocular meticulosa por parte de un oftalmólogo, que nos permita valorar la conveniencia de la realización de una prueba de imagen que objetive de forma precoz la presencia de una complicación intraorbitaria.
Meningitis neumocócica, estudio de los últimos 12 años
H. Larramona, V. Pineda, M. Domingo, J. Lozano, J. Rivera y D. Fontanals Corporación Sanitaria Parc Taulí, Sabadell.
Introducción: El Streptococcus pneumoniae es la 2ª causa de meningitis bacteriana. Es potencialmente mortal y de secuelas no despreciables. Se ha observado un incremento en las cepas resistentes a la penicilina. En España el 40% de las cepas presentan sensibilidad disminuida a la penicilina, el 10% resistencia intermedia a la cefotaxima (CTX) siendo un 5% altamente resistentes. Se han documentado fallos clínicos con retraso en la esterilización del líquido cefalorraquídeo(LCR) por cepas resistentes a la penicilina y CTX o ceftriaxona. El objetivo de este estudio es describir las características clínicas, microbiológicas, tratamiento y resultado de las meningitis neumocócicas diagnosticadas en los últimos 12 años en nuestro centro.
Material y métodos: Es un estudio observacional y retrospectivo en el que se ha cumplimentado un cuestionario a todos los niños (< 15 años de edad) diagnosticados de meningitis causada por S. Pneumoniae identificado por cultivo de LCR y/o sangre, desde enero del 89 hasta diciembre del 2000. Se ha analizado la susceptibilidad antibiótica y serotipos de todas las cepas. La CIM para los diferentes antibióticos se ha realizado por un método de agar-dilución y los serotipos por la reacción de Quelling utilizando un antisuero del Instituto Carlos III de Madrid.
Resultados: Se han diagnosticado 12 casos de meningitis neumocócica en 11 pacientes, identificándose una recurrencia en un paciente con fístula tras traumatismo craneal. La edad media fue de 2,4 años (rango: 6 meses a 11,5 años). La distribución de casos fue uniforme con una incidencia anual de 1 caso/año (rango: 0 a 3 casos/año). El 83,3% presentaron hemocultivo positivo, y en el 91,7% se aisló el neumococo en LCR con tinción Gram positiva sugestiva en el 91,6%. En 5 casos existió disminución del nivel de conciencia, uno de ellos con crisis convulsiva. Todos presentaron leucocitosis con un rango de 14.800-34.400/mm3 con predominio neutrofílico y una VSG elevada (rango 35-98). Las características del LCR fueron: leucocitosis con un rango de 40 a 9760/mm3; proteinorraquia > 0,45 gr/L (91,6% casos); glucosa < 50 gr/dl (98% casos). Los serotipos hallados fueron 6, 14, 18, 3, 8, 9, 19 y 35, siendo los más frecuentes el 6, 14 y 18 y los resistentes a penicilina el 6, 14, 9 y 19. El 41,7% de las cepas aisladas presentaron resistencia intermedia a la penicilina y el 100% fueron sensibles a la cefotaxima. En 4 de las 5 cepas con sensibilidad disminuída a la penicilina existía historia previa de toma de antibiótico, en las 7 cepas sensibles había sólo 4 con historia previa de toma de antibiótico. Coexistieron neumonía (2 casos) y otitis media (2 casos). Presentaron traumatismo craneal previo 2 pacientes, uno con fístula de seno frontal presentando 2 episodios de meningitis, el 2º episodio causado por una cepa con sensibilidad disminuída. Los tratamientos realizados fueron combinaciones de antibióticos consistentes en vancomicina a 60 mg/kg/d cada 6 h, mas una cefalosporina de 3ª generación a dosis altas (300 mg/kg/día) o ceftriaxona (100 mg/kg/d) en 5 casos, en 2 de ellos se añadió vancomicina el 2º día. Un único antibiótico fue administrado en 4 casos, consistiendo en CTX a dosis altas (2) o standard (2). En 2 casos recibieron CTX a dosis standard o altas combinadas con penicilina. Un caso recibió inicialmente CTX a dosis standard, al persistir cultivo de LCR positivo el 5º día se administró dosis altas y el 10º día persistiendo LCR positivo se cambió a vancomicina combinado con rifampicina (sensibilidad disminuida penicilina y MIC 0,5 CTX). Entre las complicaciones agudas se observó SIADH, higroma subdural, pico hipertensivo. Hubo 1 exitus. Secuelas neurosensoriales se observaron en 3 casos de los 9 pacientes a los que se realizó seguimiento (duración 2 meses a 8 años); 1 con hipoacusia severa unilateral y 2 trastornos motriz-lenguaje.
Conclusiones: La incidencia de meningitis neumocócica se mantiene constante a lo largo de los años. Presenta una morbi-mortalidad importante. El incremento mundial en las resistencias a penicilina, así como CTX obliga a estrategias terapéuticas que incluyen vancomicina como constata el fallo bacteriológico en un caso de los 12 presentados. Los serotipos con sensibilidad disminuida penicilina están incluidos en la vacuna conjugada 7-valente actual.
Echovirus 30: brote epidémico en Cantabria de meningitis vírica
M. Pacheco Cumani, C. López Vilar, M.J. López Mendía, M. Sanjuán Bilbao, C. Fernández San Julián, A. Pérez Puente, M.J. Lozano de la Torre y V. Madrigal Díez
Hospital Universitario Marqués de Valdecilla, Santander.
Introducción: Los enterovirus son los causantes del 85% de los casos de meningitis aséptica, suelen cursar de forma aguda y benigna.
Material y métodos: Revisión de los casos ingresados en el Servicio de Pediatría del HUMV, en el curso de una epidemia, de febrero a julio de 2000 con el diagnóstico de meningitis viral. Se valoraron parámetros epidemiológicos, clínicos, hemograma, PCR, estudio de LCR, cultivos de virus con identificación en centro de referencia, evolución y tratamiento.
Resultados: Se estudiaron 49 pacientes con una edad media de 6,7 años, con predominio masculino (71,9%). Máxima incidencia en abril con 19 casos.
La clínica predominante consistió en cefalea (98%) y vómitos (85%), seguido de abdominalgia (22%) y odinofagia (16%). La temperatura fue menor de 38 º en el 22,4%, superando 39,5º sólo un 4%. El signo más frecuente fue la rigidez de nuca (93,8%), presentando Kerning y Brudzinsky solamente en un 42 y 32% respectivamente. En cuanto a las exploraciones complementarias, en el hemograma se apreció una leucocitosis (> 15.000 en el 24,4%) con neutrofilia > 70% en el 92%. En el LCR se observaron glucorraquia y proteinorraquia normales, pleocitosis < 1000 con predominio de polimorfonucleares (PMN) en el 65%. La puntuación de Boyer fue ¾ 3 en el 100%.
En LCR el cultivo de enterovirus fue positivo en el 60%, mientras que el Gram y cultivo bacteriano fueron siempre negativos. Se aisló enterovirus en el 72,4% de 29 cultivos nasofaríngeos realizados. Remitidas las muestras al Centro Nacional de Toxicología en Majadahonda, se identificó en todos los casos Echo 30. La evolución fue favorable, sin secuelas, con una media de estancia de 3,6 días.
Conclusiones: Se confirman las características clínicas, de laboratorio y epidemiológicas de las meningitis víricas respecto a otros estudios, es de destacar el predominio de PMN en LCR en el 65%. Fue de gran importancia diagnóstica la extracción de cultivos nasofaríngeos específicos para enterovirus, ampliando de un 60 a un 72% la identificación del agente causal. Para el diagnóstico diferencial entre meningitis bacteriana y vírica la puntuación de Boyer nos parece de gran utilidad.
Síndrome febril en el lactante menor de 3 meses: a propósito de nuestra casuística
D. Martínez Cirauqui, M. Herranz Aguirre, M.J. Azanza Agorreta, F. Sánchez-Valverde Visus, N. Clerigué Arrieta y E. Bernaola Iturbe
Hospital Virgen del Camino, Pamplona.
Introducción: El manejo clásico del síndrome febril en lactantes menores de 3 meses incluye la realización de exámenes complementarios y su ingreso. En la actualidad, se recomienda utilizar criterios clínicos y analíticos para seleccionar lactantes con bajo riesgo de infección grave, que podrían ser controlados en su domicilio. Realizamos una revisión retrospectiva de los pacientes ingresados en nuestro Servicio por este motivo.
Objetivos: 1) Determinar la frecuencia de enfermedad infecciosa potencialmente grave en lactantes menores de 3 meses ingresados en nuestro Servicio por síndrome febril sin foco. 2) Comparar los criterios de Rochester entre los niños con enfermedad banal y enfermedad grave.
Material y métodos: Revisión retrospectiva de los pacientes ingresados por síndrome febril en el período de tiempo comprendido entre enero de 1999 y junio de 2000. Se revisan: edad, sexo, temperatura, estado general, días de ingreso, leucocitos y neutrófilos en sangre periférica , PCR, radiografía de tórax y cultivos. Los datos se analizan con el programa estadístico SPSS y las comparaciones se realizan mediante la t de student.
Resultados: Se revisan un total de 102 niños, 64 niños y 38 niñas, con edades comprendidas entre los 13 y los 89 días (media 59,19) . Días de ingreso: mínimo 1 y máximo 10 (media 3). El tiempo desde el inicio de la fiebre hasta la exploración en urgencias fue menor de 6 horas en el 57,8%, de 6-12 horas en el 6,9%, más de 12 horas en el 30,4%. Temperatura: mínima 38 y máxima 40,5 (media 38,7) . La mayoría presentaban buen estado general (80,4%) frente al 19,4% en que era regular. Datos analíticos al ingreso: Leucocitos: mín 2800, máx 33000,( media 11073, S 4939), Segmentados: mín 612, máx 19470, (media 4767, S 3886) y PCR: mín 0,1, máx 12, (media 2,27, S 2,77). Bacteriología: sólo un hemocultivo resultó positivo; 20 urocultivos (19,6%) fueron positivos; ningún cultivo de LCR resultó positivo.
Con los diagnósticos al alta se hicieron dos grupos: enfermedad banal y enfermedad grave. En enfermedad banal se incluyó síndrome febril sin foco (63,7%), infección respiratoria alta (11,8%) e infección respiratoria baja (2%), excluyendo neumonías. Enfermedad grave se consideró: infección urinaria (19,6%) y meningitis (2%). El 21,6% de los casos fueron enfermedad grave y 78,4% banal. Existen diferencias significativas entre ambos grupos en los siguientes datos analíticos al ingreso: leucocitos (p = 0,013), neutrófilos totales (p = 0,001), PCR (p < 0,001).
Comentarios: 1) Destacar la baja frecuencia de enfermedades bacterianas invasivas en nuestra serie (solo un niño presentó bacteriemia) que coincide con lo descrito en la literatura actual. 2) En nuestros pacientes, la mayor parte de enfermedades graves son infecciones urinarias. 3) Hay diferencias significativas en los leucocitos, total de neutrófilos y PCR al ingreso entre los pacientes con enfermedad grave y enfermedad banal. 4) Necesidad de utilizar otras técnicas que ayuden a diferenciar los pacientes con enfermedad banal de los que tienen una enfermedad grave, en las primeras horas, como sería la determinación de la procalcitonina.
Cluster de complicaciones cutáneas graves de varicela
M.J. Azanza Agorreta, D. Martínez Cirauqui, V. Alzina de Aguilar, P. Fiz Sánchez, E. Bernaola Iturbe, N. Clerigué Arrieta,
M. Herranz Aguirre y S. Raggio Pérez
Hospital Virgen del Camino, Pamplona, Clínica Universitaria de Navarra, Pamplona.
Introducción: Las complicaciones más frecuentes de la varicela son las cutáneas, debidas en la mayoría de los casos a Stafilococcus aureus y Streptococcus beta hemolítico del grupo A, no siendo excepcional el hallazgo de este gérmen en sangre.
Objetivo: Presentar cuatro casos de varicela, con complicaciones cutáneas graves que han requerido ingreso y que han coincidido en el tiempo.
Material y métodos: Revisión retrospectiva de los niños ingresados con varicela , en los meses de abril-mayo 2000, que han presentado complicaciones cutáneas graves.
Resultados:Caso 1: Niña de trece meses, con dermatitis atópica. Al quinto día de la erupción de la varicela, presenta fiebre, vómitos y edemas en cara, tronco y extremidades superiores. En la exploración se observan lesiones de varicela sobreinfectadas por S. Pyogenes y S. Aureus, importante celulitis subcutánea, abscesos axilares bilaterales. Hemocultivo positivo para S. Pyogenes, y S. Aureus. Recibió tratamiento endovenoso con Cefotaxima y Aciclovir. Precisó desbridamiento quirúrgico de los abscesos, con evolución tórpida, siendo dada de alta a los 20 días del ingreso. Caso 2: Niña de tres años y medio, sin antecedentes personales de interés. Al quinto día de evolución de una varicela, por la que estaba recibiendo tratamiento con Aciclovir y Cefuroxima oral, presenta fiebre alta, celulitis en la cara y absceso submentoniano con gran deformidad facial. Cultivos de sangre y contenido del absceso: negativos. Se trató con Cefotaxima, Cloxacilina, Aciclovir endovenosos y drenaje quirúrgico del absceso. La evolución fue satisfactoria, requiriendo 13 días de ingreso. Caso 3: Niño de tres años, sin antecedentes de interés. Al tercer día del comienzo de la varicela, inicia fiebre elevada, dolor abdominal y celulitis facial derecha. Hemocultivo negativo. Tratamiento con Ceftriaxona y Aciclovir endovenoso con buena evolución, tras 6 días de ingreso. Caso 4: Niña de 2 años y medio, con antecedentes de asma bronquial, Al tercer día de evolución de una varicela presenta fascitis necrotizante a nivel cervical, en ingle y pierna izquierda, osteomielitis de cadera izquierda y sepsis con coagulopatía de consumo. Los cultivos fueron negativos. Se trató con Ceftriaxona, Gentamicina , Vancomicina y Aciclovir endovenoso precisando 20 días de ingreso. La evolución fue tórpida, requiriendo desbridamiento del absceso y drenaje de la articulación de la cadera.
Conclusiones: 1) Es llamativa la coincidencia en el tiempo de casos de varicela con complicaciones cutáneas severas, pudiera deberse a serotipos de S. Pyogenes especialmente agresivos. 2) En dos de los casos había una patología de base atópica, sin estar sometidos a tratamiento con esteroides, que pudiera explicar la agresividad de la infección. 3) Es aconsejable realizar hemocultivos en los casos de complicaciones severas de la varicela (En noviembre del mismo año una lactante de 34 meses presentó una bacteriemia por S. pyogenes en el curso de una varicela). 4) Casos como estos, hacen reflexionar sobre la terapéutica con fármacos antivirales y la implantación de la vacunación sistemática antivaricelosa en nuestro medio.
Hipokaliemia grave con rabdomiolisis asociados a tratamiento con Itraconazol
R. Rodríguez Díaz, P. Carreño Guerra, P. Gómez de Quero, J.I. Sánchez Díaz, F. Mateos y J. Ruiz Contreras
Hospital 12 de Octubre , Madrid.
Antecedentes: La hipokaliemia grave ha sido descrita muy raramente como complicación del tratamiento con Itraconazol.
El objetivo de este estudio es llamar la atención sobre una complicación grave asociada al tratamiento con Itraconazol.
Presentación del caso: Paciente varón de 20 años, diagnosticado de enfermedad granulomatosa crónica ligada al X desde la edad de 2 años. Un mes antes había sido ingresado por un nódulo subcutáneo por Aspergillusfumigatus para tratamiento con Anfotericina B liposomal e Itraconazol. El tratamiento con Anfotericina se mantuvo durante los 7 primeros días sin complicaciones, con control de potasio sérico normal al final del mismo. A los 30 días del inicio del tratamiento con Itraconazol comienza con dolor en ambas pantorrillas y pérdida de fuerza en ambos miembros inferiores, proximal y distal, algo mayor en lado izquierdo que se acompaña de dificultad para la marcha. La fuerza en miembros superiores está conservada y la sensibilidad en los cuatro miembros es normal. Además existe una disminución de los ROT rotulianos y aquíleos que parecen abolidos incluso en el lado izquierdo. Las pruebas complementarias revelaron un aumento importante de la CPK (5470 U/L, CPK-MB:77 U/L)e hipoptasemia marcada (1,7 mEq/L).
En el EKG se observan ondas Q en I, II, avF, avL, V5 y V6, y T invertidas en I, II, III, V4, V5 y V6.
Inicialmente se establece tratamiento con soluciones intravenosas de K a concentraciones muy elevadas , suplementando con K oral, además de hiperhidratación para forzar la diuresis. El paciente en todo momento presenta K en orina alrededor de 20 mEq/L y pese a recibir cantidades extraordinariamente elevadas de K, el aumento en la cifra sérica es lento e incluso inexistente. A las 48 horas se suspende el Itraconazol y coincidiendo con ello, las cifras de K en sangre empiezan a aumentar de forma significativa hasta normalizarse, y al mismo tiempo comienzan a disminuir las cifras de CPK, desapareciendo la debilidad y el dolor en miembros inferiores hasta normalizarse en unos 6-7 días.
Conclusiones: Entre los posibles efectos secundarios del Itraconazol hay que tener en cuenta la hipokaliemia grave, muy raramente descrita, que puede ir asociada a rabdomiolisis y a alteraciones en el ritmo cardíaco, con la importancia que estos dos hechos suponen.
Síndrome hemofagocítico reactivo con coagulación intravascular diseminada secundario a infección por el virus de Epstein-Barr
J.R. Bretón Martínez, L. Lacruz Pérez, I. Calvo Penadés, R. Fernández-Delgado Cerdá, F. Núñez Gómez, J. Donat Colomer, R. Simó Jordá, A. Colino García, M.J. Ruiz Jiménez y R. Hernández Marco
Hospital Universitario Dr. Peset., Valencia, Hospital Clínico Universitario, Valencia.
Introducción: El síndrome hemofagocítico (SHF) es una alteración del sistema mononuclear fagocítico caracterizada por una proliferación de histiocitos que fagocitan los precusores de la médula ósea. Cursa con fiebre, bi o tricitopenia, esplenomegalia, hipertrigliceridemia y/o hipofibrinogenemia e infiltración por histiocitos de al menos un 2% de la médula ósea en ausencia de neoplasia. El SHF puede ser familiar (propio de niños menores de 2 años) o secundario sobre todo a infecciones víricas, y en menor medida a infecciones bacterianas, fúngicas, parasitarias, colagenosis, neoplasias y fármacos. Entre las infecciones víricas la causa más frecuente es el virus de Epstein-Barr (VEB). Presentamos un caso de SHF secundario a infección por VEB.
Caso clínico: Niña de 14 años que ingresó por un cuadro de fiebre máxima de 40ºC de tres días de evolución, amígdalas hiperémicas y congestivas con exudado en criptas, adenopatías laterocervicales, decaimiento y deshidratación leve. Ante la sospecha de mononucleosis infecciosa se solicitó la reacción de Paul-Bunnell que fue positiva. En su evolución posterior presentó fiebre alta persistente, deterioro del estado general, subictericia y hepato-esplenomegalia. Analíticamente destacaba: Hb 9,7 g/dl, Hto 28,1%; leucocitos 4.700/mm3 (N 46,4%, L 51,4%), plaquetas 33.000/mm3, TTPA 47s, TP 25 s, fibrinógeno 103 mg/dl, dímero D positivo; glucosa 107 mg/dl, urea 24 mg/dl, creatinina 0,5 mg/dl, triglicéridos 205 mg/dl, LDH 2156 UI/l, GOT 302 UI/l GPT 229 UI/l, GGT 131 UI/l, fosfatasas alcalinas 598 UI/l, bilirrubina total 4,7 mg/dl, proteínas totales 4,82 g/dl, albúmina 2,59 g/dl, proteína C reactiva 26,7 mg/dl, ferritina 13.272 ng/ml, b-2 microglobulina 4,6 mg/l, IgM anti-VCA positivo y IgG anti-VCA positivo. La reacción en cadena de la polimerasa en sangre fue positiva para el VEB. El aspirado medular inicial presentaba una celularidad global conservada con hiperplasia de la granulopoyesis. Se inició tratamiento de soporte con plasma fresco y se decidió su ingreso en la UCI Pediátrica. En el aspirado medular que se repitió con posterioridad se observó un aumento de la celularidad con un incremento llamativo de los histiocitos con fenómeno de hemofagocitosis. El hemocultivo y los cultivos de médula ósea fueron negativos. Dado el estado general relativamente bien conservado se decidió una actitud expectante con tratamiento únicamente de soporte. La evolución posterior fue favorable quedando la paciente asintomática sin alteraciones hematológicas.
Comentario: El SHF es una entidad de diagnóstico difícil que hay que sospechar ante un niño que padece un proceso infeccioso con deterioro del estado general, fiebre alta, pancitopenia, organomegalias, disfunción hepática, coagulopatía e hipertrigliceridemia como ocurrió en nuestro caso. Destaca la infección por el VEB como probable desencadenante del cuadro y la evolución favorable en ausencia de tratamiento.
Neurología
Estudio de diferentes aspectos de la mortalidad pediátrica debida a enfermedades neurológicas en nuestro departamento durante quince años (1985-1999)
M.T. Lacasa Pérez, J.L. Ruibal Francisco, B. de Miguel Lavisfer, O. Pérez Rodríguez y C. Martínez Hernández
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: Las enfermedades neurológicas representan la segunda causa de muerte en nuestro departamento y la primera en niños de 0 a 5 años de edad. En este trabajo se han investigado diferentes aspectos sobre la mortalidad de los niños debida a dichos procesos en el período de tiempo comprendido entre los años 1985-1999.
Material y métodos: Se han analizado de manera retrospectiva, las historias clínicas de todos los pacientes pediátricos fallecidos en relación con enfermedades neurológicas con edades comprendidas entre el mes de vida y los 18 años de edad, en el período de tiempo transcurrido entre el 1 de enero de 1985 y el treinta y uno de diciembre de 1999. Se han estudiado el número total de pacientes fallecidos, la etiología de sus enfermedades neurológicas, y su distribución por sexo y grupos de edad. También, la causa inmediata que ha originado su muerte. Los datos se han expresado como estadística básica (media, desviación típica, máximo y mínimo). Asimismo en datos absolutos y porcentajes sobre el total de los analizados.
Resultados: Los pacientes fallecidos por enfermedades neurológicas han sido 34, de los que 15 (44%) eran varones y 19 (56%) mujeres. La edad media al fallecimiento fue de 7,1 ± 6,3 (18, 0,15) años. Un 50% (17) lo hacen entre los 0 y 5 años, un 12% (4) entre los 5 y 10, un 26% (9) entre los 10 y los 15, y un 12% (4) con más de 15 años. Respecto de la etiología de dichas enfermedades, en 10 casos (29%) era de origen hipóxico-isquémico, en 9 (26%) metabólico, en 6 (18%) por procesos malformativos, en otros 6 (18%) infecciosos, y en un caso (3%) por una malformación vascular, una atrofia espinal infantil y por una epilepsia mioclónica.
Las causas inmediatas que originaron el fallecimiento de estos pacientes fueron por fracaso respiratorio por neumonías aspirativas en 19 (56%), por apneas centrales en 4 (12%), por muerte cerebral en 3 (9%), llegaron muertos a urgencias 3 (9%), mientras en 2 (6%) por fracaso hepático, en otros 2 (6%) por choque cardiogénico y en uno (3%) por una sepsis por cándida albicans que le provocó un fracaso multisistémico.
Conclusiones: La mayor parte de los niños fallecidos por enfermedades neurológicas son por procesos de etiología hipoxico-isquémica, seguidos por los de naturaleza metabólica y malformativa. El 50% fallecen entre los 0 y 5 años de edad. Un 56% de los pacientes son mujeres. La causa inmediata más frecuente de su fallecimiento es el fracaso respiratorio condicionado por neumonías aspirativas.
Glucogenosis tipo II. Presentación de un caso en su forma infantil
M.P. Bulo Concellón, R. de Andrés-Cofiño, O. Rivero Bonilla, A.M. Cordón-Martínez, G. Colli Lista y M.D. Mora Ramírez
Hospital Materno Infantil Carlos Haya, Málaga.
Introducción: La glucogenosis tipo II o enfermedad de Pompe se produce por el déficit de maltasa ácida; se transmite de forma autosómica recesiva y el gen se ha localizado en el locus 17q(23-25). Existen tres formas clínicas : infantil precoz(IIa) que se inicia en los primeros meses de vida de carácter rapidamente progresiva, infantil tardía (IIb), que comienza a partir del primer año de vida o más tardía, tiene una sintomatología lentamente progresiva y cursa sin afectación cardiaca y la forma del adulto.
Caso clínico: Niño de 4,5 años que presenta desde hace 6 meses disminución de la fuerza, caídas frecuentes y dificultad para subir escaleras. Antecedentes personales y familiares sin interés. A la exploración presenta facies miopática, amiotrofia proximal con disminución de la fuerza global y arreflexia miotática. Marcha con hipelordosis, dificultad para pasar de tumbado a sentado y para el salto, Gowers negativo. Pruebas complementarias: CPK: 1338 U/l, LDH: 1524 U/l, GOT: 176 U/l, GPT: 206 U/l, láctico y pirúvico normal. EMG: Signos de afectación miopática. Biopsia muscular: muestra acumulos de glucógeno y deficiencia de actividad alfa-glucosidasa ácida. Evolución: Progresiva presentando actualmente rectificación de columna con escápula alada e hiperlordosis y movilidad aceptable salvo para pasar la cabeza de flexión dorsal a la línea media.
Comentarios: Nuestro paciente presenta la forma tipo IIb, que es la menos frecuente y se manifiesta como una miopatía próximal lentamente progresiva con afectación de la musculatura respiratoria.. El diagnóstico se realiza mediante biopsia muscular, la cual demuestra una miopatía vacuolar PAS + y acúmulo de glucógeno. No existe un tratamiento eficaz en la actualidad.
Miopatía Nemalínica. Revisión de un caso
O. Rivero Bonilla, R. de Andrés-Cofiño, M.P. Bulo Concellón, G. Colli Lista, A.M. Cordón-Martínez y M.P. Delgado Márquez
Hospital Materno Infantil Carlos Haya, Málaga.
Introducción: La miopatía nemalínica se caracteriza por una alteración estructural del músculo. Identificada por Shy en 1963 por la existencia anatomopatológica de cuerpos nemalínicos o bastones en la fibra muscular. Se transmite de forma autosómica dominante (1q 21-q23) o recesiva y esporádica. Se caracteriza por presentar una clínica variable poco específica.
Caso clínico: Niño de 6 años que desde el nacimiento presenta retraso motor sin carácter de progresividad. En la actualidad tiene dificultad para correr y subir escaleras. Antecedentes familiares: padre fallecido por miocardiopatía. A la exploración presenta facies miopática, fenotipo marfanoide, hipotrofia muscular generalizada con hiporreflexia miotática. Disminución de la fuerza con predominio axial más acentuada en músculos cervicales anteriores y paravertebrales. Gowers incompleto. No existen signos de afectación cardiológica ni de otros sistemas.Pruebas complementarias: bioquímica normal incluida CPK y perfil metabólico. TC cerebral normal. EMG: con signos de afectación miopática. Biopsia muscular: numerosas estructuras nemalínicas formando masas en fibras tipo I. Evolución: ha sido lentamente progresiva presentando en la actualidad una mayor debilidad de la musculatura proximal en miembros inferiores.
Comentarios: La práctica de la biopsia muscular ha permitido descubrir una serie de enfermedades , que se definen por el carácter de anomalías morfológicas reveladas en el examen histoquímico y ultraestructural del músculo. En el caso de la miopatía nemalínica son diagnósticos los cuerpos nemalínicos o bastones en las fibras musculares. Dicha entidad presenta dos formas clínicas: la forma recesiva es la más grave y se presenta en la época neonatal y la forma dominante que se inicia más tardíamente y tiene mejor pronóstico.
Cefaleas: revisión de las características de la presentación orientadoras de organicidad
J.A. Balaguer, R. Jiménez, F. Pagone, O. Salvadó, G. Chumbe y C. Escofet
Hospital Universitario Sant Joan de Reus, Reus.
Introducción: Aunque la cefalea es motivo de consulta frecuente y su pronóstico la mayor parte de veces no reviste gravedad, algunas formas de presentación pueden orientar a patología orgánica subyacente. Mientras que se conocen bien algunos de estos signos de alerta, pocos trabajos determinan la fuerza predictora de estos.
Se efectúa una descripción de las cefaleas de etiología orgánica aparecidas en los últimos diez años en nuestro medio. Se identifican los aspectos de la anamnesis y exploración que mejor orientaron hacia etiología orgánica.
Material y pacientes: Se estudiaron retrospectivamente los casos de cefalea orgánica, en menores de 14 años, diagnosticados en nuestro hospital entre los años 1990 y 1999. Se excluyeron aquellos secundarios a patología infecciosa y traumática aguda o cefalea postraumática. La revisión de las historias clínicas se efectuó según un cuestionario que recogía de forma detallada 24 items agrupados en cuatro categorías: manifestaciones al inicio, características del dolor, sintomatología asociada y alteraciones a la exploración.
Resultados: Se obtuvieron un total de 13 casos, de los que los diagnósticos etiológicos fueron: tumor SNC (7), pseudotumor cerebri (2), infarto cerebral (1), hematoma subdural crónico (1), y patología sistémica -hipertiroidismo y lupus eritematoso- (2 )
De los aspectos estudiados, los que se hallaron con mayor frecuencia fueron: tiempo de aparición menor o igual de 3 meses (12/13 casos), patrón atípico (9/13) y progresividad del dolor (8/13).
Conclusiones: Ninguno de los parámetros estudiados se demuestra, por si solo, capaz de predecir organicidad. Sin embargo, para el diagnóstico precoz de la cefalea orgánica, una cuidadosa anamnesis y examen físico se siguen demostrando esenciales.
En la serie estudiada todos los casos reunieron al menos tres de los items propuestos como datos de alerta.
Síndrome de Morsier y hiperecplexia: ¿una asociación casual?
M.S. Pérez Poyato, A.D. Moreno Vázquez, I. Hernández Sousa, M.T. Guerra Pérez y E. Álvarez García
Hospital Universitario Puerta del Mar, Cádiz.
Introducción: El Síndrome de Morsier o displasia septo óptica se define por la asociación de ausencia de septum pellucidum, hipoplasia del nervio óptico, e insuficiencia hipofisaria. Clínicamente se caracteriza por retraso mental, nistagmo de persecución y déficit de agudeza visual. La sintomatología endocrina es inconstante. Se define la hiperecplexia por la aparición de reacciones de sobresalto a diversos estímulos, que simulan mioclonías estímulo-sensibles pudiendo desencadenar ,en el lactante ,hipertonía persistente con reflejos musculares profundos exaltados.
Caso Clínico: Varón de 7 meses trasladado desde otro centro para valoración neuropediátrica por presentar crisis de unos 5 minutos de duración caracterizadas por hipertonía, hiperextensión de extremidades y revulsión ocular que ceden espontáneamente y se siguen de una fase hipotónica. Antecedentes familiares: desconocidos. Antecedentes personales: ingreso al mes de vida en hospital de origen detectándose atrofia papilar bilateral. A los 6 meses de vida se realiza TAC craneal diagnosticándose de Síndrome de Morsier. Exploración neurológica: hiperpigmentación racial (ascendencia nigeriana). Motilidad global disminuida. Manos cerradas con pulgar en adducción. Tono muscular aumentado a expensas de extensibilidad. Reflejos musculares profundos exaltados, policinéticos con respuesta clonoidea. Ante pequeños estímulos tactiles y especialmente al percutir rótula o glabela, se desencadenan automatismos faciales que simulan estornudos y mioclonías de párpados y de miembros superiores. Nistagmo pendular, de búsqueda, y en resorte conjugado bilateral. No fijación ocular. Leve persecución y respuesta a la luz. Reflejos fotomotor y cocleopalpebral presentes. Desarrollo psicomotor: retraso en todas las áreas, especialmente motórica fina, adaptativa y lenguaje. Exámenes Complementarios: Analítica neurometabólica: normal. Estudio cardiológico y endocrinológico: normal. Serología de agentes neurotropos: negativa. Potenciales evocados auditivos: normal. Potenciales evocados visuales: alterados. Estudio oftalmológico: palidez papilar muy importante con cicatriz macular. EEG: foco irritativo temporo-parietal derecho. Estudio genético: pendiente de resultados. RNM: trastorno de la migración neuronal y displasia cortical tipo esquisencefalia de labio abierto en hemisferio derecho. Hipoplasia de septum pellucidum, hipófisis y de vías ópticas. Agenesia parcial del cuerpo calloso. Compatible con Síndrome de Morsier. Evolución: Durante su hospitalización no presenta crisis convulsivas. Al iniciar tratamiento con Acido Valproico mejora su estado general y disminuye la hiperexcitabilidad.
Comentarios: Revisada la bibliografía de los últimos años no hemos encontrado asociación entre el Síndrome de Morsier y la hiperecplexia. En nuestro caso coinciden ambas patologías tan infrecuentes en un mismo paciente, por lo que creemos de interés la comunicación de dicha asociación que pudiera no ser casual.
Enfermedad de Gaucher tipo III. Presentación de 2 casos
M.S. Pérez Poyato, A.D. Moreno Vázquez, I. Hernández Sousa, M.T. Guerra Pérez y E. Álvarez García
Hospital Universitario Puerta del Mar, Cádiz.
Introducción: La enfermedad de Gaucher es una enfermedad de acúmulo lisosomal de herencia A.R. producida por un déficit de la enzima beta galactosidasa ácida. Cursa con hepatoesplenomegalia progresiva, complicaciones óseas y alteraciones neurológicas (tipo 2 o infantil y tipo 3 o juvenil). El examen de médula ósea revela la presencia de células de Gaucher. El tratamiento enzimático sustitutivo con alglucerasa constituye el tratamiento de elección en la actualidad.
Caso Clínico nº 1: Varón de 2 años sin antecedentes familiares ni personales de interés que ingresa para estudio de hepatoesplenomegalia. Tras examen de médula ósea se diagnostica de Enfermedad de Gaucher tipo 3 siendo esplenectomizado. Tres años después sufre Enfermedad de Perthes bilateral. A los 9 años aparecen crisis mioclónicas rebeldes al tratamiento farmacológico, temblor intencional y de reposo, pérdida progresiva de habilidades motoras y dificultades en el lenguaje. Con 13 años inicia tratamiento enzimático sustitutivo con muy buena respuesta en los 6 primeros meses. En la actualidad continúa tratándose, si bien la afectación visceral y ósea se han controlado, persiste el deterioro neurológico progresivo, aunque existe mejoría en el control de las crisis y en el EEG.
Caso Clínico nº 2: Varón de 13 años afecto de cuadro neurológico regresivo no filiado que se nos remite por presentar crisis convulsivas diarias de difícil control farmacológico. Antecedentes familiares: epilepsia, retraso psicomotor y del lenguaje en varios familiares. Antecedentes personales: período perinatal sin incidencias. Desarrollo psicomotor: sedestación al 9º mes, deambulación a los 18 meses, adquisición lenta del lenguaje con regresión detectada a los 4 años. 1ª crisis convulsiva a los 32 meses. Exploración: facies hiposíquica. Contacto adecuado con el medio, sonrisa afectiva. Lenguaje muy pobre: frases simples. Motilidad global conservada. Reflejos musculares profundos asimétricos. Marcha con rodillas en valgo y flexo, dolor a la abducción de caderas. Hepatoesplenomegalia. Exámenes Complementarios: Analítica: anemia hipocroma. Fosfatasa ácida: 9 U/L. Hipergammaglobulinemia. Mapeo óseo y fondo de ojo: normales. Ecocardiograma: discreta hipertrofia septal. EEG: trazado enlentecido más acusado en hemisferio izquierdo. Ecografía abdominal: hepatoesplenomegalia y aumento de densidad cortical renal bilateral. Examen de médula ósea: se observan células de Gaucher.
Comentarios:
- El diagnóstico de confirmación de esta entidad se basa en la medición de la actividad de la beta galactosidasa en leucocitos aislados de sangre periférica que en nuestro caso nº 1 estaba disminuida y pendiente de resultados en el caso nº 2.
- Para la detección de portadores es imprescindible el análisis molecular de las mutaciones resultando portadora de la enfermedad la madre del paciente en el caso nº 1.
- Resaltamos la importancia del diagnóstico precoz puesto que la eficacia más completa del tratamiento se obtiene durante la época de crecimiento y previamente a la aparición de complicaciones.
Accesos de tos, manifestación de una crisis epiléptica parcial compleja: A propósito de un caso
C. Encabo Jiménez, J. Santos Borbujo, L. Monzón Corral, A.M. Hernández Alberca, I. Corral Carabias y M.S. González García
Hospital Clínico Universitario, Salamanca.
Introducción: Las crisis epilépticas autonómicas o vegetativas son debidas generalmente a una descarga en la región orbitoinsulotemporal. Se subdividen en gastrointestinales y enuréticas, pudiendo iniciarse con síntomas respiratorios, circulatorios o vasomotores. Es muy rara su presentación de forma aislada, normalmente forman parte de las crisis parciales complejas o de las crisis generalizadas.
Caso clínico: Niña de 7 años, antecedentes familiares y personales sin interés. Desde hace un mes presenta episodios de tos. En dos ocasiones, tras accesos de tos importantes, tiene pérdida de conciencia de unos veinte segundos. Tratada con antitusígenos y broncodilatadores sin mejoría. Acuden a urgencias por presentar cuatro episodios de sofocación con cianosis facial, sin pérdida de conocimiento. Tras su ingreso para estudio presenta varios cuadros. En uno de ellos, tras gran dificultad respiratoria y tos, tiene una expectoración de moco muy espeso, se sospecha aspiración de cuerpo extraño y se solicita una broncoscopia, siendo esta negativa. Otros de los episodios consisten en suspensión de la actividad voluntaria, ruidos guturales y tos, mirada perdida, palidez facial con cianosis perioral junto a movimientos orofaciales tipo deglutorio de unos 10-15 segundos de duración. Se siguen de adormecimiento. Son en vigilia y en sueño, predominando en los primeros 30 minutos del sueño. Se sospechan crisis epilépticas y se solicita EEG vigilia (normal) y EEG sueño (focalidad temporal media derecha). Juicio clínico: Crisis parciales complejas. Se le pauta carbamazepina (CBZ) a dosis 15 mg/Kg. Con niveles adecuados mantiene crisis diarias, en vigilia y en sueño, similares a las ya descritas. Solicitamos RNM: normal. Se sustituye CBZ por lamotrigina a dosis 5 mg/kg manteniéndose sin crisis hasta el momento actual. Otras pruebas diagnósticas solicitadas: hemograma, bioquímica, proteinograma e inmunoglobulinas, test sudor y escintigrafía gastro-esofágica normales; serología micoplasma y clamydia negativas; VRS negativo; Mantoux negativo
Conclusiones: Valorar como diagnóstico diferencial, en procesos con clínica respiratoria especial, las crisis epilépticas con síntomas vegetativos. Ante la sospecha clara de crisis epiléptica no conformarse con el EEG en vigilia si este resulta normal, solicitar EEG en sueño. Siempre se debe realizar RMN cerebral para descartar patología orgánica.
Hiperamoniemia: alteración del ciclo de la urea. Portadora de déficit de ornitin transcarbamilasa (OTC)
G. Espinosa Villoria, A.L. Martínez Jiménez, A.J. Guiote Torres, J.A. Delgado García, L. More y A. Martínez Bermejo
Hospital del Bierzo, Ponferrada, Hospital Universitario La Paz, Madrid.
Introducción: Las enfermedades metabólicas siguen siendo en la actualidad entidades de difícil diagnóstico. Las alteraciones del ciclo de la urea debutan clínicamente con manifestaciones neurológicas y/o digestivas en el período neonatal, aunque pueden pasar desapercibidas hasta épocas posteriores.
Caso clínico: Paciente de dos años 7/12 meses previamente sana que presenta tres meses antes del diagnóstico un cuadro de vómitos, astenia e irritabilidad en el contexto de un cuadro catarral con aumento de las cifras de transaminasas. Antecedentes familiares: tío materno fallece a los cinco meses de edad. Antecedentes personales: embarazo y parto normal. Fenotipo normal. Presentó un episodio de apnea a los dos meses con control posterior. Lactancia materna durante un mes. Alimentación complementaria regulada y bien tolerada. Desarrollo psicomotor normal. En el momento del ingreso la niña presenta facies normal con llanto continuo, escasa respuesta a estímulos y alteración de la conducta. Estudios complementarios evidencian unas cifras elevadas de transaminasas con estudios virológicos normales, alteración en las pruebas neurológicas (focalidad paroxística temporal derecha en el electroencefalograma, y lesión parenquimatosa bilateral en sustancia gris, en la resonancia magnética nuclear) y alteración metabólica (aumento de las cifras de amonio, uracilo y ácido orótico) estableciendo el diagnóstico de posible portadora de déficit de OTC. Se inicia el tratamiento dietético, disminuyendo las cifras de amonio y con evolución neurológica hacia la normalidad
Déficit de OTC: El déficit completo de OTC es letal en neonatos si no se trata. La disfunción del SNC: retraso del desarrollo, convulsiones, alteraciones sensoriales de la conducta, se observa en más de la mitad de estas enfermedades. Los vómitos inducidos por las proteínas, la disfunción neurológica e hiperamoniemía, son frecuentes en muchos trastornos del ciclo de la urea. Este déficit enzimático tiene dos características diferenciadas: herencia ligada al sexo (los heterocigotos se detectan porque presentan niveles urinarios anormales de ácido orótico o un aumento del ión amonio en sangre después de una sobrecarga proteica) y la existencia de oroticoaciduria es clave para el diagnostico y detección de portadores.
Conclusiones: En un paciente con alteraciones digestivas y trastornos neurológicos debe valorarse una enfermedad metabólica. La presencia de vómitos, irritabilidad e hiperamoniemia obliga descartar una portadora de OTC.
¿Encefalomielitis diseminada aguada (EMDA) o primer brote de esclerosis múltiple (EM)?
A.M. Cordón-Martínez, G. Colli Lista, J. Martínez Antón, M.P. Bulo Concellón, R. de Andrés-Cofiño y O. Rivero Bonilla
Hospital Materno Infantil, Málaga.
Introducción: La EMDA y la EM son dos enfermedades inflamatorias desmielinizantes del sistema nervioso central de difícil diferenciación ante un único brote. Presentamos un caso que pone en evidencia la dificultad clínica para establecer el diagnóstico inicialmente.
Caso clínico: Niña de 10,8/12 años que ingresa por cuadro de cefalea intensa, vómitos tras la ingesta y diplopía de 48 horas de evolución. A las 12 horas presenta nistagmus horizontal y marcha inestable, presentando a las 35 horas episodio de perdida de conciencia, mirada fija, hipertonía de MSD y pérdida de control de esfínteres, que cede espontáneamente, persistiendo hemiparesia e hipotonía izquierda, perdida control esfínter vesical e imposibilidad para la marcha. Proceso febril autolimitado 10-15 días antes. Antecedentes personales: origen marroquí, reside en Vélez-Málaga desde los 8 años; cefalea desde los 4 años. No otros antecedentes personales ni familiares de interés. Exploración: Peso: 33 Kg. Conciente, tendencia al sueño. Hemiparesia izquierda, parálisis pares: IV dcho, VI, VII y XI izdos., ROT exaltados, Babinsky izdo. Resto de exploración normal. Pruebas complementarias: TAC cráneo con contraste: lesión hipoatenuada de 2 cm en la sustancia blanca del centro semioval izdo con edema adyacente; RMN cráneo: numerosas imágenes nodulares en sustancia blanca periventricular y subcorticales bilaterales y asimétricas, isointensas en T1 y brillantes en T2, todas supratentoriales, salvo una en pedúnculo cerebelosos; RMN medular: normal; LCR: bandas oligoclonales positivas; Serología: VEB IgG e IgM (+); Potenciales evocados (EV): auditivos: normales, visuales: alterados, somestesicos alterados.; EEG: actividad de base enlentecida con asimetría interhemisférica. Tratamiento y evolución: mejoría progresiva tras corticoides. RMN en 20 días: nueva lesión nodular en sustancia blanca del hemisferio cerebelosos y disminución de algunas lesiones previas, con menor captación de contraste.
Conclusiones: En nuestro paciente la clínica orientaba a EMDA, mientras que la epidemiología y las pruebas complementarias apoyan el diagnóstico de EM. Clínicamente, ambas entidades son difícilmente diferenciables en el primer brote, debiendo apoyarse el diagnóstico en las pruebas complementarias, fundamentalmente la neuroimagen y los potenciales evocados, así como en el seguimiento evolutivo del paciente.
Variabilidad de expresión clínica de la siringomielina en la edad pediátrica: revisión de 20 casos
A. de Andrés, M. Martín Endrinal, J.J. García Peñas, L.G. Gutiérrez Solana y M. Luz Ruiz Falcó
Hospital del Niño Jesús, Madrid.
Introducción: La siringomielia (SM) es un proceso infrecuente en la infancia. Se caracteriza por la presencia de una dilatación quística en el interior de la médula espinal.
Objetivo: Analizar la variabilidad de expresión clínica y etiopatogénica de la SM en la infancia.
Material y métodos: Revisión retrospectiva de las historias clínicas de 20 pacientes menores de 18 años de edad, valorados en el Hospital Niño Jesús de Madrid entre enero de 1987 y enero de 2001, y diagnosticados de SM en virtud de los hallazgos de neuroimagen. En todos los casos se analizan: Edad de presentación, sexo, debut clínico, semiología neurológica, hallazgos de neuroimagen y evolución clínica. En todos los pacientes se realizaron: RMN craneal y de médula espinal, EMG, ENG y VCN.
Resultados: 15 pacientes (75%) tenían edades comprendidas entre 5-10 años. Sólo 5 casos (25%) presentaban clínica de disfunción neurológica. 6 pacientes (30%) mostraron alteraciones en la exploración neurológica. Las asociaciones etiológicas incluían: 6 anomalías de Chiari tipo I, 2 tumores de médula espinal, 2 formas postraumáticas, 2 casos tras aracnoiditis y 8 formas idiopáticas. 4 pacientes presentaban criterios de NF tipo 1. Sólo 2 casos desarrollaron semiología neurológica progresiva y requirieron cirugía.
Conclusiones: 1) En nuestra serie, la mayoría de los casos se diagnosticaron al valorar a pacientes con anomalía de Chiari tipo I, escoliosis y/o NF tipo 1, que no presentaban alteraciones en la exploración neurológica y que no desarrollaron signos de disfunción neurológica evolutiva. 2) La RM es el método de elección para diagnosticar la SM, conocer su localización y extensión y valorar las posibles asociaciones etiológicas.
Encefalitis por varicela
A. Maderuelo Sánchez, A. Rayo Fernández, M. Vázquez López, M. Llanos Carrasco, S. Fernández Fernández, B. García Cuartero, A. González Vergaz y A. Arregui Sierra
Hospital Severo Ochoa, Leganés.
La varicela es habitualmente una infección de curso benigno y autolimitado en niños sanos. Aunque las complicaciones asociadas son raras en esta población, son responsables de un importante aumento en la morbilidad y mortalidad. Tras las sobreinfecciones cutáneas, las complicaciones neurológicas son las mas frecuentes (20% de los ingresos y 10% de muertes). Estas incluyen la ataxia cerebelosa (la más característica y frecuente en los niños, con pronóstico excelente), el síndrome de Reye y la encefalitis, mas frecuente en adultos y con una elevada mortalidad. Presentamos tres pacientes con encefalitis por varicela, ingresados en nuestro centro
Caso 1: Niño de 8 años con exantema variceloso típico de 6 días de evolución, que consultó por cefalea, vómitos, sensación de inestabilidad y bipedestación. En la exploración al ingreso sólo destacaba la marcha atáxica y la imposibilidad de realizar marcha en tandem. El paciente presentó un rápido empeoramiento en las siguientes 24 horas con somnolencia intensa, aumento de la ataxia troncular, incapacidad para mantener sedestación, disartria, dismetría, vómitos incoercibles y labilidad emocional. LCR : 50 leucos con predominio mononuclear, glucosa y proteínas normales. Cultivo estéril PCR Herpes virus en LCR : negativa. TAC normal , EEG con actividad de fondo enlentecida, brotes de ondas lentas y escasa reactividad . Presentó agitación y agresividad fluctuantes con afasia expresiva desde el sexto día de ingreso. Recibió tratamiento con aciclovir iv desde el inicio, añadiéndose corticoides a partir del día 13. Mejoró a partir 18 día de ingreso. Fue dado de alta a los 21 días manteniendo ligera ataxia de la marcha que persistía una semana después. Al mes del alta, el paciente presentaba una exploración normal, con buen rendimiento escolar y sin alteraciones del comportamiento.
Caso 2: Niña de 2 años, con exantema variceloso de tres días de evolución, que consultó por somnolencia, decaimiento y reinicio de la fiebre. A la exploración estaba somnolienta, con Glasgow de 13 e hipotonia generalizada con ataxia troncular en sedestación. EEG : enlentecimiento difuso de la actividad de base. LCR: 14 leucos (mononuc), resto normal. Estudios de neuroimagen normales. Tratada desde el ingreso con aciclovir y dexametasona alternó fases de irritabilidad y somnolencia hasta el sexto día, con buena evolución posterior
Caso 3: Niño de 4 años, con varicela de 6 días de evolución, que acude por cefalea de escasas horas de evolución y dolor nucal. A la exploración se objetivó rigidez de nuca con meningeos positivos. LCR :126 leucos ( 70 % PMN ) glucosa y proteínas normales. Cultivo normal. EEG enlentecimiento generalizado. Ante la sospecha de meningoencefalitis por varicela se inicia tratamiento con aciclovir. Buena evolución, con temblor posicional y dismetría en maniobra dedo-nariz en las primeras 48 horas de ingreso y sin asociar alteración en nivel de conciencia. Alta a los 7 días con exploración normal.
Comentarios: Las complicaciones neurológicas de la varicela suponen un aumento de la morbilidad y mortalidad de esta enfermedad infecciosa tan frecuente. Este coste se evitaría con la aplicación universal de la vacunación, dado su elevado nivel de protección y su inocuidad.
Gastroenterología y Hepatología
Pancreatitis en la infancia. Revisión de nuestra casuística en los últimos 10 años
E. Aleo Luján, J.J. Borraz Torca, L. Arruza Gómez, C. Maluenda Carrillo y A. Bodas Pinedo
Hospital Clínico Universitario San Carlos, Madrid.
Objetivos: La pancreatitis se considera un cuadro de dolor abdominal poco frecuente en la infancia. La utilización de nuevas técnicas de imagen permite un diagnóstico clínico y etiológico más precoz y con ello mejorar las posibilidades de tratamiento. El objetivo de nuestro trabajo ha sido revisar los casos diagnosticados en nuestro hospital en los últimos 10 años, analizando etiología, métodos diagnósticos y manejo clínico empleado.
Material y métodos: Revisión de los casos diagnosticados de pancreatitis en niños entre 0-16 años en nuestro hospital durante el período 1990-2000. Consideramos criterio diagnóstico la clínica sugerente junto con hiperamilasemia.
Resultados: La distribución por sexos fue de 8 mujeres y 1 varón. Rango de edad: 7-16 años (mediana 11,5 años). Pruebas de laboratorio: niveles de amilasa (media 1601 U/l) y de lipasa (media 506 U/l) al ingreso. Pruebas de imagen: ecografía, TC abdominal, colangiopancreatografía retrógada endoscópica (CPRE) en 1 caso, colangiopancreatografía por Resonancia Magnética (CPRM) en 2 casos. Etiología: litiasis biliar (2 casos), tóxicos (3 casos), hipercolesterolemia (1 caso), idiopática (3 casos). Tratamiento: médico (dieta, analgesia, octreótido en 1 caso, antioxidantes en 1 caso); quirúrgico en 2 casos. Complicaciones: pseudoquiste pancreático en 2 casos.
Conclusiones: La incidencia de pancreatitis en la infancia es baja, aunque más alta de lo que se pensaba. En nuestra serie afecta predominantemente a mujeres adolescentes. Las causas predominantes son la idiopática y la secundaria a tóxicos. El diagnóstico se efectuó por la elevación de niveles de amilasa y lipasa en sangre, acompañado de pruebas de imagen (ecografía, TC abdominal, CPRE, CPRM según los casos). El tratamiento básico han sido las medidas de soporte (dieta absoluta y analgesia), acompañado de octreótido, antioxidantes o tratamiento quirúrgico en función de la etiología.
Actitud ante un paciente que ha ingerido un caústico
C. Moreno Hurtado, S. Gámez, I. Blesa, R. Núñez Núñez y E. Blesa Sánchez
Hospital Universitario Infanta Cristina, Badajoz.
Introducción: La ingestión de cáusticos en la infancia plantea la necesidad de conocer la peligrosidad del producto ingerido, decidir qué pacientes deben derivarse a un centro hospitalario e iniciar o no, tratamiento. En el Hospital es preciso conocer la actuación en urgencias, los criterios de ingreso y el tratamiento a seguir, cuándo indicar una endoscopia y el tratamiento de las lesiones graves producidas por la causticación esofágica.
Pacientes y métodos: Se revisan las fichas de urgencias de pacientes de 0 a 14 años de edad que acudieron a nuestro hospital, entre los años 1986 y 1998, por sospecha de haber ingerido productos potencialmente cáusticos. Los clasificamos en tres grupos, según que tras su atención en urgencias hubieran sido enviados a sus domicilios (Grupo A), ingresados en el Hospital sin precisar futuros ingresos (Grupo B), o ingresados varias veces (Grupo C). En todos ellos, valoramos las variables siguientes: edad, sexo, tipo de producto y medidas previas al ingreso. En el Grupo B se valoró el tratamiento tras el ingreso, realización o no de endoscopia y grado de lesión esofágica. En los pacientes del Grupo C se valoran, además el número de dilataciones necesarias y su duración, complicaciones evolutivas,0 situación final de su esófago y tipo de dieta tolerada.
Resultados: Durante el período revisado acudieron a urgencias 529 pacientes con sospecha de haber ingerido sustancias cáusticas. Los productos ingeridos han sido muy variados, siendo el más frecuente la lejía (57%), seguido de detergentes (9,5%), lavavajillas industrial (7,2%), aguarrás (5,3%), amoníaco (3,6%), sosa cáustica (3,2%), agua fuerte (1,5%), desatascador (0,7%) y una amplia miscelánea agrupada como otros (12%).
Antes de acudir a urgencias, el 43% de los niños se había sometido a algún tipo de tratamiento. Sólo 80 pacientes (15%) fueron ingresados (Grupo B) y no se realizó endoscopia en 23 de ellos, dada la práctica ausencia de síntomas y su buena evolución. En los demás, la endoscopia no reveló lesiones en 14 y en 43, fueron de Grado I o II. En los 13 pacientes del Grupo C, la endoscopia demostró lesiones esofágicas Grado III, que evolucionaron a estenosis esofágica con disfagia. En todos ellos se realizaron dilataciones esofágicas, con una duración media de 24,7 meses. A los 6 meses de la ingesta, más del 75% de los pacientes del Grupo C presentaban morfología refluyente en el tránsito digestivo. La curación en menos de 2 años se logró en 10 pacientes.
Sólo los que ingirieron sosa cáustica o lavavajillas industrial, sufrieron lesiones esofágicas graves que obligaron a varios ingresos y a tratamiento agresivo. Ningún paciente de los que ingirió lejía presentó lesiones esofágicas.
Conclusiones: En nuestra experiencia, la derivación de pacientes tras ingestión de cáusticos a un Centro de nivel 3, es innecesariamente elevada, al igual que el número de ingresos y de exploraciones endoscópicas realizadas. Todo ello puede disminuir si se tiene en cuenta la capacidad lesiva del cáustico ingerido y la valoración meticulosa de cada paciente.
Utilidad de la manometría anorrectal para la valoración y el tratamiento de la encopresis secundaria
C. Moreno Hurtado, M. Blesa, I. Vargas, R. Núñez Núñez y E. Blesa Sánchez
Hospital Universitario Infanta Cristina, Badajoz.
Introducción: Se acepta que existen alteraciones anomanométricas en niños con encopresis secundaria, aunque se discute cuáles son las variables afectadas y con qué frecuencia e intensidad. Aún se discute más la utilidad del biofeedback (BFB) basado en procedimientos anomanométricos en estos pacientes.
Pacientes y métodos: Se analizan los resultados de distintas variables anomanométricas obtenidas en 87 pacientes con encopresis secundaria, divididos en dos grupos, según hubiesen (n = 41) o no hubiesen (n = 47) sido sometidos a BFB, en dependencia de la mayor o menor gravedad clínica y afectación anomanométrica de la encopresis. Ambos grupos están constituidos por pacientes con edades (8,5 años), sexos (H/M: 3/1), edad al inicio de la encopresis (5,5 años) y tiempo de evolución de ésta similares.
Resultados: En el total de pacientes apreciamos, en relación a pacientes sin patología, acortamiento del canal anal (-15%) con discreta disminución de su presión, mayor proporción de pacientes con elasticidad disminuida del esfínter interno y con ausencia o negatividad del reflejo de continencia, así como disminución de la sensibilidad rectal (100%), aumento del umbral de relajación del esfínter interno (+35%) y mayor dificultad para la realización normal de la maniobra defecatoria, con contracción paradójica ocasional del esfínter externo. Estas alteraciones fueron mayores en los pacientes sometidos a BFB.
A pesar de la mayor afectación clínica y anomanométrica en los pacientes sometidos a BFB, de 10 de éstos, controlados a los 17,8 ± 3,22 años, sólo 1 volvió a presentar encopresis, mientras que sí la presentaron 9 de 11 no tratados mediante BFB, a los 12,64 ± 3,26 años de vida.
Conclusiones: La manometría anorrectal es útil para cuantificar las alteraciones típicas en pacientes con encopresis, del perfil del canal anal, reflejo inhibidor del ano, reflejo de continencia, umbral de relajación del esfínter interno, sensibilidad rectal y maniobra defecatoria. La reeducación mediante BFB basado en procedimientos anomanométricos es, en nuestra experiencia, muy útil para el tratamiento de la encopresis secundaria y sus indicaciones , a nuestro juicio podrían ampliarse a la gran mayoría de estos niños.
Deficiencia de hierro en las parasitosis intestinales
J.L. Olivares López, R. Fernández Fernández, M. Vázquez Olivares, J. Fleta Zaragozano y A. Clavel Parrilla
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Antecedentes y objetivos: La carencia de hierro es la causa mas frecuente de malnutrición en el mundo. Las parasitosis intestinales siguen siendo un problema de salud pública. Este estudio muestra las carencias de hemoglobina y del metabolismo del hierro en la fase aguda de la parasitación por Giardia lamblia, Enterobius vermicularis y Cryptosporidium parvum y su recuperación posterior, tras eliminación del parásito, sin aporte de hierro.
Métodos: Durante un período de 12 meses se recogieron 3036 muestras de heces de 1959 niños de los cuales 26 estaban parasitados por Giardia y 20 por Cryptosporidium. Se analizaron 939 pruebas de cello de 688 niños, de los que 42 tenían Enterobius. Fueron identificados respectivamente según técnicas de Ritchie y Graham. La Giardia fue tratada con tinidazol a 50 mg/kg/dosis única, repetida a los 14 días. Para Enterobius se utilizó pamoato de pirantel a 10 mg/kg/dosis única, repetida a los 14 días. Para Cryptosporidium sólo se hizo tratamiento sintomático. La hemoglobina se cuantificó mediante técnica de la cianometahemoglobina. El hierrro mediante el método de la ferrozina. La ferritina mediante un test inmunoenzimático. Se ha realizado análisis estadístico de normalidad con la modificación de Lilliefors, el test t-Student para las diferencias entre medias y el análisis de la varianza de una vía y de medidas repetidas para comparar su control evolutivo. Cuando no se cumple la homogeneidad de las varianzas se ha utilizado el test de Kruskall-Vallis. La significación estadística ha sido definida cuando p < 0,05. En todos los niños se ha efectuado el estudio en dos tiempos, al diagnóstico y dos o tres meses mas tarde, una vez desaparecida la parasitación.
Resultados: La edad media de los niños fue 6,85 + 4,16 años, con un rango de edad comprendido entre 10 meses y 15 años. La prevalencia de la parasitación ha sido, Giardia 2,65%, Cryptosporidium 1,53% y Enterobius 10,61%. Antes del tratamiento: Hemoglobina (< 11 g/dL), 9 (10,22%); hierro (50 ug/dL), 19 (21,59%); ferritina (< 12 ng/mL), 13 (14,77%); saturacion de transferrina (< 16%), 10 (11,36%). Después del tratamiento: Hemoglobina (< 11 g/dL), 3 (3,40%); hierro (50 ug/dL), 5 (5,68%); ferritina (< 12 ng/mL), 5 (5,68%); saturacion de transferrina (< 16%), 3 (3,40%). El análisis de la varianza muestra un incremento temporal significativo (p < 0,05), entre los dos momentos de estudio, para la hemoglobina, sideremia, ferritina, transferrina e índice de saturación de transferrina.
Conclusiones:
1) La prevalencia de la parasitosis, en nuestro medio, sigue siendo alta.
2) Los pacientes parasitados tienen deficiencias de hemoglobina, hierro, ferritina, transferrina, e índice de saturación de transferrina, en la fase aguda de parasitación.
3) El tratamiento de la parasitación, sin aporte de hierro, mejora de manera significativa las tasas de hemoglobina y los parámetros del metabolismo del hierro.
Valoración de los hábitos de alimentación y del estado nutricional en niños afectos de tumores sólidos durante seis meses de tratamiento antitumoral
C. Mata Fernández, D. Jiménez Villarreal, C. Azcona San Julián, J. Gimeno Ballester y L. Sierrasesúmaga Ariznabarreta
Clínica Universitaria de Navarra, Pamplona.
La incidencia de malnutrición proteico-energética en los niños con cáncer al diagnóstico es de un 8% a un 20% y puede alcanzar cifras de hasta el 40-60% a lo largo del tratamiento. La disminución en la ingesta de nutrientes es el principal factor que contribuye a la pérdida de peso en los pacientes con tumores malignos. Puede ser debida a trastornos anatómicos o funcionales del tubo digestivo como la obstrucción intestinal, formaciones de fístulas, disfagia y diarrea. Así mismo, las náuseas y vómitos debidas principalmente a efectos secundarios del tratamiento quimioterápico, disminuyen o hacen imposible algunas veces la ingesta de alimentos. Pero con mucha más frecuencia es la anorexia, que aparece en la mayoría de los pacientes desde el diagnóstico, la responsable de la ingesta inadecuada de nutrientes. Otras manifestaciones en los pacientes con cáncer son las aversiones alimentarias, las alteraciones en el gusto y el olfato y la depresión reactiva que aparecen a lo largo del tratamiento, y contribuyen al empeoramiento de la anorexia.
Objetivo: Valorar los hábitos de alimentación y el estado nutricional en niños afectos de tumores sólidos durante seis meses de tratamiento antitumoral, tanto en régimen de hospitalización como en su domicilio.
Pacientes y métodos: Se estudian durante seis meses de tratamiento antitumoral 12 niños elegidos al azar (5 niños y 7 niñas), afectos de tumores sólidos, de edad media 13,1 años (rango: 4-18). Durante los 6 meses que duró el estudio recibieron al menos tres tratamientos de quimioterapia y algunos también radioterapia y cirugía. Se realiza durante el ingreso hospitalario y en su domicilio un cuestionario de hábitos de alimentación, un recuento nutricional de 72 horas y de frecuencia de consumo alimentos. Se determinan al inicio del estudio el peso, la talla y el índice de masa corporal.
Resultados: Relación Peso-Talla: el 50% de los niños muestra un peso por debajo del percentil 50, y de estos el 17% están entre el percentil 3 y el 10 del correspondiente a su edad. Las niñas presentan un peso en percentiles superiores a los niños. Los pacientes de menor edad se encuentran en percentiles más bajos. En cuanto a la talla el 75% de los pacientes se encuentran por encima del percentil 50 y un 25% de estos por encima del percentil 97. Número de comidas al día: Todos los niños en su domicilio tras el tratamiento realizan cuatro comidas al día, mientras que durante el mismo sólo un 8% ingiere las cuatro comidas. El desayuno es la toma mas frecuente. Factores implicados en la disminución de la ingesta de alimentos durante el ingreso hospitalario: los más frecuentemente referidos son: pérdida de gusto, náuseas y vómitos, molestias orales, odinofagia y aversión alimentaria secundaria a quimioterapia. También está implicada la anorexia relacionada con factores psicológicos (depresión, disputas continuas con la familia por la comida, expresión de la ira miedo y confusión que sienten utilizando la comida como elemento de regateo...). Tipo de alimentación: La distribución de los alimentos ingeridos (en porcentajes) es: leche y derivados (16%), bollería (15%), patatas fritas y frutas (10% cada uno), bocadillos, huevos y pasta (9% cada uno), carne (6%) legumbres (5%), embutido y verduras (4% cada uno), pescado (2%) y frutos secos (1%).
La ingesta energética total (Kcal) es inferior a la recomendada (en todos los grupos de edad, tanto niños como niñas). La distribución energética de los principios inmediatos es inadecuada ya que se observa un aumento en el porcentaje de hidratos de carbono y proteínas y una disminución en el aporte lipídico.
Conclusiones: Este grupo de pacientes muestra un mal hábito alimentario incluso antes de confirmarse la enfermedad, que se acentúa durante el tratamiento. Su alimentación es inadecuada tanto en cuanto a su composición como al aporte energético. Dado que el desayuno es la comida del día mejor realizada convendría reforzarla.
Reeducación del esfínter anal en los pacientes con mielomeningocele
C. Moreno Hurtado, G. Blanco, I. Blesa, I. Vargas, R. Núñez Núñez y E. Blesa Sánchez
Hospital Universitario Infanta Cristina, Badajoz.
Introducción: En el mielomeningocele se producen trastornos de la defecación consistentes en incontinencia fecal asociada a grados variables de estreñimiento.
Con objeto de conseguir mejorar la continencia fecal para llevar una vida social lo más normal posible, han surgido diversas opciones terapéuticas con resultados variables. Con las técnicas de biofeedback (BFB) pueden conseguirse en muchos casos evacuaciones fecales completas de una forma más fisiológica que con otros métodos y una continencia más parecida a lo normal. Presentamos nuestra experiencia con esta terapia, basada en procedimientos anomanométricos, en pacientes con mielomeningocele.
Pacientes y métodos: Hemos estudiado todos los pacientes afectos de mielomeningocele remitidos a nuestra Unidad de Diagnóstico Instrumental para valoración funcional anorrectal o estudio de estreñimiento y/o incontinencia. En todos se realizó un primer estudio funcional mediante manometría anorrectal utilizando sondas tipo Arhan para lactantes y niños mayores y una sonda elaborada en nuestra unidad para los recién nacidos, polígrafo "Elige® 130 T de tres canales" y PC Polygraf® de 4 canales. Se realizó biofeedback a todos los pacientes que reunieron los criterios siguientes: 5 años o más, umbral de percepción < 60 ml, presencia de reflejo inhibidor del ano, contracción de glúteos, colaboración del niño y su familia. Se realizan sesiones que consisten en la distensión dentro del recto del balón de la sonda de manometría. Esta distensión debe ser percibida por el paciente y ante ella debe intentar aumentar la presión sobre el margen anal ayudado por la musculatura glútea. Como los cambios de presión son vistos por el paciente en una gráfica, se le invita a mejorar progresivamente su potencia y duración. Como complemento se realizan maniobras defecatorias, consistentes en hacer que el niño expulse un balón inflado en el recto, utilizando el diafragma y la prensa abdominal.
Resultados: Valoramos 24 pacientes de 4,37 ± 4,16 años de edad (13 días a 15 años). Doce de ellos reunieron los criterios exigidos para realizar BFB. Se realizaron entre 4 y 65 sesiones (35,143 ± 19,97) tras las cuales se consiguió una disminución del volumen de percepción rectal de 24.667 ± 14.133 a 3,33 ± 2,95 ml y un aumento de la presión sobre el margen anal que pasó de 45,625 ± 17,82 a 114,375 ± 13,99 mmHg. Estas diferencias fueron altamente significativas (p < 0,001). La maniobra defecatoria se normalizó en la mayoría de pacientes. Así mismo, se consiguió una mejoría clínica clara en 10 de los 12 pacientes que pasaron a tener una continencia fecal completa la mayor parte del tiempo.
Conclusiones: Consideramos que el biofeedback basado en procedimientos anomanométricos es un medio más útil que otras terapias conservadoras, para los problemas defecatorios de los pacientes afectos de mielomeningocele, puesto que consigue una mejoría clínica en la mayoría de los pacientes, con una continencia adecuada y sin los inconvenientes de otros procedimientos.
Hiperplasia nodular focal múltiple hepática en mujer adolescente
A. Millán Jiménez, J.L. Sarasa Corral, D. Vallejo Díaz, G. Pérez Tejerizo, A. Leal Orozco, J.A. Fernández Merchán y M. Ruiz Moreno
Fundación Jiménez Díaz, Madrid.
Antecedentes: La presencia de múltiples lesiones ocupantes de espacio (LOES) en el parénquima hepático es un hallazgo poco frecuente en niños y adolescentes, sin patología hepática previa. La hiperplasia nodular focal múltiple es una posibilidad diagnóstica de etiología benigna, que suele cursar de forma asintomática.
Caso clínico: Se trata de una adolescente de 14 años, previamente sana, que consulta por dolor en hipocondrio derecho irradiado a escápula homolateral, y náuseas, de 6 horas de evolución. No refiere alteración del tránsito intestinal, síntomas urinarios ni traumatismo local. Afebril. Niega consumo de anovulatorios u otros fármacos o drogas. Ecografía-doppler hepática: ligera hepatomegalia, con múltiples lesiones focales de diverso tamaño, de ecogenicidad irregular y sin signos de hipertensión portal. Hemograma, VSG, estudio de coagulación y proteinograma: en rango normal. Alfa-fetoproteina: 4 ng/ml ( < 20). ASAT: 17 UI/L, ALAT: 21 UI/L, GGTP: 29 UI/L, FA: 124 UI/L. Anticuerpos no órgano específicos: negativos. Estudio negativo de hepatitis virales. Gammagrafía hepática con tecnecio 99: múltiples lesiones ocupantes de espacio que no captan el trazador. Resonancia y TAC con contraste: hepatomegalia con múltiples imágenes sólidas, hiperintensas en la fase arterial e isointensas o hipointensas en la fase portal y tardía. Biopsia, con aguja dirigida por laparoscopia, de una lesión hepática: proliferación hepatocelular sin signos de malignidad compatible con hiperplasia nodular focal.
Evolución: Sin tratamiento activo, a los 7 meses del diagnóstico está asintomática y un control con ecografía-doppler muestra lesiones de tamaño y características similares a las observadas inicialmente.
Comentarios: La hiperplasia nodular focal múltiple es de etiología idiopática, tiene pronóstico favorable y sólo requiere un manejo terapéutico expectante. El diagnóstico diferencial con adenomas múltiples, hiperplasia nodular regenerativa, hemangiomas o lesiones tumorales malignas exige un exhaustivo estudio diagnóstico, que debe incluir biopsia dirigida por laparoscopia, con objeto de garantizar la obtención de tejido patológico.
Anticuerpos antitransglutaminasa tisular, diagnóstico y seguimiento de la enfermedad celiaca
C. Encabo Jiménez, J. de Manueles Jiménez, P. Guevara Ramírez, C. González Rodríguez, A. Grande Benito e I. Corral Carabias
Hospital Clínico Universitario, Salamanca.
Objetivo: Valorar si el análisis de Ac antitransglutaminasa tisular (tTG-Ab) es útil en el diagnóstico y seguimiento de la enfermedad celiaca, frente a determinaciones de Ac antigliadiana, Ac antiendomisio, Ac antireticulina y la biopsia de intestino delgado.
Material y métodos: Análisis de 70 sueros de pacientes de edades comprendidas entre 114 años. 36 varones y 34 mujeres. Se determinan:
- Ac antigliadina IgA-IgG: método de ELISA. (+ > 10 U/ml)
- Ac antireticulina R1 IgA: inmunofluorescencia indirecta (+ > 1/20)
- Ac antiendomisio IgA: inmunofluorescencia indirecta (+ > 1/5)
- Ac antitransglutaminasa tisular IgA: método ELISA. Se han utilizado cuatro equipos comerciales (1- Ag purificado + > 15 U/ml; 2- Ag purificado + > 25 U/ml; 3- Ag purificado + > 25 U/ml; 4- Ag recombinante + > 8 U/ml )
Los pacientes, todos ellos con valores de Ig A dentro de límites normales, han sido distribuidos en cuatro grupos para su estudio: a) no enfermedad celiaca.(55); b) enfermos celiacos en período de provocación con gluten (3); c) enfermos celiacos con dieta exenta de gluten.(8); d) enfermedad celiaca no confirmada (4).
Resultados:
SensibilidadEspecificidad
Ac antiendomisio100%100%
Ac anti liadina I100%93,3%
Ac tTG1 - 100%1 - 96,7%
2 - 100%2 - 100%
3 - 100%3 - 98,3%
4 - 100%4 - 100%
Ac anti liadina I G85,7%66,6%
Ac antireticulina R 185,7%100%
Conclusiones: Determinaciones de Ac antiendomisio y tTG-Ab son útiles en el estudio de la enfermedad celiaca (ambas presentan una alta sensibilidad). Variando el punto de corte de alguna de los equipos para determinar tTG se llegaría a una concordancia total con los resultados en los Ac antiendomisio.
Determinaciones de Ac antigliadina Ig G y de Ac antireticulina presentan resultados pobres, por lo que se podrían no solicitar de forma rutinaria. Valorar Ac antigliadiana Ig G cuando haya un déficit de Ig A.
Siempre realizar biopsia de intestino delgado para el diagnóstico exacto de enfermedad celíaca.
Comparación de parámetros antropométricos y nutricionales entre dos poblaciones de características similares
I. Rodríguez Ruiz, A. Mariño Gil, B. Ferriz Mas, M.P. Bulo Concellón, E. Meléndez Bellido y M. Casanova Bellido
Universidad de Cádiz (Cátedra de Pediatría), Cádiz, Hospital Universitario de Puerto Real, Puerto Real.
Memoria y objetivos: La aceleración secular, el estilo de vida y los hábitos alimenticios son los factores responsables de las modificaciones que se producen en el status nutricional de las poblaciones. El objetivo de este estudio ha sido conocer si existen diferencias entre parámetros antopométricos y nutricionales de dos poblaciones de similares características en un intervalo de cinco años.
Material y métodos: La población A compuesta por 745 niños de los que 349 eran varones y 396 mujeres, distribuidos por grupos de sexo y edades desde los 6 a los 14 años. La población B formada por 365 niños de los cuales 198 eran varones y 177 mujeres, distribuidos en grupos. En todos ellos se determinó el peso (P), la talla (T), el índice de masa corporal (IMC), los pliegues subescapular (PS ) y tricipital (PT) y el cociente p. subescapular/p. tricipital (PS/PT). Estadística: se calculan las medias y las diferencias mediante el test de la T de Student para datos no pareados.
Resultados: Se han incrementado los siguientes parámetros de la serie B con diferencias significativas (p < 0,05). En los niños: IMC a los 12 y 13 años (3,18 y 3,13 kg/m2). PT a los 7, 8, 10 y 13 años (3,26; 2,47; 2,32; 5,57 mm). PS a los 10 y 13 años (3,78 y 3,49 mm). PS/PT a los 7 y 11 años (0,142 y 0,12 mm). En las niñas: P a los 6 y 14 años (3,45 y 5,61 g). IMC a los 6 y 12 (1,67; 2,68 kg/m2). PT a los 6 años (3,79 mm). PS a los 3 años (2,56 mm). PS/PT a los 7 y 9 años (0,064; 0,019).
Comentarios: No encontramos uniformidad en las variaciones halladas en los diferentes parámetros para los distintos grupos. Sin embargo existe la tendencia en algunos de ellos al incremento de los indicadores de adiposidad general (IMC) sola o combinada con los de adiposidad periférica (PT) o troncular (PS, PS/PT).
Estudio comparativo de las gatroenteritis en lactantes y escolares ingresados en un hospital general
A.I. Jiménez Moya, M.T. Penela Vélez de Guevara, R. Gracia Remiro, C. Santana Rodríguez, S. Jiménez Casso, D. Romero Secos y M. Herrera Artín
Hospital General de Segovia, Segovia.
Introducción: La gastroenteritis aguda (GEA) sigue siendo un importante motivo de ingreso hospitalario en la edad pediátrica. Su incidencia, frecuencia y etiología, varían según la edad del paciente, época del año y región geográfica. Aunque con las actuales pautas de rehidratación oral se logra una respuesta favorable, numerosos niños siguen siendo hospitalizados por GEA.
Objetivos: Valorar los datos epidemiológicos, clínicos y microbiológicos relativos a las GEA en lactantes y escolares que se producen en nuestra provincia y que requieren ingreso hospitalario, y contrastar los datos obtenidos de ambos grupos etarios.
Material y métodos: Realizamos un estudio retrospectivo de todos los casos de GEA ingresados en nuestro hospital, de referencia para la provincia, en las secciones de lacatantes (1-24 meses) y escolares ( 2-14 años). El período de estudio ocupa desde enero-1996 a septiembre-1998, ambos inclusive. Revisamos las historias clínicas de dichos pacientes, recogiendo datos analíticos, clínicos, epidemiológicos y microbiológicos.
Resultados:
LactantesEscolares
Ingesos (% sobre el total)25%8%
Razón varón/mujer1,01,2
Razón urbano/rural2,01,4
Incidencia estacionalFeb-Mar-Abr-May-AgoJun-Jul-Ago-Sep
Toxoinfección alimentaria1%5%
Vómitos60%65%
Diarrea85%75%
Densidad urinaria > 103022%31%
PH < 7,3011%4%
Germen más frecuenteRotavirusSalmonella enteritidis
Dieta absoluta inicial60%88%
Fluidoterapia endovenosa87%87%
Estancia media4 días3 días
Los cuadros de GEA continúan siendo un importante motivo de ingreso, sobre todo en lactantes. Se presentan con mayor incidencia en los meses del período estival. Los datos clínicos más frecuentes fueron: diarrea y vómitos. El germen causal detectado con mayor frecuencia en lactantes fue Rotavirus y en escolares Salmonella enterítidis. Resaltamos la escasa relevancia de los datos obtenidos con el coprocultivo a la hora de instaurar el tratamiento. Llama la atención el elevado porcentaje de ingresos por GEA en comparación con otros centros, pudiendo estar justificado por la ausencia de área de observación pediátrica en nuestro hospital, así como por la dispersión geográfica de la población infantil que atendemos.
Rehidratación oral con bebidas refrescantes. Riesgo de yatrogenia en la infancia
I. Vitoria Miñana, X. Castells Piera, E. Ochoa Avila y T. Arias Jordá
Hospital Lluis Alcays, Xàtiva, Centro de Salud Padre Jofré, Valencia.
Antecedentes: Un aspecto básico del tratamiento de la diarrea aguda es la solución de rehidratación oral (SRO).Sin embargo, cada vez se emplean más las bebidas refrescantes (BR) como alternativas al SRO en lactantes y niños. El objetivo de este estudio es comparar las recomendaciones de ESPGHAN para la SRO en Europa con la composición de BR españolas.
Métodos: Se ha analizado la composición de glucosa, sodio, potasio y osmolaridad de 61 BR comercializadas en España. La concentración de sodio y potasio se ha determinado por potenciometría indirecta con electrodo selectivo. La glucosa se ha analizado por el método de la hexocinasa y la osmolaridad por osmometría basada en el punto de congelación.Los aparatos utilizados han sido un autoanalizador modular (Roche Diagnostics) y Microosmómetro 3MO (Advanced Instruments).
Resultados: En las siguientes tablas se expresan las concentraciones de sodio, potasio, glucosa y osmolaridad de los 61 refrescos agrupados en 7 tipos de bebida así como las recomendaciones de la ESPGHAN sobre la composición de la SRO. (véase tabla supeiror)
De los refrescos analizados, las bebidas isotónicas son las que más sodio contienen (valor medio 17,5; valor máximo de 33 mmol/l para Isostar®), pero muy inferiores a los 60 mmol/l recomendados para SRO. Respecto al potasio, sólo los zumos de frutas contienen cifras aceptables aunque insuficiente sodio y excesiva osmolaridad. En cuanto a la glucosa, tanto las gaseosas como las colas light prácticamente no la contienen. La relación glucosa/sodio es excesiva en todos los grupos de bebidas excepto las colas light por contener escasa glucosa. La osmolaridad es inacepable es todos los grupos de bebidas (en 5 por exceso y en 2 por defecto). Los riesgos del empleo de BR como SRO son la falta de reposición electrolítica (de sodio fundamentalmente) y la persistencia de la diarrea tanto por la excesiva carga osmolar así como por la falta de reversión de la bomba de sodio acoplada a la glucosa.
Conclusiones: Es inadecuado el uso de refrescos como SRO en el tratamiento de la diarrea aguda del niño por: a) Insuficiente glucosa, sodio y osmolaridad (colas light y gaseosas); b) Excesivas osmolaridad y relación glucosa/sodio con sodio insuficiente (colas, zumos, isotónicas, tés y otros)
Estudio de marcadores serológicos de hepatitis B en niños saharauis en régimen de acogida
J. Sánchez Gimeno, J.J. Ventura Juara, J. Martín Carpi, E. Lomba y L. Ros Mar
Hospital Materno Infantil Miguel Servet, Zaragoza.
Introducción: La hepatitis B es una enfermedad actualmente casi erradicada en la población infantil de nuestro país tras la instauración de la vacunación sistemática en el período neonatal. Por el contrario no ocurre lo mismo en otras poblaciones con origen en áreas de bajo nivel socio-económico que residen temporalmente en nuestras ciudades como es el caso de los niños saharauis en régimen de acogida.
Objetivos: Estudio de los patrones serológicos de hepatitis B en una población de niños saharauis y valoración de la posible utilidad de medidas profilácticas a realizar.
Material y métodos: Determinación de marcadores serológicos (HbsAg, HbeAg, Anti HBc IgG, Anti HBc IgM, Anti HBs) en una población de 48 niños saharauis (27 mujeres y 21 varones) de edades comprendidas entre 6 años y 11 meses y 17 años, con una edad media de 11 años y 9 meses, en régimen de acogida en familias aragonesas. Así mismo se realizó la determinación de enzimas hepáticas.
Resultados: De los 48 pacientes estudiados, encontramos negatividad frente a los marcadores de hepatitis B en 32 de ellos (66,66%). En los 16 pacientes restantes (33,33%) alguno de los marcadores fue positivo, siguiendo los siguientes patrones serológicos:
Anti HBc IgG + (contacto con Hepatitis B)1
Anti HBs y Anti HBc + (Hepatitis B pasada)9
HBsAg, HBeAg y Anti HBc + (portador crónico o hepatitis crónica)5
HBsAg, Anti HBs y Anti HBe + (portador sano)1
En todos los casos estudiados los valores de las enzimas hepáticas se encontraron dentro del rango de normalidad.
Conclusiones:1) En nuestro estudio hemos objetivado una elevada seroprevalencia de marcadores de hepatitis B en esta población, con un 18,75% de niños que habían pasado la enfermedad, un 10,41% en estado de portador crónico o afectos de hepatitis crónica y un 2,08% en estado de portador sano (a la espera de confirmación con la determinación del DNA viral). 2) Consideramos que unas mínimas mediadas higiénicas evitarían la posibilidad de contagio en las familias de acogida. 3) Dado el aumento de los programas de acogida de estos niños en nuestro país, consideramos necesaria la realización de estudios serológicos encaminados a la detección de pacientes seropositivos y la adopción de las medidas terapéuticas adecuadas. 4) En los pacientes que se demuestre seronegatividad creemos necesaria la puesta en marcha de una campaña de vacunación. 5) Así mismo queremos llamar la atención de la autoridades sanitarias y de las diferentes ONGs de cara a promover programas de vacunación precoz en sus núcleos de origen.
Genética y Dismorfología
Diagnóstico prenatal de nefromegalia en el SDR. de ojo de gato
M. Puerta Vílchez, J.A. Peña Ortega, E. Fernández Tardáguila, M.J. Montes González, J,L. Barrionuevo Porras y M. Pérez
Hospital Universitario Virgen de las Nieves, Granada.
Introducción: El sdr. de ojo de gato o de Schmid-Fraccaro, se define por coloboma de iris, atresia anal y otras alteraciones. La alteración cromosómica causante del cuadro consiste en la existencia de un segmento tri-o cuadriplicado del cromosoma 22 consistente en satélites, el brazo corto entero, el centrómero y un trozo del brazo largo (22p ter-22q11). Puede cursar con afectación de múltiples sistemas: alteraciones cardíacas, digestivas, urogenitales y un fenotipo característico.
Caso clínico: RNAT. EG: 40+2. Peso: 3.510 gr. Talla: 50 cm. PC: 34 cm. Apgar: 9-9. Reanimación tipo I. En Ecografía prenatal al 7º mes se diagnosticó de Nefromegalia. Al nacimiento se aprecia atresia anal y apéndice preauricular derecho. A los dos meses de edad se diagnostica de coloboma de iris y retina. A los cinco meses sufre crisis de cianosis, sudoración y cansancio con las tomas. Precisó ingreso en UCIP. Se diagnostica entonces de drenaje venoso anómalo total supradiafragmático en vena imnominada. Se realiza RMN donde se evidencia agenesia del cuerpo calloso y estudio citogenético, con el resultado de 46XY + un marcador. En estudio gammagráfico se aprecia riñón multiquístico derecho afuncionante.
Evolución: A los 7 meses fenotípicamente presenta: cabello escaso, fontanela anterior amplia, linea capilar anterior en "V" con vello en la frente, cejas anchas, mirada amaurótica con nistagmo horizontal, coloboma bilateral, pabellones auriculares con antehélix prominente, CAE estrecho, tórax prominente, miembros delgados, dedos largos en manos y pies, anomalías en los pliegues palmares. Peso 5,78 (M, -3SDS), T: 68,7 (M), PC: 42,5 (-1, -2SDS). Aún no había alcanzado la sedestación. A los 15 meses: P: 9,75 (-1, -2SDS), T: 80,2 (M, +1SDS), PC: 470,5 (M). Se repite estudio citogenético comprobándose que el marcador corresponde a un cromosoma 22 parcial (47XY + 22p ter-22q11 en todas las metafases). A los 27 meses su crecimiento sigue normal con un pronóstico de talla genética de 177,5 ± 8 y una talla por la curva de crecimiento de 181. Deambulación alcanzada a los 23 meses.
Conclusiones:1) Es muy importante un detenido examen ocular en el RN, pues pueden pasar desapercibidas numerosas anomalías (coloboma, cataratas congénitas, etc) que se asocian a determinados síndromes dismórficos. 2) Se debería realizar estudio citogenético de líquido amniótico ante el hallazgo ecográfico de nefromegalia intraútero para descartar su asociación a cromosomopatías. 3) Aunque las alteraciones citogenéticas de este síndrome se pueden presentar sin manifestaciones clínicas también pueden existir casos de gran expresividad fenotípica como es el caso que aquí presentamos.
Síndrome de Lejeune. Presentación de un caso
E. Navarro Hinojosa, L. Ortiz González, J.F. Mesa, A. Vilela, E. Piñán, J.M. Arroyo Fernández y J.L. Paz
Hospital General de Mérida, Mérida.
Introducción y objetivo: El Síndrome fue descrito por Lejeune y cols en 1963, esta producido por una delección del brazo corto del cromosoma 5, que incluye la región 5p-14-15, zona critica para que se manifieste clínicamente. La prevalencia oscila 1/20000 y 1/45000 nacidos vivos, siendo mas frecuente en hembras. Hasta en el 85% de los casos la lesion es producida de "novo" en el propio paciente y solo en 10-15% es heredable a partir de un portador equilibrado. Los rasgos clínicos comunes en la mayoría de los afectos son dismorfia facial, microcefalia, retraso mental severo y de recién nacido un característico llanto agudo y quejumbroso que semeja el maullido de un gato, denominándose por tal motivo síndrome de "Cri du chat". Actualmente ha pasado de ser una deficiencia profunda con muy mal pronostico a ser una deficiencia moderada severa al beneficiarse de la estimulación precoz. Presentamos el caso de un recién nacido varón afecto de una delección parcial del cromosoma 5.
Resultados: Caso clínico. Recién nacido que ingresa en nuestro servicio con 9 horas de vida por hipotermia, rasgos dismórficos y un llanto débil y mantenido característico. Fruto de 1ª gestación controlada de madre de 24 a. y padre de 28 a. sin patología. Parto espontáneo, cefálico a las 39 semanas de e.g, presentando un APGAR de r.n de 8/10. El examen físico revelo: hipotonía global moderada, llanto agudo mantenido, reflejo de recién nacido disminuidos. Peso 2.950 g. (P 10-50), Talla 47,5 (P10-50), P C: 32,5cm (P3). Cara redondeada, orejas grandes malformadas de baja implantación. Oblicuidad palpebral antimongoloide y micrognatia. Puños cerrados con anteposición de ambos pulgares. Surco simiesco bilateral Clic de cadera bilateral y pie equino-varo. El resto de exploración por aparatos fue normal.
Se realizan exámenes complementarios para descartar patología asociada: EKG, EEG, Ecografía cerebral y abdominal y ecocardio normales. Estudio ORL revela una epiglotis globulosa en forma de omega. Se realiza Cariotipo presentando 46 XY del (5) pter 15.
Desarrollo evolutivo. El desarrollo ponderoestatural ha sido normal para su edad y sexo. El PC ha sido el parámetro somatométrico mas afectado, al 3 mes de vida cae por debajo del P3 no recuperándolo en ningún momento de su desarrollo. El seguimiento realizado desde su nacimiento ha presentado un retraso psicomotor evidente, sedestación al año, deambulacion a los 3 años. Con 4 años come y bebe solo, iniciando minimo lenguaje basado fundamentalmente en gesto y signos, interesándose por todo lo que le rodea. Se realiza RNM por incoordinación motora de mmss presentando ligera hipoplasia vermiana. Con 5 años buena comprensión, lenguaje mínimo. Actualmente con 7 años controla esfínteres es sociable se comunica fundamentalmente con gesto y signos, ausencia de frases, articula palabras sueltas, realiza dos ordenes sencillas seguidas. Desde el punto de vista psicoterapéutico inicia estimulación precoz desde los primeros meses de vida y desde del 5º año acude a un centro de educación especial donde continua aprendiendo y adquiriendo nuevas habilidades.
Discusión: El factor educativo es fundamental, llevado a cabo por psicólogos, pedagogos, logopedas y psicomotricistas que consiguen mejorar las expectativas de futuro de los afectos de este síndrome. El pronostico, no obstante, va a depender de la extensión de la deleccion y de las patologías asociadas al síndrome.
Asociación de déficit de GH con síndrome velocardiofacial con microdelección 22q11
J.A. Peña Ortega, J.L. Barrionuevo Porras, M. Puerta Vílchez, M.J. Montes González, E. Fernández Tardáguila, R. Espigares Martín, F. Ruiz-Cabello Osuna y L. Ortega Martos
Hospital Universitario Virgen de las Nieves, Granada.
Introducción: El síndrome velocardiofacial se describió por primera vez en 1978 por Shprintzen como un síndrome polimalformativo caracterizado por hendidura palatina, defectos cardiacos y un fenotipo facial característico, con una herencia autosómica dominante. Posteriormente se descubrió la presencia de la microdelección 22q11 en gran parte de estos pacientes, lo que los incluye dentro del síndrome CATCH-22: defectos Cardiacos, facies Anómala, hipoplasia Tímica, Cleft del paladar e Hipocalcemia; asociados a microdelección en 22q11.
Objetivo: Presentar un caso de síndrome velocardiofacial con estudio genético molecular de síndrome CATCH-22 positivo en el que se asocia un déficit clásico de GH.
Caso clínico: Paciente que es remitido a la consulta para valoración dismorfológica por Cardiología con 9 años y 7 meses. Antecedentes personales: al nacer se detecta fisura palatina, presentando a los dos días de vida cianosis, frialdad y rechazo del alimento, diagnosticándose de sd. de Fallot. Se le realizó cirugía paliativa a los 3 meses , y corrección definitiva a los 6 años. Inicialmente succión mala, con crecimiento muy lento, retraso psicomotor, con deambulación a los 2 años. Lenguaje tardío con mala pronunciación, voz nasal. Miopía severa desde la primera infancia y escoliosis después de la cirugía cardiaca definitiva. Exploración física: peso 21,2 Kg (-2DS); talla 122,7 cm (-2DS); PC 54 cm (-1-2DS). Hendidura palpebral 2,5 cm. Distancia intercantal 3,7 cm. Distancia interpupilar 5,5 cm. Dolicocefalia. Cara estrecha, con hendiduras pequeñas hacia abajo. Retrognatia. Paladar estrecho con cresta central. Paladar blando restaurado, no úvula. Dientes mal alineados. Dorso nasal convexo, prominente, con punta engrosada. Alas nasales hipoplásicas. Pabellones auriculares despegados y rotados, con concha en U. Torax carinatum. Asimetría de hombros. Cicatriz de esternotomía media.
Inicialmente es diagnosticado de síndrome caracterizado por dismorfia craneofacial, cardiopatía y alteraciones esqueléticas, compatible con sd. velocardiofacial. Seguimiento posterior: con 11 años 3 meses, peso 21,6 Kg; talla 129,3 cm; PC 54; retraso escolar, no signos puberales. Con 12 años 2 meses: peso 24,6 Kg; talla 133 cm; PC 54 cm . Edad ósea 9 años 6 meses. Test de clonidina: basal 0,5; pico 3,8 a los 90´. Se repite test de GH mediante hipoglucemia insulínica, presentando basal de 0,30 y pico de 3,85 con una glucemia de 39. RM craneal: pequeña zona de atrofia cortical, localizada a nivel parieto-occipital derecha. A los 12 años 8 meses: peso 25,5 Kg; talla 134,3 cm; PC 54,8 cm. A los 13 años 2 meses: peso 26,6 Kg; talla 137,6 cm; PC 54,5 cm. Se autoriza tratamiento con GH con 2 unidades al día de forma continuada. A los 13 años y 8 meses: peso 32,2 Kg; talla 142 cm; PC 55,5 cm. Velocidad de crecimiento 4,4 cm en 6 meses. Se realiza estudio genético molecular de sd. CATCH-22 que es positivo
Conclusiones:1) Los estudios de genética molecular permiten confirmar diagnósticos clínicos que anteriormente sólo podían basarse en la descripción fenotípica. 2) Estos estudios permiten comprender el origen común de diferentes síndromes anteriormente caracterizados, con las implicaciones clínicas y terapéuticas que conlleva. 3) Presentamos en nuestro caso la asociación de déficit de GH con el sd. velocardiofacial, asociación descrita en un número reducido de casos y que permite ampliar el fenotipo.
Trisomía parcial 8 q como consecuencia de una translocación parental
M. de Felipe Jiménez-Casquet, C. Fuentes Gutiérrez, J.A. López Medina, J.L. Barrionuevo Porras, A. Gallego Torres, M.R. Benavides Román, P. Cárdenas Guerrero y J. Moreno Martín
Hospital Universitario Virgen de las Nieves, Granada.
Antecedentes: La trisomía 8 presenta una incidencia del 0,45/1000 recién nacidos vivos, siguiendo en frecuencia a las tres autosomías clásicas (Sd. Down, Sd. de Edwars, Sd de Patau). El 85% de los casos son genotípicamente mosaicismos, alternando las células trisómicas con las normales. La trisomía completa es aparentemente mortal durante la vida intrauterina. Existen trisomías parciales, la mayoría de los casos descritos por duplicación del brazo corto del cromosoma 8. Presentamos un caso excepcional en la literatura pediátrica de trisomía parcial del brazo largo del cromosoma 8, como consecuencia de una translocación recíproca entre cromosomas 8 y 14 maternos.
Caso clínico: Madre 32 años, multípara, Fórmula obstétrica 20222, polihidramnios por eco prenatal al 6º mes de vida. Recién nacida a término de 40 semanas de gestación, peso 3640 g (P75), talla 52 cm(P75-90), perímetro cefálico 36 cm (P90). Apgar al minuto de 8 y a los 5 minutos de 9. A la exploración presenta braquicefalia, cara plana, hipertelorismo, ojos hundidos, angiomas planos en glabela, párpados, filtrum labial y occipital. Raiz nasal ancha con narinas antevertidas y anchas, labio inferior grueso y evertido, paladar ojival, anquiloglosia, micrognatia, pabellones auriculares de implantación baja y ligeramente plegados. Cuello corto. Mamilas hipoplásicas y separadas, tórax y abdomen normal. Genitales femeninos normales, angiomas en región sacra, sinus pilonidal y surcos palmares y plantares profundos. En las pruebas complementarias realizadas se obtiene un hemograma, gasometría y bioquímica completa normal. Radiografía de tórax y pelvis, cráneo y serie ósea normal. TAC y RMN craneal normal. Fondo de ojo sin hallazgos. Ecografía cardíaca: ductus permeable y comunicación interventricular muscular mínima. El estudio citogenético en sangre periférica con bandas G: fórmula cromosómica 46 XX, add (14)(q32), en todas las metafases analizadas, con material extra en el cromosoma 14 identificándose éste como procedente del cromosoma 8. Estudio citogenético paterno: normal. Estudio citogenético materno: 46 XX, t(8;14)(q22;q32).
En sucesivos controles se aprecia retraso pondo-estatural y psicomotor, desde su diagnóstico se realiza estimulación psicomotriz precoz y seguimiento periódico.
Conclusión: El síndrome de la trisomía 8 se define como dismorfia cráneo-facial y retraso psicomotor con signos característicos como los pliegues palmo-plantares profundos y labios gruesos con labio inferior evertido. La mejoría de las técnicas de diagnóstico citogenético han permitido describir nuevos casos de trisomía autosómica, como el caso presentado: trisomía 8 parcial con material extra del brazo largo del cromosoma 8 debido a translocación recíproca equilibrada 8q 14q materna, con fenotipo similar a los mosaicismos y trisomías 8 p descritos. El diagnóstico en el período neonatal nos permite una vigilancia estrecha y detección precoz de las posibles complicaciones asociadas (anemia hipoplásica, neutropenia, infecciones, neoplasias hematológicas y de otras localizaciones).
¿Tercera familia con braquidactilia y displasia esquelética o un nuevo tipo de braquidactilia?
M.J. Sánchez Álvarez, G. Rodríguez Criado, M. Taguas-Casaño y A. Carranza
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción: Las braquidactilias son un heterogéneo grupo de malformaciones, en ocasiones difícil de encuadrar. La clasificación de Bell (1951) es de gran utilidad para su estudio. El objetivo de nuestro trabajo es investigar el diagnóstico de una familia con braquidactilia.
Métodos: Exploración clínica y estudio radiológico de la paciente índice, su madre y su abuela y hemos recogido los antecedentes de su bisabuela y de un tío abuelo de la rama materna también afectos. Se ha realizado el árbol genealógico de la familia y se ha revisado la literatura sobre el tema.
Resultados: El caso index muestra braquidactilia de manos y pies con pulgares en forma de autoestopista y dedos gordos en valgo marcado, montando sobre los segundos que como el resto de los dedos de los pies están en varo. La abuela los tiene igual. La madre sólo tiene cortedad del segundo metacarpiano. Las Rx del caso índice y de su abuela muestran cortedad de los metatarsianos y de las falanges proximales de los dedos 1-2 y 3 de manera variable, junto con discretas anomalías de sus epífisis y metáfisis proximales, así como de los metacarpianos y de las falages proximales de los dedos gordos. Los peronés son largos. No muestran otras anomalías morfológicas ni intelectuales.
La amplia y actualizada revisión bibliográfica muestra similitud de nuestros pacientes con la braquidactilia E2 (McKusick y Milch 1964) con metacarpianos y metatarsianos cortos con variable afectación falángica y herencia AD, pero nuestros casos presentan elongación de los peronés y formas características de los dedos gordos y pulgares. Estas formas son similares a las de las familias descritas por Mononen (1992) junto con discretas anomalías metafisarias y epifisarias con el nombre de braquidactilia con displasia esquelética, ligada a X dominante. Nuestros enfermos no tienen las anomalías carpianas y tarsianas de las familias de Mononen. La familia que mostramos tiene un patrón hereditario que parece AD, pero no se puede descartar una herencia ligada a X.
Conclusiones: Presentamos la tercera familia publicada con el síndrome de Mononen o alternativamente estamos ante un nuevo tipo de braquidactilia con un presumible patrón AD, no pudiendose descartar la herencia ligada a X.
Diagnóstico presintomático de enfermedad de Charcot-Marie-Tooth tipo 1 en un niño de 10 años
L. Postigo García, P. Lozano Puente, L. Palacín González, M. Cruz Hermosilla, M.J. López, I. Bueno, F. Palau y F.J. Ramos
Hospital Clínico Universitario Lozano Blesa, Zaragoza, Hospital Universitario La Fe, Valencia.
La enfermedad de Charcot-Marie-Tooth tipo 1 (CMT-1) constituye el tipo de neuropatía hereditaria crónica más frecuente en la infancia. Se caracteriza por debilidad y atrofia lentamente progresivas con afectación distal de pies y piernas, arreflexia osteotendinosa generalizada de predominio en extremidades inferiores, alteraciones de la sensibilidad vibratoria y propioceptiva y alteraciones electroneurográficas. Son autosómicos dominantes alrededor de un 70-80% de los casos, y la mayoría de ellos son debidos a la duplicación de un segmento de 1,5 Mb del cromosoma 17p 11,2 que engloba al gen de la proteína de la mielina periférica PMP-22 (forma CMT-1ª).
Presentamos el caso de un paciente de 10 años que acude a la consulta para estudio genético, por historia familiar de neuropatía hereditaria tipo CMT-1. En el momento de la primera visita está asintomático y realiza actividades deportivas diversas sin dificultad. A la exploración general se aprecia hipotrofia discreta bilateral de la musculatura de pantorrillas predominantemente en compartimento anteroexterno y pies cavos. Dificultad para caminar de talones. En la exploración neurológica arreflexia rotuliana y pequeñas alteraciones de la sensibilidad poco valorables.
En el Electroneurograma (ENG) se objetiva afectación muy avanzada con disminución importante de la velocidad de conducción motora y sensitiva, compatible con neuropatía periférica. Se realiza estudio molecular de la duplicación del locus CMT-1 A/NHPP en nuestro paciente así como en su padre (sintomático), abuela paterna (sintomática), hermana de 19 años (sintomática) y hermana de 15 años (asintomática). Son portadores de la duplicación y por tanto enfermos, todos los estudiados excepto la hermana menor.
Nuestro paciente se controla periódicamente en consultas de genética y neuropediatría y acude a rehabilitación para paliar posibles deformidades progresivas. En el momento actual tiene 15 años, clínicamente presenta dificultades para correr y a la exploración ha aumentado levemente su deformidad en pie cavo.
Este paciente constituye un caso de diagnóstico presintomático; queremos destacar la importancia de los estudios neurofisiológicos y de los estudios moleculares de ADN para detectar portadores asintomáticos, y establecer un seguimiento y tratamiento adecuados así como asesoramiento genético.
Síndrome Branquio-oto-renal. Una nueva familia
M. Taguas-Casaño, G. Rodríguez Criado, M.J. Sánchez Álvarez, A. González-Meneses López y A. Sánchez Moreno
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes: El síndrome otobranquiorrenal (SBOR) es una enfermedad autosómica dominante con una gran variabilidad clínica, incluso intrafamiliar. Se caracteriza por un defecto en la audición (en el 93% de los casos) que puede ser de transmisión, neurosensorial o mixta; malformaciones a nivel del oido externo, medio e interno (36-82%); anomalías branquiales consistentes en fístulas o quistes (50%) y anomalías renales que varían desde aplasia bilateral a leve hipoplasia (67%). Sólo en una parte de los pacientes se ha encontrado una alteración genética consistente en la mutación del gen EYA 1, localizado en 8q13.3. Nuestro objetivo es investigar una familia afecta de este síndrome a partir de un caso índice.
Método: Estudio clínico, analítico, radiológico y citogenético del caso índice, así como estudio de la familia y comparación con la bibliografía actualizada.
Resultados: Paciente varón, nacido el 27-10-96. Fruto único de embarazo con oligoamnios, diagnosticado al mes de vida de insuficiencia renal. Porta botón de gastrostomía desde 1997 por dificultad en la alimentación. En tratamiento desde 1998 con hormona de crecimiento por talla baja. Actualmente el paciente está en programa de diálisis por insuficiencia renal crónica terminal y en lista de espera de transplante. Tiene además una sordera neurosensorial. Visto por primera vez a los cuatro años en la Unidad de Dismorfología. Exploración: peso 16 Kg (p10), talla 98 cm (p3), PC 49,5 cm. Orejas en forma de copa, gachas, sin apéndices preauriculares ni quistes ni fístulas branquiales. Paladar hendido. Genu valgo. Estrabismo convergente. Pruebas complementarias: Ecografía abdominal: severa hipoplasia renal bilateral con pequeños quistes en su interior. CUMS: normal. Potenciales evocados auditivos: respuesta mal estructurada y umbral de 50 dB en ambos oídos. TAC de mastoides y estudio molecular genético pendiente de realización. Antecedentes familiares: madre sana; padre con insuficiencia renal en diálisis y sordera neurosensorial; dos hermanos sanos; tía paterna con sordera neurosensorial; primo paterno con senos preauriculares; abuela paterna con sordera neurosensorial, y bisabuela paterna con sordera.
Los criterios diagnósticos de SBOR son sordera, apéndices preauriculares, orejas gachas o en copa, fístulas branquiales y anomalías renales; siendo necesario tres de estos criterios o dos con antecedentes familiares de 2 o más afectos. La alteración genética asociada con el SBOR se ha comprobado en otros síndromes que comparten características con éste como son el otobranquial, otobranquiofacial, otobranquioureteral; esto, junto a que dentro de una familia pueden existen miembros con alguno de estos síndromes, corrobora la hipótesis establecida de que todos ellos son la expresión diferente de una misma alteración. También se han descrito fenotipos BOR con anomalía genética no ligadas a 8q13.3 (Stratakis y cols 1998).
Conclusión: Nuestro caso índice y su familia reúnen los criterios diagnósticos necesarios para encuadrarlo dentro del síndrome BOR.
Agenesia renal y alteraciones de la fusión de los conductos de Müller
J.J. Borraz Torca, L. Arruza Gómez, B. Nievas Soriano, B. Agúndez Reigosa, B. Sánchez de la Vara y B. Losada Pinedo
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: Se presenta el caso de una niña de 13 años con agenesia renal izquierda y otras malformaciones del aparato urinario que asociaba útero doble con hemivagina ciega izquierda.
Caso clínico: Adolescente mujer de 13 años que acude a nuestro servicio de urgencias por dolor en fosa iliaca izquierda (FII) a punta de dedo, de 16 horas de evolución. En la exploración al ingreso presentaba buen estado general, febrícula, normocoloración mucocutánea, acné e hirsutismo. El abdomen era blando, depresible, sin defensa, con dolor selectivo a la palpación en FII y discreto rebote. Genitales externos femeninos normales. El resto de la exploración no mostró otros datos de interés.
Como antecedentes de interés destacaba la presencia de agenesia renal izquierda diagnosticada en la infancia por infecciones de repetición, y ureterocele congénito derecho. Epigastralgias recurrentes de un año de evolución. Se realizó ecografía abdominal ginecológica en la que se evidenció útero doble con colección de 5,2 cm de diámetro compatible con hematocolpos y hematometra, con hemivagina ciega izquierda. El estudio por RMN realizado posteriormente confirmó la presencia de estos hallazgos, así como la agenesia renal izquierda.
Se realizó una histerectomía izquierda simple mediante laparotomía.
Discusión: La incidencia global de malformaciones congénitas del aparato genital femenino es del 5 al 30/1000. No son un diagnóstico habitual de la edad pediátrica, ya que no suelen presentar clínica hasta edades medias de la vida. No obstante, es importante tenerlas presentes en adolescentes con patología uro y ginecológica o dolor abdominal. Hasta un 35-90% de los casos las malformaciones genitales están asociadas a anomalías urinarias, y en el caso de los úteros bidelfos, con hemivaginas obstructivas, las agenesias renales ipsilaterales suelen ser la norma. Por tanto, ante cualquier malformación urinaria es importante descartar otras ginecológicas asociadas, y viceversa.
Rubinstein-Taybi y Glaucoma congénito bilateral. Importancia de la orientación inicial
G. Villar Villar, B. Nievas Soriano, R. Gómez Casares, V. San Antonio Arce, A. Retuerta Oliva y R. Jiménez Baos
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: El síndrome de Rubinstein-Taybi es un síndrome polimalformativo con anomalías de la cara, pulgares, dedos gordos del pie, asociado a retraso mental y otras posibles malformaciones, entre las cuales una de las menos frecuentes pero más importantes por sus posibilidades terapéuticas es el glaucoma congénito bilateral. Presentamos un caso de asociación de ambos procesos diagnosticados precozmente.
Caso clínico: Niña de 3 meses que acude remitida de su hospital diagnosticada clínicamente de síndrome de Rubinstein-Taybi, para realizar estudio oftalmológico. Como antecedentes personales presentaba un embarazo y parto normales, con ictericia neonatal fisiológica, screening metabólico normal, vacunación incorrecta y sin alergias conocidas. A la exploración al ingreso presentaba peso 4150 (< P3), 57 cms (P25) de talla y perímetro craneal de 37 (< P3). En dicha exploración destacó una facies peculiar con nevus flameus en frente, raíz nasal y nuca. Nariz en pico de loro, hipertelorismo. Paladar ojival. Hipertricosis de dorso y raíz de miembros superiores. Dedos pulgares mal implantados, desviados y anchos. Clinodactilia. Neurológicamente presentaba escaso sostén cefálico y leve hipotonía axial. El resto de la exploración no mostró otras alteraciones significativas.
Evolución: A su ingreso se realizó exploración oftalmológica bajo anestesia general que confirmó la presencia de un glaucoma congénito bilateral, realizándose goniotomía de 120º cursando ésta sin complicaciones. Fue dada de alta y actualmente acude a revisiones periódicas.
Discusión: El síndrome de Rubinstein-Taybi se caracteriza por la asociación de las malformaciones clínicas descritas, y la posible afectación de otros órganos, como corazón, riñón, pulmón, testículos y sistema óseo. Sin embargo, y a pesar de que el glaucoma congénito es una asociación infrecuente, hoy en día se plantea en la literatura revisada la importancia de realizar un estudio oftalmológico en todos los casos en los que la clínica pudiera orientar a este síndrome, debido a las posibilidades terapéuticas de esta patología oftalmológica. Esta exploración debe ser detallada y completa, a ser posible en centros con experiencia en el tratamiento de dicha patología y siempre orientada por el pediatra general.
Neumología
Discinesia ciliar primaria: caso clínicode diagnóstico tardío
E. Esteban Torné, S. Herrero Pérez, N. López Segura, M. Viñolas Tolosa, R.M. Busquets Monge, G. Figueras Figueras y J. Lloreta Trull
Hospital del Mar de Barcelona, Barcelona.
Antecedentes y objetivos: La discinesia ciliar primaria (DCP) es una enfermedad de origen congénito caracterizada por la disfunción total o parcial de las células ciliadas o flageladas. El diagnóstico precoz es fundamental para un mejor control de los síntomas.
Material: Paciente mujer de 18 años que ingresa por atelectasia masiva del pulmón derecho sin fiebre ni sintomatología respiratoria previa. Antecedentes familiares de asma. Antecedentes patológicos: Crisis de asma repetidas desde los 5 meses de edad que requieren tres ingresos por reagudizaciones y tratamiento intercrítico con corticoides inhalados y ß2 agonistas de acción prolongada. Sinusitis y rinitis de repetición. Diagnosticada de alergia a ácaros, polen de olivo y epitelio de gato. IG E elevada. Fumadora desde los 17 años de 10 cigarrillos/día. A los 17 años ingresa por atelectasia de lóbulos medio e inferior derechos por tapón mucoso. Se realizó broncoscopia con desobstrucción del árbol bronquial derecho y normalidad anatómica. Citología y baciloscopia de BAS negativos. En el mismo ingreso se realizó serología de Aspergillus que fue negativa. Funcionalismo respiratorio: patrón obstructivo. Test del sudor negativo.
Evolución: Tras nueva desobstrucción del árbol bronquial derecho es dada de alta con tratamiento antiinflamatorio inhalado. Posteriormente se realiza cepillado de epitelio nasal en el que se evidencia pérdida del brazo interno de la dineína en el 70% de los cilios observados sin alteraciones ultraestructurales que sugieran componente secundario. Test de la sacarosa positivo. La paciente es diagnosticada de discinesia ciliar primaria y sigue tratamiento antiinflamatorio y fisioterapia respiratoria. Actualmente presenta una mejor evolución clínica y ha dejado de fumar.
Discusión: El diagnóstico de la DCP se realiza mediante en estudio funcional y estructural de los cilios. En el caso presentado ambos diagnósticos son confirmatorios. Es muy importante el diagnóstico precoz de esta entidad para un mejor control de la sintomatología y para intentar disminuir la morbilidad de esta entidad. El caso que hemos presentado es un ejemplo de la evolución tórpida que pueden seguir estos pacientes sin un control adecuado.
Neumopatía asociada a Hidatidosis
C. Moreno Hurtado, E. Blesa Sánchez, R. Núñez Núñez, R. Cabrera García y J.I. Santamaría Osorio
Hospital Universitario Infanta Cristina, Badajoz.
Introducción: La Hidatidosis pulmonar cuando se presenta en forma típica no suele plantear problemas diagnósticos. Sin embargo, con cierta frecuencia puede presentarse con formas complicadas, como ocurrió en los pacientes motivo de esta comunicación, en los que la neumopatía asociada dificultó enormemente su diagnóstico.
Pacientes y métodos: Revisamos las hidatidosis pulmonares tratadas en nuestro centro durante los últimos 20 años. Valoramos la edad, sexo, procedencia, manifestaciones clínicas y métodos diagnósticos. Consideramos especialmente las dudas diagnósticas surgidas y la enorme dificultad del diagnóstico diferencial cuando la neumopatía asociada es grave.
Resultados: Del total de 46 pacientes intervenidos de hidatidosis pulmonar, 11 presentaron neumopatía asociada grave. La sintomatología predominante en 9 pacientes, fue de dolor torácico, dificultad respiratoria y fiebre elevada. Además, 4 de ellos presentaron tos irritativa de larga evolución, acompañada de esputos hemoptoicos en 2 y vómica en 3. Los otros 2 pacientes sólo presentaron tos recurrente, 1 de ellos con hemoptisis. A todos se les realizó radiografía simple de tórax, encontrándose: hidroneumotórax a tensión en 3, neumoquiste en 6 (todos con condensación pulmonar periquística importante, que hacía difícil su interpretación) y en 2, derrame pleural simple. 3 pacientes presentaron 2, 3 y 5 quistes en el mismo pulmón y uno, afectación bilateral. Los hallazgos radiológicos nos hicieron sospechar la existencia de hidatidosis, en 5 pacientes en los que existían imágenes sugerentes de membrana hidatídica en el interior de una cavidad aérea. Dos de ellos con hidroneumotórax y 3 con neumoquiste. En los otros 6, el diagnóstico fue de mayor dificultad. La TAC sugirió hidatidosis, al detectar membranas incarceradas en 2 de ellos. En otros 2 se practicó toracotomía siendo la hidatidosis una de las varias causas sospechadas. En los 2 pacientes con derrame pleural, la radiografía de control posdrenaje mostró el quiste hidatídico.
Conclusiones: Las formas complicadas de hidatidosis pulmonar exigen un alto índice de sospecha y experiencia previa para realizar un diagnóstico preoperatorio correcto. Su confusión es especialmente probable con otras causas de hidroneumotórax, abscesos pulmonares, neumonías masivas y derrames pleurales. En éstos es especialmente peligrosa la punción, ya que se puede ocasionar la rotura del quiste hidatídico.
Bronquiolitis y asma ¿es la eosinofilia un factor pronóstico?
A. Rayo Fernández, C. Calvo Rey, M.L. García García, M.R. Albañil Ballesteros y M. Tomico del Río
Hospital Severo Ochoa, Leganés, Centro de Salud Cuzco, Fuenlabrada.
Objetivos: Conocer la evolución en el tiempo de los pacientes menores de 2 años que presentaron un episodio de bronquiolitis y analizar los factores de riesgo de desarrollar asma posterior, con especial atención a la presencia de eosinofilia en el episodio agudo.
Pacientes y métodos: Se analizaron los lactantes < 2 años que precisaron ingreso por bronquiolitis durante tres temporadas epidemiológicas (1990-1993), recogiendo valores de eosinófilos durante el episodio agudo y su evolución en el tiempo (asma a corto plazo < 5 años de edad, asma a largo plazo > 5 años o no desarrollo de asma). Se valoraron otros factores de riesgo de asma (historia familiar de asma, tabaquismo pasivo, edad y aislamiento VRS).
Resultados: Se estudiaron 170 pacientes (100 niños y 70 niñas) que sufrieron un episodio de bronquiolitis (edad inicial 5,4 ± 4,2 meses y edad actual (7,15 ± 0,78 años). Desarrollaron asma a corto plazo 113 pacientes (66,5%) y asma a largo plazo 60 pacientes (35,3%).
Los valores medios de eosinófilos fueron más elevados en los pacientes que desarrollaron asma en algún momento de su evolución (p = 0,028). El número de pacientes con eosinófilos > 1% resulta significativamente diferente entre los tres grupos de pacientes estudiados (p = 0,029). El porcentaje de eosinófilos > 1% en el momento del ingreso se asoció con asma a corto plazo (p = 0,013). No se encontró relación entre eosinofilia y asma a largo plazo. La historia familiar de asma se asoció significativamente con el desarrollo de asma a largo plazo (p = 0,033).
Conclusiones: En nuestro medio la eosinofilia > 1% durante el episodio de bronquiolitis en lactantes < 2 años se asocia con un mayor riesgo de presentar asma en los primeros años de la vida, así como la historia familiar de asma con el desarrollo de asma a largo plazo.
Incidencia de ingresos por sibilancias en lactantes menores de 2 años: 6 años de estudio
A. Díaz Moro, S. Lapeña López de Armentia, L. Castañón López, J.P. Martínez Badás, I. Fernández Natal, J. Mallo Castaño y E. Alvaro Iglesias
Hospital de León, León.
Objetivo: Conocer la incidencia de ingresos por sibilancias en lactantes menores de 2 años en nuestro medio y su relación con factores perinatales.
Materiales y métodos: Se realiza estudio de cohortes retrospectivo de todos los lactantes menores de 2 años, ingresados en nuestro Servicio por sibilancias desde el 1-1-95 hasta el 31-12-00 y que hayan nacido en nuestra maternidad.
Resultados: Entre 1995-1998 nacen en nuestra maternidad un total de 7.766 neonatos, 4.064 varones (52,3%) y 3.702 mujeres (47,7%). Ingresan por presentar sibilancias (bronquitis, bronquiolitis) 204 lactantes, con una incidencia acumulada de 2,6% (IC95%: 2,2%-2,9%), siendo VRS+ 106 (52%). La edad media es de 6,1 ± 5,1 (mediana: 5 meses) y el 89% ingresan antes de los 12 meses de vida. De los 204 pacientes hospitalizados, 120 son varones (no significativo; RR:1,3; IC95%: 1,0%-1,7%). El riesgo relativo (RR) más elevado lo encontramos para edad gestacional ¾ 30 semanas (6 ingresos de 27, siendo 3 VRS+; P < 0,0001; RR: 8,7; IC95%: 4,2-17,8), y peso al nacimiento < 1200 gr (3 ingresos de 15, siendo 1 VRS+; P < 0,001; RR: 7,71; IC95%: 2,8-21,4). Menor influencia tienen otros factores analizados como mes de nacimiento, mes de alta de la maternidad, o estancia en Neonatología.
Conclusiones: 1º- El 2,6% de los neonatos nacidos en nuestra maternidad ingresan antes de los 2 años por presentar sibilancias, de los cuales el 52% son VRS+. 2º- El 89% de los ingresos se producen antes del año de vida. 3º- Presentan mayor riesgo si son prematuros o con bajo peso al nacimiento.
Budesonida inhalada como tratamiento de mantenimiento en la hemosiderosis pulmonar idiopática . A propósito de un caso
P. Sevilla Ramos, A.M. Carrasco Torres, J.A. Gómez Carrasco, E. Vegas Muñoz, M. Alonso Cristobo y E. García de Frías
Hospital Universitario Príncipe de Asturias, Alcalá de Henares.
Introducción: La hemosiderosis pulmonar idiopática es un transtorno raro de etiología desconocida. Se caracteriza por la presencia de hemorragia pulmonar crónica o recurrente con acumulo de hemosiderina y fibrosis intersticial a largo plazo. Presentamos el caso de una paciente diagnosticada de esta enfermedad después de tres años y medio de evolución.
Caso clínico: Niña de 12 años con astenia, tos leve y fiebre máximo 38º de 3 días de evolución que acude a urgencias por vómitos hemáticos en últimas 12 horas. Como antecedentes destaca ingreso en nuestro hospital dos meses antes por anemia ferropénica (Hb 5 mg/dl) sin que se hallara el origen del sangrado. Presenta un hemograma con Hb 5 mg/dl, bioquímica, coagulación, PCR, inmunoglobulinas y sistemático de orina normales. Radiografía de tórax con patrón intersticial difuso bilateral. Ingresa con la sospecha clínica de hemosiderosis pulmonar. Se realiza broncoscopia hallándose numerosos siderófagos en el lavado broncoalveolar y acúmulo de macrófagos cargados con hemosiderina en la biopsia transbronquial con lo que se confirma el diagnóstico y se inicia tratamiento con prednisona oral 1,5 mg/kg/día durante 15 días hasta el alta. Los anticuerpos antimembrana basal glomerular fueron negativos. Desde el diagnóstico han pasado 3 años y medio, se han llevado a cabo controles periódicos con hemogramas, espirometrías y TAC torácicos sin que hayan aparecido signos de fibrosis ni nuevas recaídas de la enfermedad. Durante el primer año y medio se administraron corticoides orales a dosis decrecientes hasta total supresión. En los dos últimos años ha recibido tratamiento de mantenimiento Budesonida inhalada a dosis de 800 mcg/día, apreciándose un leve descenso en las cifras de Hb durante los primeros meses del tratamiento manteniendo a partir de entonces cifras estables.
Comentarios: La hemosiderosis pulmonar es una enfermedad rara de etiología desconocida. La triada típica es sintomatología pulmonar crónica, anemia ferropénica y alteraciones variables en la radiografía de tórax. El uso de corticoides asociados o no a inmunosupresores durante el proceso agudo parece mejorar el pronóstico. Los últimos datos publicados parecen evidenciar un aumento de supervivencia asociado al uso de tratamientos prolongados con corticoides e inmunosupresores. Los corticoides inhalados podrían ser una opción terapéutica en el tratamiento de mantenimiento de la hemosiderosis pulmonar primaria.
Carcinoma Mucoepidermoide Bronquial. Presentación con hemoptisis leve repetida en un asmático
A. Retana Castán, M.J. Elizari Saco, C. Salvador Goyanes Sotelo, N. Pardo García, C. León González, S. Bagué Rosell y C. Puzo Ardanuy
Hospital Sant Pau, Barcelona.
Introducción: El carcinoma mucoepidermoide bronquial es un tumor que deriva del tejido de las glándulas salivares del árbol traqueobronquial. Aunque sean de bajo grado de malignidad, también se han de extirpar por la posibilidad de metástasis en los ganglios linfáticos pulmonares.
Caso clínico: Paciente 15 años. En el curso de 2 meses consulta en 3 ocasiones por tos, disnea y expectoración ligeramente hemoptoica,catalogadas de crisis asmáticas leves.
Antecedentes patológicos: Bronquitis obstructivas de repetición, que se iniciaron a los 2 años. Pruebas cutáneas (4 años) positivas a polvo, ácaros, gramíneas y pelos de animales. Tratamiento habitual: broncodilatadores y antibióticos. Inmunoterapia durante dos años. A partir de los 8 años, mejora y se retira el tratamiento. Única crisis a los 12 a., con expectoración hemoptoica..
Remitido a consulta externa para nueva valoración alergológica y funcional respiratoria. Las crisis de disnea se repiten siempre con hemoptisis leves.
Exploración física normal. Peak flow superior al teórico
Pruebas complementarias: Rx tórax, analítica general y coagulación, PPD, O.R.L., prueba de esfuerzo y espirometría normales. P. cutáneas idem. IgE alta.
TC pulmonar Lesión en la pared anterior del bronquio principal izquierdo a pocos cm. de la carina.
Biopsia por broncoscopia: Carcinoma mucoepidermoide de bajo grado.
Intervención quirúrgica. Resección cuneiforme del bronquio principal izquierdo con conservación del pulmón. La anatomía patológica confirma carcinoma mucoepidermoide de bajo grado con ulceración superficial de la mucosa, los límites de la resección y los ganglios de nivel V, VI, VII estaban libres.
Comentarios: El carcinoma mucoepidermoide es raro en niños menores de 8 años. Es más frecuente en adultos. La sintomatología es inespecífica (tos y dolor torácico). Si hay obstrucción bronquial, aparecen neumonitis o atelectasias persistentes, La hemoptisis es más frecuente en el adulto. En ocasiones la clínica imita las crisis de asma bronquial, tanto en asmáticos confirmados como no.
Leiomioma endobronquial como causa de patología respiratoria de repetición
A. Pérez Palomino, C. Miranda Cid, A. Belanger-Quintana y C. Lozano Jiménez
Hospital Ramón y Cajal, Madrid.
Introducción: El leiomioma es un tumor benigno del músculo liso, extremadamente raro en la infancia. Se localiza principalmente en el aparato digestivo (27%), vejiga (15%), tejidos blandos (15%), cabeza y cuello (11%) y subcutáneo (9%). La localización pulmonar es excepcional estando documentados sólo 5 casos en la infancia, y de ellos solamente 2 en pacientes sin patología predisponente como son los estados de inmunodepresión (infección por HIV, trasplante). En mujeres jóvenes se han relacionado con la toma de anticonceptivos orales. Dentro de los pulmonares los endobronquiales sólo supondrían el 33% mientras que los parenquimatosos ocupan un 47%. A nivel traqueal aparecen en un 18%. Presentamos el caso de una paciente con leiomioma endobronquial.
Caso Clínico: Niña de 4 años y 5 meses, sin antecedentes de interés, que en los últimos 24 meses tuvo dos episodios de bronquitis, dos episodios de dificultad respiratoria y tos irritativa y una neumonía de la língula, por la que ingresa. A la exploración presenta fiebre, hiporexia, tos y dificultad respiratoria con sibilancias en hemitórax izquierdo. El hemograma, bioquímica, VSG y coagulación fueron normales con BAAR en esputo y serología TBC negativas. Hemocultivos estériles. Electrocardiograma normal. En la radiografía de tórax y TAC se objetivó atelectasia con condensación en pulmón izquierdo con presencia de adenopatías mediastínicas. Inició tratamiento con amoxicilina clavulánico intravenoso y broncodilatadores con buena evolución clínica. Se realizó broncoscopia visualizando tumoración endobronquial en bronquio principal izquierdo con obstrucción casi completa de su luz. Se extirpa por broncotomía posterior y se biopsian adenopatías, que son informadas como linfadenitis reactiva. La masa tumoral corresponde a una proliferación subepitelial constituida por células de aspecto mesenquimal quiescente, que corresponde a músculo liso, compatible con leiomioma endobronquial.
Conclusiones: Los leiomiomas pulmonares son entidades muy poco frecuentes que pueden debutar como episodios de dificultad respiratoria y de bronconeumonía si son de origen endobronquial.Los traqueales pueden producir tos irritativa. En el caso de los parenquimatosos el diagnóstico suele ser casual al realizar radiografías de tórax, ya que suelen ser asintomáticos.
Apnea del sueño tratada con presión positiva continua nasal en paciente afecto de mucopolisacaridosis
C. Trillo Belizón, S. Broncano Lupiañez, A. Rodríguez Leal, J.A. Martín García, J.M. Gómez Vida, A. Valenzuela Soria, E. Fernández Segura, B. Martín Castaño, J. Rubiño Hodar y A.M. Martín Polo
Hospital General Básico de Motril, Motril.
Antecedentes: Las mucopolisacaridosis son un grupo de enfermedades hereditarias causadas por la degradación incompleta y el depósito de mucopolisacáridos. Las manifestaciones clínicas se deben a la acumulación de éstos en diversos órganos. Hay publicados casos de acúmulo a nivel del tracto respiratorio superior, que junto a la macroglosia y retrognatia propias del fenotipo, favorecen la aparición de apneas del sueño de origen periférico que pueden ser controladas con presión positiva continua en las vías respiratorias (CPAP nasal).
Caso clínico: Niño de 10 años con fenotipo compatible con una mucopolisacaridosis tipo Hurler-Scheie que presenta somnolencia diurna que le impide la realización de su actividad habitual.
En la exploración física destaca entre sus rasgos dismórficos una gran macroglosia y microretrognatia. Opacificación corneal derecha. Otorrea purulenta bilateral. AR. Disminución de murmullo vesicular bilateral. AC: soplo sistólico III/VI en foco pulmonar y mitral. Abdomen: gran hepatoesplenomegalia. Rigidez articular de todas las extremidades. Inteligencia conservada.
Entre las pruebas complementarias destacan alteraciones radiológicas típicas de la enfermedad de Hurler, así como la excreción urinaria de dermatán sulfato y en menor proporción de heparán sulfato. En ecocardiografía se constata miocardiopatía hipertrófica con insuficiencia mitral y aórtica.
Durante una noche se realizó estudio polisomnográfico, demostrando apneas con desaturaciones prolongadas y frecuentes con patrón obstructivo. Se inició tratamiento con CPAP nasal, controlándose la sintomatología con CPAP nasal a 6 cm H2O.
Conclusiones: La CPAP nasal tiene una indicación clara en este tipo de pacientes mejorando considerablemente su calidad de vida.
Heliox: Una nueva opción terapéutica en la bronquiolitis aguda
F. Martinón-Torres, M. Picón Cotos, S. Fernández Cebrián y A. Rodríguez Núñez
Hospital Clínico Universitario de Santiago, Santiago de Compostela.
Antecedentes: Una característica central en la fisiopatología de la bronquiolitis es el incremento de las resistencias en la vía aérea y de los flujos gaseosos turbulentos, lo que origina un aumento del trabajo respiratorio del paciente, que finalmente puede conllevar fatiga y fracaso respiratorio. En este contexto, las propiedades físicas del HELIOX, podrían ser beneficiosas en estos pacientes.
Objetivo: Evaluar los efectos terapéuticos de la administración de una mezcla gaseosa de baja densidad (HELIOX: 70% helio/ 30% oxígeno) en lactantes con bronquiolitis por virus respiratorio sincitial (VRS).
Diseño: Estudio clínico prospectivo, intervencionista, comparativo.
Escenario: Unidad de Cuidados Intensivos Pediátricos, Hospital Clínico Universitario.
Pacientes: Dieciseis lactantes entre 1 mes y 12 meses consecutivamente admitidos en la UCIP para el tratamiento de una bronquiolitis aguda por VRS.
Intervención: Los 8 primeros pacientes fueron designados como grupo control y recibieron el tratamiento de soporte utilizado habitualmente en nuestra unidad (adrenalina). Los siguientes 8 pacientes recibieron además HELIOX calentado y humidificado a través de mascarilla con reservorio y válvula, siguiendo nuestro protocolo.
Mediciones: Se registraron score clínico respiratorio, frecuencia respiratoria, frecuencia cardíaca, carbónico espirado, saturación arterial de oxígeno y FiO2 basales, a intervalos horarios las primeras 4 horas, y después cada 8 horas hasta la suspensión del tratamiento con heliox o su traslado a planta. Se analizaron con fines comparativos los datos obtenidos en las primeras 4 horas. Igualmente se recogieron en cada paciente datos demográficos, edad, tiempo de evolución previa del cuadro, duración de estancia en UCIP, y duración del tratamiento con heliox.
Resultados principales (media ± sd): Basalmente no existían diferencias entre el grupo heliox y el control en edad (4,75 ± 3,4 vs 5,13 ± 3,4 meses), tiempo previo de evolución (49,5 ± 20,7 vs 45 ± 18,9 hrs), score clínico (6,44 ± 1,2 vs 6,56 ± 1,3), frecuencia cardíaca (162 ± 24,6 vs 168,8 ± 15,3 lpm), frecuencia respiratoria (63,5 ± 8 vs 60 ± 7,6 rpm), SatO2 (91,8 ± 2,9 vs 91,3 ± 2 %), etCO2 (35,7 ± 6,7 vs 33,3 ± 5,6 mmHg) y FiO2 (0,32 ± 0,1 vs 0,31 ± 0,08). Después de una hora, el score clínico disminuyó en ambos grupos, siendo el descenso significativamente mayor en los pacientes tratados con heliox (3,56 ± 1,27 vs 5,69 ± 1,19, p < 0,005). Igualmente, la frecuencia cardíaca y respiratoria eran significativamente menores en el grupo HELIOX tras una hora: 127,2 ± 12,36 vs 160,75 + 15,83 bpm (p < 0,005) y (40,25 ± 6,65 vs 56,13 ± 7,64 rpm (p < 0,005), respectivamente. Estas diferencias seguían siendo significativas transcurridas 4 horas. No se encontraron diferencias en SatO2, EtCO2 ni FiO2. La duración del tratamiento con heliox osciló entre 36 y 90 horas (59 ± 24,05 hours) y no se detectaron afectos adversos. La estancia media en UCIP fue menor en el grupo tratado con heliox, aunque no de forma significativa (4 ± 1,07 vs 5,38 ± 1,6 días).
Conclusiones: Nuestros resultados sugieren que el heliox es una opción no invasiva segura, sencilla, y eficaz en el tratamiento de la bronquiolitis aguda por VRS. El heliox mejoró globalmente el estado respiratorio de los pacientes, no solo por la marcada disminución en el score clínico sino tambien por la disminución de la taquicardia y taquipnea acompañantes. La aparición de los efectos beneficiosos ocurrió en la primera hora de tratamiento en todos los casos y se mantuvo mientras se continuó el tratamiento con heliox. Creemos que el heliox podría ser considerado un tratamiento de primera línea en el manejo de los pacientes con bronquiolitis aguda por VRS ingresados en UCIP, si bien son necesarios estudios prospectivos más amplios para establecer su lugar concreto en el esquema terapéutico de la bronquiolitis.
Neumonía de evolución tórpida como presentación de enfermedad granulomatosa crónica
M.T. Herráiz Culebras, M. Valle Velasco González, C. Casanova Macario, J.L. Aparicio Sánchez y H. Armas Ramos
Hospital Universitario de Canarias, La Laguna, Hospital Nuestra Señora de la Candelaria, Santa Cruz de Tenerife.
Introducción: La enfermedad granulomatosa crónica, raro trastorno genético con una incidencia 1/1.000.000, comprende un grupo de inmunodeficiencias debidas a defectos del complejo enzimático NADPH oxidasa de los fagocitos. Se caracteriza por infecciones recurrentes causadas por microorganismos catalasa positivos y tiene mayor expresión en varones.
Caso clínico: Varón de 5 años, en cuyos antecedentes personales destacan la necesidad de cirugía menor a los 7 meses por bultoma en muslo izquierdo, y a los 4 años, por bultoma axilar izquierdo cuyo estudio microbiológico fue negativo y objetivandose en el anatomopatológico un infiltrado granulomatoso. Presenta síndrome febril con condensación radiológica persistente en LSD, resistente a poliantibioterapia. En los exámenes complementarios destaca la elevación de reactantes de fase aguda, estudios microbiológicos negativos (cultivos ordinarios, hongos y micobacterias en sangre, jugo gástrico, broncoaspirado, lavado broncoalveolar y biopsia pulmonar abierta, serología de bacterias atípicas y Mantoux), estudio inmunológico normal (hemograma, poblaciones linfocitarias, niveles de complemento e inmunoglobulinas), y estudios para inmunodeficiencias secundarias negativos. TAC torácico con infiltrado parenquimatoso en LSD con broncograma y adenomegalias paratraqueales, subcarinales e hiliares derechas hipodensas. En anatomía patológica de biopsia pulmonar, infiltrado intersticial parcheado de linfocitos B.
Evolución: Tras buena respuesta inicial al añadir glucocorticoides, se objetiva empeoramiento al descenso de la dosis de los mismos, mostrando en esta ocasión Burkholderia cepacia en cultivo de LBA y observándose en nueva muestra obtenida por PAAF la presencia de granulomas con necrosis central. El estudio de inmunidad celular granulocítica (capacidad oxidativa por citometría) confirma la EGC probablemente en su forma autosómica. Se realiza tratamiento antibiótico dirigido y profilaxis diaria con trimetropin-sulfametoxazol con estabilidad clínica y radiológica en los 3 últimos meses.
Conclusión: En todo cuadro infeccioso de evolución tórpida, se debe excluir la existencia de una inmunodeficiencia primaria, incluyendo como primer escalón, pese a su rara incidencia, el estudio de inmunidad celular granulocítica, evitando así la realización de técnicas diagnósticas invasivas.
Hemoptisis masiva y embolización terapeútica en un paciente afecto de fibrosis quística
M. Valle Velasco González, M.T. Herráiz Culebras, T. Pérez Arvelo, J.L. Aparicio Sánchez, R. Castro Conde y P. Lupiani Castellanos
Hospital Universitario de Canarias, Santa Cruz de Tenerife.
Introducción: Si bien la hemoptisis de pequeña cuantía es común entre los enfermos afectos de fibrosis quística, su forma masiva es infrecuente (1% / año) y representa una emergencia médica con un alto índice de mortalidad (50 - 85%).
Caso clínico: Varón de 9 5/12 años, conocido afecto de fibrosis quística desde la edad de 2 años, genotipo Delta F508 homozigoto, insuficiente pancreático, con colonización respiratoria por Haemophilus influenzae, Staphylococcus aureus y Pseudomona aeruginosa, portador de gastrostomía y de reservorio central (port a cath), mostrando en su última prueba de función respiratoria un FEV1 del 55% respecto al valor teórico.
Coincidiendo con reagudización respiratoria presenta un episodio de hemoptisis masiva (> 240 ml/día), autolimitado, que recidiva durante su manejo conservador (reposo, antibioterapia intravenosa agresiva y suspensión del tratamiento inhalado y de la fisioterapia respiratoria) y requiere ingreso en la unidad de cuidados intensivos pediátricos donde se practica la embolización arterial. Previamente a ella, se realiza fibrobroncoscopia que muestra sangre proveniente del árbol bronquial derecho y una TAC torácica de alta resolución que objetiva bronquiectasias quísticas difusas, adenomegalias mediastínicas e hiliares e infiltrado de características alveolares en LSD.
Tras la embolización dirigida de dos ramas arteriales bronquiales del LSD con microcoils, cesa el sangrado.
Conclusiones: Los pacientes afectos de fibrosis quística con hemoptisis masiva deben ingresar en Centros dotados de Radiología Intervensionista. Dada la afectación pulmonar difusa de estos pacientes, la aproximación previa al punto sangrante puede favorecer el éxito de la embolización.
Neonatología
Hemocromatosis neonatal familiar
J.A. López Medina, C. Rubio Quevedo, M.D. Linares Gálvez, M. Samaniego Muñoz, J.M. Fenández Lorenzo, R. Sánchez Navarro y R. López-Jurado Romero de la Cruz
Hospital Universitario Virgen de las Nieves, Granada.
Antecedentes: La hemocromatosis neonatal (HN) es una enfermedad rara de la que se han descrito menos de 60 casos. La enfermedad tiene un curso clínico rápido y progresivo con muerte en el período neonatal precoz.
Presentamos 2 casos ocurridos en nuestra unidad neonatal:
Caso 1 (año 1998): Varón de 3190 g nacido a las 38 semanas de gestación, de una primigesta, sana de 27 años de edad. La puntuación de Apgar fue de 9 al minuto y 9 a los 5 minutos. A las 2 horas de vida el niño presentó respiración ruidosa, distensión abdominal y rechazo del alimento y de forma progresiva en 48 horas ictericia, oliguria, edemas y hemorragias diseminadas. La analítica inicial mostraba severas alteraciones en la coagulación y las proteínas totales. Las transaminasas estaban moderadamente elevadas y el hemograma se apreciaba una una ligera plaquetopenia. Tabla 1.
Caso 2 (año 2000): Hermano del anterior, Varón de 1440 g nacido a las 32 semanas de una tercera gestación, un hermano sano. La puntuación de Apgar fue de 6 al minuto y 9 a los 5 minutos precisando reanimación tipo 2. En la exploración inicial leve hipotonía con distress respiratorio. En las siguientes 24 horas se desarrollo ictericia y hemorragias diseminadas. La analítica la exponemos en la Tabla 1.
BrrdBrr indASTALTLDHAct ProtTPTAPT
(mg/dl)(mg/dl)UI/LUI/LU/L(%)(sg)(mg/dl)
Caso 18,37,62057183628734,3
Caso 25,43,5653040025684,0
Tabla 1: analítica de ambos hermanos a su ingreso. (C: caso clínico; Brr d: Bilirrubina directa; Brr ind: Bilirrubina indirecta; AST: aspartato aminotransferasa; ALT: alanina aminotransferasa; LDH: lactato deshidrogenasa; Act Prot: Actividad de Protrombina; TPTA: Tiempo parcial de Tromboplastina activado; PT: Proteínas totales).
La evolución en el caso clínico número 1 y 2 es mala con muerte a las 2 semanas de vida y 4 días de vida respectivamente.
Anatomía patológica: En ambos casos se demuestra depósito de hierro en hígado, páncreas exocrino y estómago. Asimismo este depósito también aparece en parte del sistema endocrino (tiroides, paratiroides y suprarrenales), corazón, timo y plexo coroide. El bazo los ganglios linfáticos y la médula ósea están libres de depósitos de hierro.
Conclusiones: La hemocromatosis neonatal es una entidad distinta y no asociada a la hemocromatosis del adulto. El defecto no se debe a un aumento de la absorción intestinal de hierro. La ausencia de depósitos de hierro dentro de los macrófagos en la hemocromatosis neonatal sugiere una función reticuloendotelial alterada que provocaría estos depósitos en diversos órganos. Creemos que el daño hepático en la hemocromatosis neonatal puede mejorar con tratamiento antioxidante y quelantes del hierro de forma precoz.
Tetatoma congénito cráneo-cervical gigante
P. Cárdenas Guerrero, J.A. López Medina, L. Fidel Moltó Ripoll, M. de Felipe Jiménez-Casquet, C. Fuentes Gutiérrez, M.R. Benavides Román y R. López-Jurado Romero de la Cruz
Hospital Universitario Virgen de las Nieves, Granada.
Antecedentes: El teratoma es el tumor más frecuente del período neonatal, siendo la localización sacrococcigea la más habitual en todas las series. Aparece en la mayoría de los casos en niñas, y la anatomía patológica describe componentes de las tres hojas blastodérmicas de tipo maduro. La ecografía prenatal se presenta como el método diagnóstico más fiable intraútero. Presentamos un teratoma que por su tamaño y localización creemos de especial interés
Caso clínico: Mujer embarazada de 35 años de edad, con antecedente de aborto en el primer trimestre de gestación, realiza controles ambulatorios adecuados de su embarazo. En la ecografía correspondiente a la semana 28 de gestación se describe un feto vivo, que presenta una masa en la zona cráneo-cervical de 12 x 12 cm, de consistencia mixta, y un polihidramnios de aproximadamente 9 litros. Ante estos hallazgos y tras reunión conjunta de los servicios de Obstetricia y neonatología se decide practicar cesárea en la semana 29 de edad gestacional. Nace un feto de sexo femenino con una tumoración blanco grisácea, de localización cráneo-cervical derecha, que produce gran deformidad craneal y facial que hace imposible cualquier maniobra de reanimación cardio-pulmonar. El test de Apgar es de 3 al minuto y de 0 a los cinco minutos.
La necropsia informa de un feto de sexo femenino de 1980 gramos de peso (> P90)- incluyendo la tumoración-; longitud cráneo-caudal de 36 cm (P20); perímetro cefálico de 24 cm (< P10); perímetro torácico de 20,5 cm (P20); y perímetro abdominal de 22 cm (P20). Presenta una tumoración en la zona cráneo-cervical derecha de 16 x 13 cm con agenesia de maxilares superiores y del esfenoides. Hay extensión a masa encefálica por la zona esfenoidal afecta. Asocia una importante hipoplasia pulmonar. La microscopía óptica describe una neoformación constituida por tejidos inmaduros de las tres hojas blastodérmicas (cartílago, elementos neurales primitivos y elementos glanduloides), acompañado de áreas de necrosis y calcificación. Todos estos datos son compatibles con un teratoma inmaduro.
Conclusión: En la literatura pediátrica son excepcionales los teratomas congénitos gigantes de localización cráneo-cervical. La anatomía patológica no es capaz de determinar en muchos de estos casos el origen exacto del tumor. La ecografía prenatal es el método diagnóstico de elección para el teratoma, siendo los de origen intracraneal los que más dificultades de diagnóstico precoz presentan. El pronóstico de estos tumores es sombrío y la actitud debe ser consensuada entre obstétras y neonatólogos para una adecuada actitud.
Abstinencia neonatal por Opiáceos. Casuística
J. Arenas Vilà, F. Bastida Fenoy, C. Esporrín Gállego, G. Perkal Rug, P. Adroher Tarrés y H. Dergham Dergham
Hospital Santa Caterina, Girona.
Introducción: Desde la existencia de un programa de mantenimiento con metadona, al que se acogen muchas mujeres toxicómanas, hemos visto incrementado el número de casos de Síndrome de Abstinencia Neonatal (SAN). Los recién nacidos con SAN tienen que recibir unas atenciones y unos cuidados especiales dada la sintomatología que presentan durante los primeros días o semanas.
Material y métodos: En estos últimos 5 años hemos ingresado en la unidad de neonatología un total de 13 recién nacidos, hijos de madre toxicómanas por opiáceos (8 hembras y 5 varones). De las 13 madres, 11 seguían programa de mantenimiento con metadona (PMM), las dos restantes eran adictas a la heroína.
El sistema de valoración del SAN ha sido la escala de Finnegan.
Iniciábamos tratamiento cuando la puntuación de la escala de Finnegan era mayor de 8 en dos determinaciones sucesivas.
El tratamiento administrado ha sido en todos los pacientes fenobarbital oral.
Comentarios: De los 13 pacientes, fue necesario efectuar tratamiento en 10 de ellos, y todos eran hijos de madre en PMM. El SAN fue prolongado, durando más de 10 días en 9 de los pacientes.
Conclusiones: El PMM en las gestantes toxicómanas produce generalmente una SAN más intensa y prolongada.
Es interesante estudiar nuevos tratamientos sustitutivos para utilizar en estas pacientes gestantes para no producir un cuadro de SAN tan intenso.
Monitorización de la velocidad de crecimiento en pretérminos valorado mediante knemometría
M.P. Bulo Concellón, A. Mariño Gil, I. Rodríguez Ruiz, M. Mohmed Ahmed, S. Rico de Cos y M. Casanova Bellido
Hospital Universitario de Puerto Real, Puerto Real, Universidad de Cádiz (Cátedra de Pediatría), Cádiz.
Introducción: El recién nacido prematuro constituye el grupo de edad menos estudiado desde el punto de vista auxológico. Con la aparición del knemometro manual es posible el estudio de la velocidad lineal del crecimiento de la pierna asi como la detección precoz de alteraciones en la misma difícilmente constatables con somatometría convencional.
Objetivos: Valorar la utilidad de la knemometría en la monitorización del crecimiento en recién nacidos (RN) pretérminos y comparar los datos obtenidos mediante esta técnica con los de la somatometría convencional.
Material y método: Se estudian 16 Rn de 36 semanas de edad gestacional corregida con un peso inferior a 2.000 gramos (gr) sin ninguna patología asociada salvo su prematuridad. Se recogen los siguientes datos: peso (gr), longitud (cm), perimetro craneal y torácico (cm) y longitud rodilla-talón (mm) mediante knemometría, con un intervalo de tiempo de 72 horas. Se realiza un estudio longitudinal de la velocidad de crecimiento determinándose mediante la diferencia de longitud al inicio y al final del estudio y diviendo por el número de días.
Resultados: Obtenemos los incrementos de velocidad de crecimiento por knemometría (VC), del peso y de la longitud en cada niño en intervalos de 72 horas. En la siguiente tabla se exponen los valores medios de velocidad de crecimiento de la pierna en mm/día, asi como los incrementos medios de peso (IP) en gr/día y de longitud (IL) en cm/día.
R n 1Rn 2Rn 3Rn 4Rn 5Rn 6Rn 7Rn 8
VC0,440,340,320,500,490,260,410,42
IP27,638,0720,824,3722,2214,3328,1215,45
IL0,170,160,130,180,140,120,100,13
R n 9Rn 10Rn 11Rn 12Rn 13Rn 14Rn 15Rn 16
VC0,410,360,340,360,610,500,710,72
IP19,0628,619,4117,8910,7611,7810,7130
IL0,110,120,080,070,100,100,110,17
Comentarios: La Knemometría neonatal gracias a su precisión resulta una útil para la valoración de la velocidad de crecimiento lineal de la pierna en Rn prematuros durante cortos períodos de tiempo.
Hiperqueratosis epidermolítica en recién nacido
D. Martín Fernández-Mayoralas, M. Moro Serrano, B. Sánchez de la Vara, V. San Antonio Arce, A. Retuerta Oliva y J.J. Borraz Torca
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: Se presenta el caso de una niña con lesiones cutáneas de ictiosis al nacimiento que eran compatibles clínicamente con hiperqueratosis epidermolítica, en una forma moderada-leve, lo que se confirmó posteriormente mediante biopsia.
Caso clínico: Recién nacido a término que ingresa en la Unidad de Cuidados Medios de Neonatología de nuestro hospital por presentar eritema diseminado de la piel con lesiones queratósicas.
Como antecedentes personales presentaba un embarazo que precisó tratamiento antihipertensivo, sin otros problemas asociados. El parto fue por cesárea por bradicardia fetal, con Apgar 9/10 al nacimiento. Como antecedentes familiares cabe destacar la presencia de dermatitis atópica en la familia materna y de máculas hipopigmentadas compatibles con vitíligo en la rama paterna.
A su ingreso presentó una exploración dentro de límites normales, con un peso y desarrollo adecuados para su edad gestacional, en la que destacó la presencia de lesiones hiperqueratósicas lineales a lo largo del brazo derecho, bien delimitadas, que afectaban a la palma de la mano y uñas. Presentaba hiperqueratosis palmar y eritema diseminado en el resto de la piel, sin afectación mucosa.
Durante su estancia presentó como incidencia un episodio de hipoglucemia leve que remontó con aporte intravenoso de glucosa, sin mayor incidencia.
Fue dado de alta y citado para posterior biopsia de piel, en la cual se encontraron hallazgos compatibles con hiperqueratosis epidermolítica.
Discusión: La hiperqueratosis epidermolítica es un proceso poco frecuente, estimado en 1 de cada 300.000 recién nacidos aproximadamente. Su aproximación diagnóstica inicial no es fácil debido a su variabilidad en la forma de presentación y a que evoluciona durante las primeras semanas de vida. El diagnóstico de certeza se establece mediante el estudio dermatológico con biopsia.
Distrofia muscular congénita por déficit parcial de merosina: a propósito de un caso
C. Polo Mellado, M. de Alaiz Rojo, P. Ruiz del Arbol Sánchez, B. Curros Novo, B. Fernández Colomer y J. Blas López Sastre
Hospital Central de Asturias, Oviedo.
Introducción: La distrofia muscular congénita se incluye dentro de un grupo de enfermedades cuya expresión clínica es similar, predominando la hipotonía. La debilidad muscular es más prominente en los grupos musculares proximales y si esta debilidad es de comienzo prenatal puede dar lugar a hipoquinesia fetal con artrogriposis. Las dificultades postnatales a la succión-deglución y la tendencia a los accidentes de aspiración recurrentes pueden ser igualmente característicos.
Caso clínico: RN varón fruto de una gestación de 40 semanas sin incidencias y parto eutócico. A las pocas horas de nacer presenta episodio de atragantamiento con parada cardiorrespiratoria posterior que precisó maniobras de reanimación; tras el mismo, el niño presenta hipotonía e hiporreflexia severas y alteración de la succión-deglución. A lo largo de su ingreso, la succión-deglución fueron mejorando hasta su normalización, el niño estaba más despierto, con buena conexión con el medio y buena respuesta a estímulos; sin embargo persistía una hipotonía severa en tronco y extremidades. Ante la sospecha de miopatía congénita se determina CK en varias ocasiones siendo siempre superior a 1000 U/L. Se realiza electromiografía y neurografía con resultados compatibles con miopatía. Se realiza biopsia muscular (deltoides) demostrando severas alteraciones en el músculo consistentes en variabilidad en el tamaño de fibras con tendencia a la forma redondeada, fibrosis peri y endomisial con sustitución adiposa importante; la inmunohistoquímica objetiva, alfa 2-laminina negativa y merosina irregular y tenue. Otras pruebas realizadas con resultado normal fueron: aminoácidos en sangre-orina, cuerpos reductores en orina, amonio, ácido láctico, estudio oftalmológico, electroencefalograma, ecografía craneal y RMN craneal. Nuestro paciente está con tratamiento rehabilitador desde el mes de vida y presenta bastante buen control del tronco, manteniendo una adecuada sedestación, maneja bien los antebrazos aunque muestra debilidad de cintura escapular; así mismo no controla el apoyo de las piernas aunque la movilidad es buena; su desarrollo psíquico es normal.
Conclusiones: Aportamos un nuevo caso de distrofia muscular congénita con déficit parcial de merosina que no asocia alteraciones en la sustancia blanca cerebral y cuya evolución ha sido favorable a pesar de la gravedad inicial de cuadro.
Enterocolitis necrotizante aguda: Casuística en un Servicio de Neonatología
R. Santiago Gómez, M. Domínguez Salgado, B. Jiménez Esteban, C. Fresno Calle, M.L. Jiménez Esteban y M.C. Arrabal Teran
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: La enterocolitis necrotizante aguda (NEC) es la urgencia digestiva neonatal más frecuente (0,3-2,4/mil RN). Se asocia a una gran tasa de mortalidad (11-37%). Nuestro propósito es estudiar los datos del servicio con el fin de determinar su repercusión, y compararlos con los existentes en la bibliografía actual.
Material y métodos: Llevamos a cabo un estudio retrospectivo de los niños diagnosticados de NEC desde 1990 hasta 1998 en nuestro servicio. Se evaluaron en cada caso: factor causal, presentación clínica y complicaciones, diagnóstico y tratamiento.
Resultados: La incidencia de NEC en nuestro servicio fue de 24 casos en los 8 años transcurridos, correspondiente al 1,46/mil recién nacidos. Se identificaron 79% cateterismos umbilicales, 22% de sepsis, tratamiento esteroideo materno en el 33% y ventilación mecánica en el 58% de los casos, alimentación enteral con fórmula en el 71%. El 73% de los casos desarrollaron el cuadro más allá del décimo día de vida. Fallecieron el 32%. De ellos, el 28% habla recibido tratamiento médico y el 4% quirúrgico. Del total, 89% eran grandes pretérminos y un 77% pesaron menos de 1.500 g al nacimiento. A lo largo de su evolución desarrollaron complicaciones de tipo renal (4%), cardíaco (29%), neurológico (21%) o respiratorio (25%) y de tipo digestivo (8% absceso hepático, litiasis biliar, estenosis e IPLV y 7% peritonitis).
Conclusiones: La mayor parte de los resultados obtenidos concuerdan con las series bibliográficas. A pesar una mayor precocidad en el diagnóstico y una mejor atención terapéutica, las tasas de mortalidad no han disminuido en los últimos años probablemente debido a la gran inmadurez de los niños ingresados actualmente en las unidades neonatales.
Encefalopatía hipóxico isquémica - Revisión de casos clínicos en un Servicio de Neonatología
R. Santiago Gómez, M. Domínguez Salgado, M.L. Jiménez Alamo, B. Jiménez Esteban, C. Fresno Calle, M.C. Arrabal Teran y J. Campos Castello
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: La Encefalopatía Hipóxico Isquémica (EHI) es una patología relativamente frecuente que asocia una gran morbimortalidad. Presentamos los datos estadísticos del servicio y el algoritmo diagnóstico-terapéutico que empleamos.
Material y métodos: Se revisaron las historias de los 50 pacientes ingresados en el Servicio de Neonatología entre 1991 y 1997 que cumplían los criterios de EHI.
Resultados: El número de pacientes estudiado equivale a un 1,6% de los RN vivos ingresados en el Servicio y al 0,4% de los RN vivos del hospital. Un 68% nacieron en nuestro centro y el 32% restante fueron trasladados desde otros centros. La edad media de las progenitoras fue de 30 ± 5 años y la edad gestacional de los niños de 40 ± 3 semanas. Todos, salvo uno que fue diagnosticado de CIR, poseían un peso adecuado para su edad gestacional. En un 64% de los casos el embarazo finalizó por cesárea siendo el sufrimiento fetal la indicación en el 37% de ellos. Un tercio de los niños presentó un pH umbilical menor de 7,10 y en el 91% de los casos el registro CTG fue patológico. El líquido amniótico fue meconial en el 36%. Se observó patología funículo-placentaria en todos los casos (vuelta o nudo de cordón, placenta previa). El 84% de los recién nacidos precisó reanimación tipo II o superior. Todos los RN padecieron accidentes vasculares de tipo isquémico, en ningún caso hemorrágico. Fallecieron e1 20%.
Conclusiones: Los datos obtenidos no difieren significativamente de las series publicadas en la bibliografía, aunque nuestra incidencia queda sobrevalorada al incluir los traslados procedentes de otros centros. Es precisa una correcta coordinación entre el obstetra y el neonatólogo, dada la importancia de los acontecimientos perinatales en la génesis de los eventos isquémicos.
Meningoencefalocele: revisión de 6 casos
R. Vera-Medialdea, L. Olivares, M. González-López, R. Maese Heredia, A. del Pino de la Fuente, S. Robles-Febrer, M. Urbano-Pérez, F.J. Rivas-Moreno, J. Lucena-Travé y A. Jurado-Ortiz
Hospital Materno Infantil, Málaga.
Introducción y objetivos: Los defectos del tubo neural son responsables de la mayor parte de las malformaciones del SNC. El defecto craneal se presenta con una frecuencia 10 veces menor que la de los defectos de cierre del tubo neural en el raquis. El objetivo de esta revisión es conocer la incidencia y posibles asociaciones del meningoencefalocele con otras anomalías en nuestro medio.
Material y métodos: Revisión retrospectiva de historiales de recién nacidos con el diagnóstico al alta de meningoencefalocele, durante los últimos 6 años.
Resultados: (véase tabla inferior)
Conclusiones:
- Nuestros resultados coinciden en que la localización más frecuente es la occipital.
- Entre las anomalías asociadas que hemos encontrado destaca la hidrocefalia postquirúrgica y la microcefalia en 2 casos. En 1 caso se ha asociado con anomalía de Poland. Otras asociaciones descritas en la literatura, como S. de Meckel, quiste dermoide, paladar hendido, labio leporino, siringomielia, lipomas, teratomas, craneosinostosis, agenesia del cuerpo calloso y mielomeningocele, no han estado presentes en ninguno de nuestros casos.
Anomalía de Poland: un caso asociado a meningoencefalocele
M. González-López, L. Olivares-Ramos, R. Vera-Medialdea, C. Vida Fernández, T.J. Martínez Arán, J. Blasco Alonso, M. Urbano-Pérez, F.J. Rivas-Moreno, J. Lucena-Travé y A. Jurado-Ortiz
Hospital Materno Infantil, Málaga.
Antecedentes y objetivos: La anomalía de Poland y el meningoencefalocele son hallazgos poco frecuentes en Neonatología. Presentamos un caso de anomalía de Poland en un recién nacido asociado a meningoencefalocele occipital.
Caso clínico: Recién nacido varón que ingresa en nuestro servicio procedente de otro hospital, por presentar una tumoración a nivel occipital. A su ingreso se observa dicha tumoración con un tamaño de 13x11cm, íntegra, pediculada, redondeada, de consistencia blanda, con contenido líquido a la transiluminación, cubierta de piel y pelo y, una asimetría torácica por hipoplasia del hemitórax derecho. En el TAC de cráneo se observa un defecto de cierre occipital en la línea media, del que protruye una bolsa de contenido líquido con imágenes más densas en su interior. Se realiza un mapa óseo en el que se evidencia ausencia de 3ª, 4ª y 5ª costillas derecha en su porción anterior. En el TAC torácico se confirma la deformidad de la pared torácica derecha, con ausencia de los músculos pectorales sugestivo de anomalía de Poland. Se interviene quirúrgicamente practicandose exéresis de la tumoración occipital y reconstrucción posterior. El diagnóstico anatomopatológico fue de meningoencefalocele. Posteriormente presenta un aumento progresivo del perímetro craneal, observándose en la ecografía una hidrocefalia que precisa de válvula de derivación ventrículo peritoneal. La evolución fue favorable.
Comentarios: Las características más comunes de la anomalía de Poland son la ausencia del pectoral menor y de la porción esternal del pectoral mayor, ademas de la sindactilia ipsilateral de la mano. Han sido descritas en la literatura variantes de esta anomalía y distintas asociaciones. No hemos encontrado ningún caso de anomalía de Poland asociado a meningoencefalocele en el recién nacido.
Aplasia cutis congénita y cutis marmorata telangiectásica congénita. Un caso clínico
M.A. Zafra Anta, M. de Troya y A. Núñez
Hospital Pozoblanco, Pozoblanco.
Antecedentes: La aplasia cutis congénita (ACC) puede presentarse de forma aislada, asociada a diversas anomalías físicas, principalmente de extremidades, o formando parte de diversos síndromes malformativos. El cuero cabelludo es la localización preferente de la ACC. La incidencia estimada es 3 de cada 10.000 nacidos vivos.
La cutis marmorata telangiectásica congénita CMTC- (flebectasia generalizada congénita) es una anomalía vascular benigna, con dilatación de capilares y venas superficiales. Se parece a la cutis marmorata fisiológica, aunque más pronunciada, y relativamente constante. La evolución tiende a la mejoría gradual, con resolución parcial o completa al llegar a la adolescencia.
Presentamos un caso de ACC y CMTC asociados, sin otras malformaciones.
Caso clínico: Recién nacido varón a término de peso 3.270 gramos, nacido en parto normal. Padres no consanguíneos. La madre, 35 años, tercigesta, obesa leve, con hipotiroidismo subclínico en tratamiento con hormona tiroidea, antecedente de serología lúes RPR falso positivo en embarazo previo y alopecia tipo androgénica. Embarazo controlado, con serología Rubéola (+ IgG) Toxoplasmosis (-) Lúes (-), Antígeno de hepatitis HBs negativo. No lesiones vesiculosas ni exantemáticas durante embarazo, no otra medicación. La placenta era normal. Una hermana del caso padece hipertiroidismo. No hay otros antecedentes.
El recién nacido presenta en el cuero cabelludo dos zonas de defecto cutáneo cerca de línea media, de unos 5 y 8 mm de diámetro. Además, en una extremidad hay una lesión vascular plana, no continua, desde glúteo a pie derecho. El resto de la exploración física fue normal.
Los estudios complementarios fueron normales: hematimetría y fórmula bioquímica básica, serología TORCH, anticuerpos antinucleares negativos, electrocardiograma, radiografía de tórax, ecografía cerebral y abdominal, radiología de manos y pies, cariotipo. El estudio oftalmológico no objetivó anomalías.
La evolución de la ACC fue sin complicaciones, epitelizándose ligeramente en las primeras 3 semanas de vida. La cutis marmorata evoluciona en regresión en los primeros meses de vida.
Conclusión: Tanto la ACC pequeña, sin alteración ósea subyacente, como la CMTC son generalmente de buen pronóstico. Se debe realizar una valoración dermatológica y una exploración cuidadosa para excluir anomalías asociadas.
Cardiología
Síndrome de Catch 22. Estudio de 25 pacientes con cardiopatías troncoconales
J. Grueso Montero, N. Menéndez González, J. Santos de Soto, B. García, A. Moruno Tirado, A. Delicado, J.L. Gavilán Camacho, C. Casas y M. Gil-Fournier Carazo
Hospital Universitario La Paz, Madrid, Hospital Universitario Virgen del Rocío, Sevilla.
Gracias al desarrollo de la genética molecular con técnicas que incluyen análisis cromosómico de alta resolución e hibridación in situ mediante inmunofluorescencia (FISH), se han llegado a detectar microdelecciones de genes contiguos, causantes principales de defectos cardiacos aislados o sindrómicos. De tal forma ha sido posible comprobar que la delección 22q11 es la base genética común que comparten una serie de síndromes , actualmente agrupados en el acrónimo CATCH 22 . Todos ellos presentan entre otros, defectos cardiacos troncoconales o del arco aórtico. El defecto se hereda de forma autosómica dominante, encontrándose una frecuencia del 25% en los padres de los pacientes con la delección, aunque no pueden descartarse mosaicismos, no detectables con los métodos actuales.
Desde principios del año 1996, en que diagnosticamos mediante FISH al primero de nuestros pacientes, iniciamos un estudio prospectivo en los pacientes que presentaban el tipo de cardiopatía previamente descrita y que además presentaban algún rasgo dismórfico y/o hipocalcemia. A todos ellos además del estudio genético, se realiza un estudio de la inmunidad celular , se investigan rasgos dismórficos y los niveles de calcio.Si existe hipocalcemia, se determinan fósforo y paratohormona.
El grupo lo constituyen 25 pacientes y 6 parejas de padres en los que el resultado de sus hijos resultó positivo. Se ha detectado la delección 22q11 en 6 casos ( 4 con Interrupción del Istmo Aórtico tipo B y 2 pacientes con T. De Fallot ) y en la madre de uno de los pacientes con T. De Fallot.
El interés del trabajo radica en la posibilidad de brindar consejo genético a los padres y en cuanto a los pacientes, prevenir las complicaciones derivadas de la alteración inmunitaria, evitando las transfusiones de sangre no irradiada e impidiendo así la enfermedad de injerto contra huésped, ya que casi todos ellos precisan cirugía paliativa o correctora en el período neonatal.
Tratamiento quirúrgico de coartación de aorta en niños menores de tres meses: 1993-1999
J. Grueso Montero, A.I. Rodríguez Becerra, J. Santos de Soto, C. Campaña Marchal, J.A. Castillo Camacho, A. Álvarez Madrid, A. Descalzo Señorans y M. Gil-Fournier Carazo
Hospital Universitario Virgen del Rocío, Sevilla.
El propósito de esta revisión es comprobar los resultados de la cirugía de la Coartación de Aorta en recien nacidos y lactantes menores de 3 meses.
Se han analizado retrospectivamente los datos de 32 pacientes consecutivos, diagnosticados en el período neonatal, que se han clasificado en 3 grupos: 14 en el Grupo I (Coartación simple, CoAo) , 3 en el grupo II ( Coartación asociada a Comunicación Interventricular (CoAo+CIV) y 15 en el grupo III ( Coartación asociada a Cardiopatías Complejas ( CoAo+CC).
El procedimiento quirúrgico más frecuentemente utilizado ha sido la aortoplastia con parche de la subclavia (56,3%) y en los análisis por grupos, esta técnica ha sido la más utilizada en las coartaciones asociadas a C.C (60%). En 5 casos se hizo Banding de la Arteria Pulmonar y en 1 caso cierre de la CIV con parche, en el mismo acto quirúrgico. Las complicaciones preoperatorias han sido menos frecuentes en los pacientes operados más precozmente desde el diagnóstico y sobre todo en neonatos menores de 30 dias (73,9%) (p = 0,043). La mortalidad precoz total (18,8 %) ha sido 0 en el grupo I , 1 paciente en el grupo II y 5 pacientes en el grupo III. En el seguimiento, la incidencia de recoartación ha sido del 18,8% (6 pacientes), 2 de cada grupo. La mortalidad tardía (12,5%) ha sido más frecuente en los pacientes con coartación asociada a C.C y en los que su situación al ingreso fue más grave, no obstante las diferencias no han sido significativas (p = 0,5).
Como conclusiones destacamos que los factores de riesgo para mortalidad precoz han sido, la gravedad de los síntomas al ingreso, la hipoplasia del arco aórtico (50%), la asociación a C.C (83,3%) y las complicaciones postoperatorias. (100%). . Para recoartación, aunque ha sido más frecuente en los intervenidos con resección término-terminal, no ha habido diferencias significativas en cuanto a la técnica empleada (p = 0,1). Ha sido más frecuente en los que tenían hipoplasia del arco. En cuanto a mortalidad tardía, los factores de riesgo están relacionados con la complejidad de la cardiopatía (75%) y con la gravedad de los síntomas al ingreso.
Repermeabilización de fístula sistémico-pulmonar trombosada con terapia fibrinolítica en el neonato
A. González-Meneses López, J. Santos de Soto, J. Grueso Montero, J.A. Soult Rubio, A. Moruno Tirado y A. Descalzo Señorans
Hospital Universitario Virgen del Rocío, Sevilla.
La terapéutica trombolítica con activador tisular del plasminógeno recombinante (r-TPA) en fístulas quirúrgicas sistémico-pulmonares es excepcional en la infancia, con muy escasas publicaciones en la literatura internacional.
Presentamos el caso de una niña de 9 días de vida afecta de cardiopatía compleja con oligohemia pulmonar a la que se practicó una fístula modificada de Blalock-Taussig, sin ligar el ductus. Al 6º día postoperatorio presentó hipoxemia severa comprobándose por Eco 2D-Doppler y cateterismo cardiaco trombosis total de la fístula. A continuación se coloca un catéter en el extremo aórtico de la fístula y se instaura, a su través, tratamiento con r-TPA en perfusión continua, comprobándose la repermeabilización total de la fístula a las 14 horas del tratamiento.
El paciente fue dado de alta a los 23 días de vida, permaneciendo actualmente (4 meses de edad) la fístula permeable.
El tratamiento trombolítico con r-TPA aplicado localmente en caso de trombosis aguda de fístula sistémico-pulmonar, puede ser una buena opción terapéutica, dados los riesgos de una nueva intervención quirúrgica.
Ventana Aorto-Pulmonar: Nuestra experiencia
A. Moruno Tirado, J. Santos de Soto, J. Grueso Montero, E. Martínez Montero, J.L. Gavilán Camacho, J.A. Castillo Camacho, J. Toro Ortega, M. Gil-Fournier Carazo y A. Descalzo Señorans
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivo: El defecto del septo aorto-pulmonar (ventana AoP),es una cardiopatia congénita rara,de la que hay tan solo unos 300 casos publicados desde su primera descripción en 1830. Presentamos nuestra casuística, resaltando la importancia de su reparación precoz para evitar el desarrollo de enfermedad vascular pulmonar irreversible.
Métodos: Entre los años 1979-2000, 7 pacientes fueron intervenidos de dicha cardiopatia en nuestro centro. Edad media en el momento de la cirugia: 8 meses (rango 11 dias a 30 meses). Peso medio: 6,2 Kgs (rango 2,7 a 14,8 Kgs). Cuatro (57 %) se asociaron con anomalias complejas: Interrupción del arco Ao (IA Ao) tipo A: 2, Hipoplasia del istmo Ao: 1 y D-TGV con CIV amplia:1.
Un paciente (14%) presentó el tipo I de ventana AoP (proximal), 5 (70%) el tipo II (distal) y el restante, el raro tipo III (defecto total del septo AoP).
El diagnóstico de presunción fue realizado en 5 casos (70%) por ecocardiografia. En todos los casos el diagnóstico se confirmó mediante Cateterismo cardiaco.
Resultados: No hubo ningún fallecimiento durante la intervención quirúrgica. El enfermo con D-TGV y CIV, falleció 22 dias después por crisis de hipertensión pulmonar. Tras un seguimiento medio de 69 meses, los restantes se encuentran asintomáticos sin medicación. Solo uno, afecto de IAAo, presenta cierto grado de retraso madurativo.
Conclusiones: A pesar de ser una entidad rara, se debe pensar en ella, ante toda cardiopatia congénita compleja con severa hipertensión pulmonar.
La corrección quirúrgica precoz, evitará el desarrollo de enfermedad vascular pulmonar irreversible, sobre todo, en estos casos asociados a otras anomalias cardiacas complejas.
Cierre percutáneo de la comunicación interauricular. ¿Ha llegado el momento?
J.L. Zunzunegui Martínez, E. Maroto Álvaro, J.I. Zabala Argüelles, E.J. García Fernández, C. Maroto Monedero, N. Domínguez Garrido, M.T. Tomé, Almudena Castro, R. Greco y J.C. de Agustín
Hospital General Universitario Gregorio Marañón, Madrid.
La CIA tipo II es una de las cardiopatías más frecuentes cuyo tratamiento clásico ha sido el cierre quirúrgico con circulación extracorpórea. Una alternativa atractiva es el tratamiento mediante cateterismo.
Métodos: Desde Febrero de 1998 a Diciembre de 2000 49 niños con CIA tipo II fueron seleccionados para cierre percutáneo. La edad y el peso fueron 5,5 ± 2,5 años y 21 ± 5 Kg. Se empleo anestesia general y ecografía transesofágica (ETE) y/o intravascular (EIV) durante el procedimiento. La vía de acceso vascular fue la vena femoral y los dispositivos empleados fueron: Angel-Wings y Amplatzer. Se administró AAS (5mg/Kg/d) durante 6 meses.
Resultados: Los datos hemodinámicos fueron: presión pulmonar media 13 ± 2 mm Hg, Qp/Qs 2,2 ± 0,6 y resistencias pulmonares 1,3 ± 0,3 U/m2. El diámetro del defecto medido por ETE y/o EIV fue de 14 ± 4mm y por oclusión con balón de 15 ± 4 mm. Se emplearon 59 dispositivos, 4 Angel-Wings y 55 Amplatzer. Diez dispositivos Amplatzer se retiraron, sin complicaciones, por no ajustar en la CIA. En 45 niños se implanto correctamente el dispositivo y en cuatro no fue posible por lo que se indicó cierre quirúrgico electivo. La duración del cateterismo fue de 78 ± 31 minutos y el de escopia de 18 ± 12 minutos. La mayor parte de los niños se dieron de alta al día siguiente, media de 27 ± 16 horas, y sólo un paciente permaneció ingresado 5 días por una crisis asmática. No se produjeron complicaciones y no se administro hemoderivados en ningún niño. El seguimiento medio es de 11,4 ± 9 meses. Se realizaron revisiones los días 1, 7, y 180 tras el procedimiento con radiografía de tórax y ecocardiografía transtorácica. La Rx tórax mostró en todas los casos la correcta implantación del dispositivo. En la ecocardiografía existía un cortocircuito residual en cinco niños el día 1º, y en tres 3 el día 7º. A los seis meses sólo un paciente presentaba un cortocircuito trivial.
Conclusiones: El cierre percutáneo de la CIA es una técnica segura con nula mortalidad y sin complicaciones clínicas importantes. Respecto a la cirugía clásica disminuye el tiempo de hospitalización, no precisa circulación extracorpórea, no necesita administrar hemoderivados y tiene mínimas consecuencias estéticas en el niño. Cuando se realiza una adecuada selección de los pacientes la frecuencia de implantación es del 92% y el cierre completo si cortocircuito 98%.
Complicaciones de la atrioseptostomía de Rashkind bajo control ecocardiográfico: análisis 102 casos
J.L. Zunzunegui Martínez, J.I. Zabala Argüelles, García Fernández, E. Maroto Alvaro, C. Maroto Monedero, M.M. Sánchez, N. Domínguez Garrido, A. Castro, M.T. Tomé y M.L. Franco
Hospital General Universitario Gregorio Marañón, Madrid.
La atrioseptostomía de Rashkind (AR) consiste en la creación de una comunicación interauricular (CIA) no restrictiva mediante un catéter balón. Es necesario para estabilizar al paciente en cardiopatías congénitas cianóticas con hipoxemia severa y mezcla inadecuada o cuando es necesario disminuir la presión de la aurícula derecha o izquierda.
Métodos: Entre enero de 1990 y diciembre de 2000 se realizaron 1926 cateterismos cardíacos en niños, 698 fueron procedimientos intervencionistas y de estos 104 fueron AR. Las cardiopatías incluyeron 71 D-TGA, 22 hipoplasia de ventrículo izquierdo, 9 atresias tricúspide y 2 atresias pulmonares. La edad y el peso en el momento de la AR fueron de 2,8 ± 0,8 Kg (1,9-4,5) y 1,6 ± 11 días (1-120). La vía empleada fue la vena femoral (VF) mediante un introductor del 7F en 100 y la vena umbilical en dos. Los catéteres empleados fueron Rashkind (5,5F, 5F, 4F) y Miller 5F. La técnica se realizó en la UCIP o en la UCIN y se monitorizó el procedimiento con ecocardiografía transtorácica en proyección subxifoidea.
Resultados: La AR se completó en 102 niños (98%), en dos neonatos con hipoplasia de ventrículo izquierdo aunque se pudo cruzar el foramen oval la aurícula izquierda era demasiado pequeña para el inflado del balón. En dos recién nacidos (1,9%) no se pudo canalizar la VF, realizándose la AR por la vena umbilical, y en cuatro se canalizó inadvertidamente la arteria femoral hololataral (3,8%). Cinco (4,8%) presentaron trastornos del ritmo con repercusión hemodinámica: 3 TQSV, un flutter auricular y un BAVC que preciso marcapasos endocavitario transitorio durante12 horas. En 101 (99%) niños se utilizo sólo ecografía y en uno fue necesario trasladarlo al laboratorio de hemodinámica por presentar una pequeña disección de la VCI que dificultaba la progresión del catéter. En ningún caso se produjo perforación cardíaca definida como la aprición de derrame pericárdico. Un neonato de 12 horas de 1.900 gr de peso con SDR y portador de una D-TGA falleció al as 18 horas de AR debido a un desgarro hepático con hematoma subcapsular.
Conclusiones: La AR por vía femoral bajo control ecocardiográfico es un procedimiento seguro que evita traslados innecesarios en recién nacidos gravemente enfermos. Las complicaciones más frecuentes incluyen las relacionadas con el acceso vascular y con los trastornos del ritmo. En la actualidad la mortalidad del procedimiento es rara, < del 1%, y afecta a pacientes con patologías no cardíacas asociadas.
Impacto de la conexión ventrículo derecho - arteria pulmonar (CVA) en el crecimiento de las arterias pulmonares en la atresia pulmonar con comunicación interventricular (AP+CIV)
M.D. Herrera Linde, F. Villagra Blanco, M. Cazzaniga, J.I. Díez Balda, R. Gómez, P.A. Sánchez, J.I. Herráiz, M. San José, M.L. Polo y J. Pérez León
Hospital Ramón y Cajal, Madrid.
Objetivo: No hay acuerdo sobre el tratamiento quirúrgico más adecuado en la (AP+CIV), y que procedimientos proporcionan un mayor crecimiento en los casos con hipoplasia de arterias pulmonares. Valoramos el impacto de la CVA en el crecimiento y desarrollo de las arterias pulmonares y en que medida las hace viables para la corrección definitiva.
Metodología: Entre 1978 y mayo 2000 hemos realizado CVA en 33 pacientes con AP+CIV. Su edad es de 41,6 meses (0,8-102) y 1su peso de 12,4 Kg (3,2-25,5). 17 son varones y 16 hembras. La CVA consistió en el implante de un parche transanular en 14 casos y de un conducto avalvulado en los 18 restantes. Otras técnicas asociadas fueron el cierre de una CIA en 1, unifocalización de MAPCAS en 3 y arterioplastias en 3. El tiempo de circulación extracorpórea fue de 74,8 minutos (44-145) y el clampaje aórtico de 32 minutos (0-100). EN 8 no se clampó la aorta. Antes y después de la CVA medimos angiográficamente el diámetro de ambas arterias pulmonares, expresándolo con diferentes fórmulas: % del diámetro normal según la edad, valor de Z, indice de Nakata e Índice de McGoon. Valoramos también antes y después de la CVA la presencia de estenosis significativas en ambas arterias pulmonares.
Resultado: 2 fallecieron precozmente (6%). En un seguimiento medio de 41,9 meses (1-129) fallecen 4 y se pierde el seguimiento en 2. El diámetro de la arteria pulmonar derecha (APD) e izquierda (API) antes y después de la CVA fue: % valor normal: 59,8% vs 87,7% (APD) (p < 0,0001), 59,1% vs 79,6% (API) (p < 0,0005). Valor Z: -3,4 vs -1,4 (APD) (NS) y -3,5 vs -3,15 (API) (NS). Índice Nakata: 125 vs 261 mm2/m2 (p < 0,02). Índice McGoon: 1,56 vs 2,02 (NS). El número de casos con estenosis significativa antes y después de la CVA fue: Estenosis en APD 8 vs 10 y estenosis en API 4 vs 9. Finalmente 12 (36,3%) de los 33 pacientes iniciales alcanzaron la corrección total definitiva posteriormente.
Conclusión: La CVA proporciona un crecimiento significativo de las arterias pulmonares, viabilizando la corrección total en al menos 1/3 de los casos. Sin embargo incrementa también el número de casos con estenosis arteriales pulmonares significativas.
Hematología
Trombocitopenia severa y rotura esplénica asociada a mononucleosis infecciosa
J.A. Fernández Merchán, V. Barajas Sánchez, D. Vallejo Díaz, N. Domínguez Garrido, R. Escorihuela Esteban y M. Bernácer Borja
Fundación Jiménez Díaz, Madrid.
Introducción: Son conocidas las diferentes formas de presentación de la mononucleosis infecciosa (MNI) así como el distinto grado de complejidad de las complicaciones asociadas. Una de las complicaciones es la trombopenia severa que, aunque se presenta con una incidencia muy baja, es conveniente tenerla en cuenta dada su potencial gravedad. En ocasiones la púrpura trombocitopénica (PTI) puede ser la primera manifestación de la enfermedad vírica. La rotura esplénica es excepcional en esta enfermedad. Presentamos el caso de un adolescente afecto de MNI que debutó con una púrpura trombocitopénica severa y que en el transcurso de la evolución sufrió una rotura esplénica.
Caso clinico: Paciente de 17 años de edad que consulta en el Servicio de Urgencias por la aparición progresiva de hematomas y sangrado de encías. Desde hacía una semana presentaba fiebre y odinofagia. Antecedentes personales, no contributorios. Antecedentes familiares, sin antecedentes de interés. Exploración física.- Peso 60 Kg (P 10-25); Temp: 37,2ºC; TA: 64/130; FC: 77 lpm; FR: 20 rpm. Afectación del estado general. Palidez de piel. Sangrado gingival. Hematomas generalizados en miembros y tronco con púrpura petequial asociada. Adenopatía laterocervical izquierda de 2x2cm de consistencia dura, no adherida a planos profundos. ACP: normal. Abdomen: esplenomegalia de unos 6 cm. No hepatomegalia. ORL: Amígdalas hipertróficas con exudado puntiforme. Neurológico normal. Pruebas complementarias: Hemograma: Htíes 4.370.000/mm3, Hb 14,3 g/dl, Hto 42,4%, VCM 97, HCM 32,8; leucos 10.200/mm3 (17%S, 12%C, 46%L, 6%M, 19% cianófilas); plaquetas 1.000/mm3. Estudio de coagulación, normal. Grupo Sanguíneo: A Rh +.
Bioquímca: glucosa 108 mg/dl, ionograma normal, ALAT 419 U/L, LDH 480 U/L. Serología: Anticuerpos heterófilos (Paul Bunell), positivo. Tratamiento.- Gammaglobulina I.V a dosis de 400 mg/Kg/día. Metilprednisolona a dosis de 10 mg/Kg/día.
Evolución: El quinto día de tratamiento el paciente sufre deterioro hemodinámico asociado a intenso dolor abdominal. Se realiza ecografía abdominal en la que se aprecia gran cantidad de líquido libre peritoneal. En la TAC abdominal se objetiva rotura esplénica de polo superior con hematomas intraparenquimatosos y subcapsulares e importante hemoperitoneo. Se realiza esplenectomía. La evolución posterior fue satisfactoria.
Conclusiones y comentarios:1) En los pacientes con PTI debe realizarse estudio de anticuerpos heterófilos (Paul Bunell) dado el papel de la infección por Epstein Barr en la etiología de este proceso. 2) El tratamiento de una PTI severa (< 10.000 plaquetas/ml) se basa en la administración de corticoides y/o gammaglobulina a altas dosis (0,4-0,8 gr/Kg/dosis). En algunos casos de PTI asociada a MNI descritos en la literatura se ha utilizado el tratamiento antiviral con aciclovir. 3) Ante una rotura esplénica asociada a MNI la mayoría de autores están de acuerdo en que la esplenectomía es la mejor opción terapeútica excepto en aquellos casos en los que la lesión esplénica se limita a un hematoma subcapsular en pacientes hemodinámicamente estables.
Un caso de leucemia congénita
R. Ávila Villegas, F.J. Garrido Torrecillas, J.A. López Torres, E. Ocete Hita, J.M. Ramos Navas-Parejo, A. Ruiz Extremera, R. Santana Delgado y E. Pérez Bonilla
Hospital Universitario San Cecilio, Granada.
Introducción: La leucemia aguda congénita es excepcional y el tipo más frecuente es la mieloide, a diferencia de lo que ocurre a otras edades. La clínica puede ser muy llamativa al nacimiento: petequias, equímosis, visceromegalias, masas nodulares tipo fibroma por la infiltración por células leucémicas. El dato analítico más frecuente es la hiperleucocitosis (cifras de > 150.000) con predominio de células blásticas que pueden contener bastones de Auer y se consideran patognomónicos de ésta enfermedad. El pronóstico es muy malo, falleciendo los neonatos fundamentalmente por hemorragias e infecciones.
Caso clínico: Recién nacido mujer de 39 semanas de edad gestacional con peso al nacimiento de 3.315 g y 51 cm. Apgar a los 1, 5 y 10 minutos de 4-5-9 respectivamente y reanimación tipo III. Antecedentes obstétricos: madre de 34 años, secundigesta. Embarazo controlado que cursa con diabetes gestacional. Parto: cesárea urgente por sufrimiento fetal agudo. Exploración: mal estado general con hematomas y elementos purpúricos generalizados. Auscultación C-R: hipoventilación bilateral y taquicardia de 180 l/m. Abdomen globuloso con hepatomegalia de 3,5 cm y esplenomegalia de 2 cm.
Exploraciones complementarias: hemograma: hematíes 2.880.000, Hb 10,3, Hto 31,6, leucocitos 219.200 con blastos en sangre periférica de > 200.000, plaquetas 166.000. Coagulación: actividad de protrombina 6, T.P.T.A 2 minutos y 17 segundos, fibrinógeno 22, dímero D 5.491. Bioquímica: GOT 329, resto normal. Cultivos centrales y periféricos negativos. Serología TORCH ampliada: negativa.
Evolución: Se traslada el neonato a la UCIP y N con la sospecha de coagulación intravascular diseminada (CID). Se instaura ventilación mecánica convencional por dificultad respiratoria progresiva y canalización de arteria y vena umbilical. A la media hora de vida se produce hemorragia digestiva y respiratoria, siendo necesaria la reexpansión de la volemia con cristaloides y posteriormente con hemoderivados. El S. de hematología nos informa de la presencia de abundantes blastos, sugerentes de leucemia neonatal. No se consigue la estabilización clínica, produciéndose el exitus de la paciente a las 6 horas de vida. La necropsia confirma el diagnóstico de leucemia aguda congénita mieloide (promielocítica M3).
Comentarios: La leucemia congénita neonatal mieloide promielocítica M3, cuando debuta como una C.I.D. tiene un pronóstico fulminante.
Linfohistiocitosis hemofagocítica. Presentación de un caso clínico
F.J. Garrido Torrecillas, R. Ávila Villegas, J.A. López Torres, R. Santana Delgado, C. Trillo Belizón, E. del Moral Romero, A. Sánchez Marenco, F. Contreras Chova y A.E. Jerez Calero
Hospital Universitario San Cecilio, Granada.
Introducción: La linfohistiocitosis hemofagocítica, poco frecuente en pediatría, se produce por proliferación de las células del sistema fagocítico mononuclear. Se encuentra englobada dentro de las histiocitosis (clase II). Existen dos formas de presentación, familiar (autosómica recesiva) y esporádica (asociada a procesos infecciosos).
Caso clínico: Varón de 17 meses que ingresa por cuadro de fiebre y deposiciones líquidas de más de 10 días de evolución. Estaba siendo seguido en consulta por adenopatías inguinales derechas, resto de antecedentes personales y familiares sin interés. Exploración física: mediano estado general, palidez cutáneo-mucosa, hidratación conservada. Peso 10.400 g (< p10). Exantema petequial en abdomen y eccematoso petequial en región del pañal, dermatitis seborreica en cuero cabelludo. Abdomen globuloso timpanizado, hepatoesplenomegalia. Adenopatías inguinales derechas, microadenias laterocervicales. ACR: soplo sistólico, ventilación normal. Exploraciones complementarias: Hemograma: anemia y trombopenia, en controles sucesivos pancitopenia. Fibrinógeno, transaminasas, colesterol, inmunoglobulinas, ferritina, transferrina, fosfatasa alcalina, LDH, c3 y c4 normales. Hipoproteinemia con hipoalbuminemia, hipertrigliceridemia. PCR y Bilirrubina al principio normales posteriormente elevadas. RX tórax y cráneo normales. Serologías: VHB, VHA, VHC, VEB, TABM, VIH, VVZ, Toxoplasma, rubeola, sarampión, adenovirus, mycoplasma, influenza y parainfluenza negativas, CMV VHS IgG Positiva. Mantoux negativo. Coprocultivo positivo a rotavirus, urocultivo negativo, hemocultivo positivo staf. coag. neg. y cándida. Bioquímica, citología y cultivo LCR negativos. Eco abdominal adenopatías inguinales, lesión hipoecoica hepática. TAC: lesión hepática calcificada. Biopsia ganglionar: infiltrado histiocítico con eritrofagocitosis, siendo los macrófagos de morfología típica sin evidencias de malignidad.
Confirmado el diagnóstico de linfohistiocitosis hemofagocítica se instaura tratamiento con Vp-16, corticoides y metotrexate intratecal. Presenta evolución no favorable con alteraciones hidroelectrolíticas y cuadro séptico falleciendo a las 2 semanas.
Comentarios: El diagnóstico se basa en la sospecha clínica y se confirma por la anatomía patológica de médula ósea, hígado, pulmón, bazo o ganglios linfáticos, presentando infiltrado histiocitario, de características morfológicas e inmunofenotípicas normales, sin evidencias de malignidad y por la presencia de eritrofagocitosis (permite diagnóstico diferencial con resto de histiocitosis). La anatomía patológica no permite diferenciar entre la forma familiar y la esporádica, debiendo basarnos en la historia clínica, antecedentes familiares y exploraciones complementarias. El pronóstico es nefasto, especialmente si se retrasa el tratamiento, basado en administración de VP-16, corticoides y metotrexate intratecal, seguidos de trasplante de médula ósea alogénico.
Patología asociada a anticuerpos antifosfolipídicos
A.M. Hernández Alberca, D. Fernández Álvarez, M. Muriel Ramos, C. Encabo Jiménez, I. Corral Carabias y G. Carlone Martín
Hospital Clínico Universitario, Salamanca.
Introducción: Los anticuerpos antifosfolipídicos (aFL) en la edad pediátrica se asocian a múltiples patologías. Se observan habitualmente en pacientes con lupas eritematoso sistémico, aunque también pueden aparecer en pacientes con patologías no- inmunes como procesos infecciosos siendo en este caso transitorios. Independientemente de la etiología provocan en el niño un estado de hipercoagulabilidad, que asociado o no a otros factores protrombóticos, pueden ocasionar trombosis arteriales o venosas.
Material y métodos: Estudio retrospectivo de 6 pacientes ingresados en los 3 últimos años con aFL positivos. Estudiamos: tiempo de tromboplastina parcial activado (TTPA); inmunidad: ANA, DNA, ENA, crioaglutininas, factor reumatoide y complemento (C3, C4); aFL: anticoagulante lúpico (ACL), anticuerpos anticardiolipina (aCL), antifosfatidilserina (aFS) y ß2 glicoproteína (ßG); clínica asociada con aFL y diagnóstico clínico.
Resultados:
EdadTTPAE. inmunidadaFLClínica asociada aFLDiagnóstico
13 45 segCrioagluninas aCL+Livedo reticularis,Fiebre Q años positivaslesiones purpúricas,crónica (a)epilepsia y hemicránea
2 a 44 segnormalaCL +Proceso viral
3 a 61 segnormalACL +Mononucleosis
5 a 32 segnormalaCL + Trastorno isquémico Síndr. Antifos aFS + en ambos piesfolipídico primario
4 a 22 segnormalaCL +Miocardiopatía dilatada
9 a 76 seg normalACL +Gastroenteritis
En nuestro caso los aFL están asociados más frecuentemente a patología infecciosa que autoinmune. El TTPA no estaba alargado en 2 de los casos. Hay manifestaciones clínicas relacionadas con la presencia de aFL en 2 casos, en el resto no repercusión clínica.
Conclusión: Ante la presencia de manifestaciones clínicas que sugieran la presencia de aFL se deben determinar a pesar de tener un TTPA normal.
Síndrome antifosfolipídico primario
A.M. Hernández Alberca, M. Muriel Ramos, D. Fernández Álvarez, M.S. González García, A. Remesal Escalero y J.A. Martín García
Hospital Clínico Universitario, Salamanca.
Introducción: El síndrome antifosfolipídico (SAF) se caracteriza por la asociación de trombosis venosas o arteriales y la presencia de anticuerpos circulantes dirigidos contra los fosfolípidos amónicos.
Podemos hablar de SAF secundario cuando aparece en pacientes con lupus eritematoso, síndromes semilúpicos o asociado a otras patologías no autoinmunes y SAF primario en aquellos sujetos que no presentan evidencia de enfermedad sistémica.
Caso clínico: Niña, de 5 años que acude por presentar de forma brusca color eritematoso en ambos pies, a nivel digital, con sensación de frialdad y dolor. En el transcurso de media hora la coloración pasa a ser violácea, hasta hacerse totalmente cianótica. Antecedentes personales: ingresada 8 días antes por cuadro de gastroenteritis aguda con coprocultivo negativo. Antecedentes familiares sin interés.
Se consulta a cirugía vascular (doppler) con resultado de hipoperfusión embolismos dístales. El resto de la exploración fije completamente normal. Se comienza tratamiento con heparina de bajo peso molecular (1 mg/kg). A las 4-5 horas la coloración se restablece, persistiendo durante 36 horas en los dedos una zona eritematosa con patrón reticular. Este tratamiento se mantiene durante 14 días. Analítica realizada: hemograma normal (167.000 plaquetas); tiempo de protombina 98%, TTPA 32 seg, DD 1,17 µg/ml, PDF > 20 µg/ml; complemento y factor reumatoide normales; anticuerpos ANA, DNA y ENA negativos; crioaglutininas negativos; estudio de trombofilia:heterocigota para la variante termolábil de la MTHRF, anticuerpos anticardiolipina IgG negativo, IgM 36,1 MPL (positivo moderado); antifosfatidilserina IgG 28,4 GPS/ml (0-16). Bacteriologías y serologías negativas.
Conclusión: La asociación de anticuerpos antifosfolipídicos con la variante MTHRF pudo ocasionar en nuestro caso la manifestación trombótica. No hay consenso respecto a una terapia anticoagulante prolongada., salvo cuando coexistan otros factores de riesgo trombótico.
Anemia secundaria angiomatosis neonatal difusa
E. Pérez Monjardin, A. Rodríguez Varela, C. Tomas Rates, C. Lecuona López, J. Ardit Lucas y V. Álvarez Angel
Hospital Universitario La Fe, Valencia, Hospital Clínico Universitario, Valencia, Hospital General de Valencia, Valencia.
Introducción: Las enfermedades hemorrágicas por alteración de los vasos constituyen un extenso grupo de procesos, de importancia variable, según la intensidad de sus manifestaciones clínicas y la frecuencia de su presentación ofrecen como característica común desde el punto de vista analítico, una alteración no constante de las pruebas de fragilidad vascular, con normalidad de las pruebas trombocíticas y factores plasmáticos.
Caso clínico: Lactante varón de 2 meses de edad que ingresa por úlcera en área genital sobre angioma. presenta desde el nacimiento angiomas múltiples en número de nueve, distribuidos por cara, tronco, y extremidades, todos de menos de 1 cm salvo uno en talón y el de área genital, que son de mayor tamaño. Destacar como antecedente ingreso al nacimiento por prematuridad y a las dos semanas de vida por hemorragia digestiva baja. A la exploración destaca escara escrotal con úlceras serpenginosas vecinas y erosión perianal, así soplo sistólico y palidez cérea. A su ingreso se realiza analítica evidenciando anemia severa Hb 5,6 gr/dl, Htco: 17,8%) que requiere transfusión. posteriormente se realiza estudio de coagulación, fondo de ojo, eco craneal y abdominal y serie ósea sin hallazgos patológicos. Para descartar la presencia de angiomas en otras localizaciones se realiza RNM evidenciando lesión ocupante de espacio a nivel perineal y genital, sin extensión a cavidad pélvica.
Conclusiones: La existencia simultánea de angiomas cutáneas, sobre todo cuando se asocia a anemia severa, nos obliga a descartar, a pesar de su rareza, angiomas en otras localizaciones.
Trombosis venosa profunda como complicación infrecuente de la varicela
C. Trillo Belizón, J.M. Gómez Vida, J.A. Martín García, A. Rodríguez Leal, S. Broncano Lupiañez, E. Ortega Páez, A. Valenzuela Soria, E. Fernández Segura, J.A. Rubiño Torres y A. Jiménez Raya
Hospital General Básico de Motril, Motril.
Antecedentes: Se han comunicado defectos transitorios de proteínas S y C secundarios a la varicela. Estos déficits pueden ocasionar complicaciones como trombosis venosa profunda, embolismo pulmonar e incluso coagulación intravascular diseminada.
Caso clínico: Niña de 13 años con lesiones residuales de varicela (10 días de evolución) que presenta dolor intenso en pantorrilla izquierda, que le impide la deambulación, y fiebre de 38ºC. A la exploración destaca dolor a la palpación de los gemelos izquierdos con tumefacción, rubor y calor de la zona y signo de Homann positivo. Entre las pruebas complementarias destacamos: Dímero D > 1600, PDF 160, resto de coagulación normal (incluyendo anticoagulante lúpico y antitrombina III). Discreta leucocitosis, serie roja y plaquetas normales. VSG 18 mm 1ª hora. PCR 5,2 mg/dl. Bioquímica básica, transaminasas, CPK, LDH e inmunoglobulinas normales y anticuerpos antinucleares negativos. C3 164 y C4 17 mg/dl. Proteína C 109%, proteína S total 62% (valor normal 85%). Ecografía doppler venosa de miembro inferior izquierdo con signos de trombosis venosa profunda por debajo de la bifurcación de la vena poplítea izquierda. Se inició tratamiento con heparina de bajo peso molecular, siendo su evolución satisfactoria.
Conclusiones: La trombosis venosa profunda es una entidad rara en la edad pediátrica y se suele relacionar con defectos congénitos o conectivopatías. En el caso que presentamos, el antecedente de varicela puso de manifiesto un defecto transitorio de proteína S que se tradujo clínicamente en una trombosis venosa profunda.
Secuestro esplénico agudo
A. Rayo Fernández, S. Fernández Fernández, C. Calvo Rey y R. Díaz-Delgado
Hospital Severo Ochoa, Leganés.
Niña de 13 meses que acude a Urgencias por irritabilidad intensa. Afebril. No vómitos ni diarrea. No hiporexia.
AF: Procedentes de Guinea Ecuatorial.
AP: Anemia drepanocítica diagnosticada 2 meses antes. Desde entonces ha precisado ingreso por cuadro de neumonía por neumococo resistente a penicilina. No otros procesos intercurrentes. Vacunación incompleta.
EF: Tª 37,2ºC TA 122/56 FC 138 lpm.
REG. Irritable. Intensa palidez mucocutánea. Precordio hipercinético. Latido y pulso celer. No exantemas ni petequias. Meníngeos negativos. Abdomen blando y depresible. Esplenomegalia dolorosa a 5 cm de reborde costal. No dolor articular.
PC: Hemograma: Hb 5,3 Hto 15,9 VCM 75,1 Leucocitos 27.260 (32% N, 54% L, 3,5%M, 3,7%E) plaquetas 145000 Reticulocitos 364000/mm3. Frotis: dianocitos, aislados esquistocitos. PCR 33,2. BQ: Glucosa 101 creatinina 0,4 Na 136 K 5,1 Bilirrubina total 1,62 mg/dl, indirecta 1,34, directa 0,28 LCR BQ: normal Rx tórax: condensación residual en LSD. ECO abdominal: esplenomegalia de 9x5 cm de ecogenicidad homogénea sin otros hallazgos. Cultivos de sangre, orina y LCR negativos.
Evolución: Ante la sospecha de secuestro esplenico agudo se inicia hidratación i.v. adecuada, analgesia con cloruro mórfico para el tratamiento del dolor y transfusión de concentrado de hematíes. Evoluciona satisfactoriamente, con normalización de los niveles de hemoglobina, disminución progresiva de la esplenomegalia y desaparición del dolor abdominal.
Comentarios
- Dado el aumento de casos de anemia falciforme en nuestro país, debido al incremento de población susceptible de padecerla (raza negra), es preciso conocer las manifestaciones clínicas de dicha enfermedad y sus complicaciones.
- El secuestro esplénico agudo es una causa importante de muerte en estos pacientes. Debemos sospecharlo ante un aumento brusco y masivo del bazo con dolor intenso, en el contexto de una anemia.
- Con un seguimiento clínico estrecho y la instauración precoz del tratamiento podemos disminuir la morbimortalidad asociada.
Neutropenia transitoria del lactante. Tratamiento con G-CSF
M.I. Illán Rueda, J. de la Cruz Moreno, R. Ibáñez Ruiz, A. Solas Beltrán, C. García Llopis, B. Espín Jaime y F. González Rivera
Hospital General de Especialidades Ciudad de Jaén, Jaén.
Introducción: Se define la neutropenia como el recuento absoluto de neutrófilos (RAN) < de 1500/ mm3. La neutropenia se presenta de forma transitoria que dura días o semanas y que, en general es adquirida; o crónica con una duración de seis meses o superior) que suele ser de causa inmunitaria o congénita. El riesgo de infección es directamente proporcional al RAN y a la reserva de neutrófilos en médula ósea (diez veces mayor), suelen ser asintomáticas o presentarse como infecciones leves en piel y mucosas (< de 1000 neutrófilos) o ser potencialmente mortales (< 500 neutrófilos).
Caso clínico: Se presenta una niña de dos años, sin A.F. de interés, que presenta celulitis subcutánea tras picadura de insecto, con cultivo positivo a S. Aureus, coincidente con neutropenia < 1000. A los tres meses de este episodio presenta adenoflemón submandibular izquierdo con cultivo positivo a S. Aureus y neutropenia severa y semanas más tarde adenoflemón derecho de las mismas características del anterior; siempre preciso tratamiento antibiótico intravenoso y drenaje quirúrgico. Estudio inmunitario: neutropenia de 183/ mm3; monocitosis y eosinofilia; inmunoglobulinas normales; IgE 119,5; C3, C4 y CH50 normales; linfocitos totales 4774; subpoblaciones linfocitarias normales; test de transformación linfoblástica frente a mitógenos, con canavalina A, fitohemaglutinina y Pokweed positivo; anticuerpos IgG antitétanos en suero 3,3 UI/ml (inmune); test de NBT 12% (normal); Anticuerpos anticitoplasma de neutrófilo en suero, negativo; aspirado de médula ósea con hiperplasia de la serie mieloide con algunos rasgos megaloblásticos; recuento de necrófilos en ambos padres normales. Se practican recuentos seriados, descartándose neutropenia cíclica. Con diagnóstico de neutropenia crónica del lactante se inicia tratamiento con G-CSF, precisando dosis de 2,5 mcgr/kg dos veces en semana para mantener RAN > 3000.Se practican RAN mensuales, detectándose a los 7 meses de tratamiento neutrofilia (15.000) sin infección intercurrente. Con este dato se suspende el tratamiento con G-CSF, continuando seguimiento en controles periódicos, manteniéndose un número normal de neutrófilos.
Discusión: La neutropenia transitoria del lactante es de etiología autoinmne en la mayoría de los casos. Las complicaciones infecciosas graves son raras en estos pacientes, a pesar de RAN muy bajos.
Presentamos una niña con 3 procesos infecciosos graves y neutropenia severa, motivo por el que se decidió, tras descartar otras causas de neutropenia, el tratamiento con G-CSF.
La dosis necesaria para lograr un RAN normal fue baja, y la duración del tratamiento fue de siete de meses, lo que confirma el carácter transitorio de ésta entidad.
El tratamiento fue eficaz con normalización del recuento de neutrófilos en sangre periférica, y ausencia de infecciones graves en la evolución. No hubo efectos secundarios importantes, salvo dolor óseo que fue de baja intensidad. Consideramos el G-CSF terapéutica útil en enfermos con neutropenia grave y procesos infecciosos severos de repetición.
Fiebre y dolores óseos en paciente con Ataxia-Telangiectasia
M. Cañellas Fuster, M. Guibelalde del Castillo, N. Nieto del Ricón, P. Bastida, M.I. Llull Ferretjans, A. Roca Jaume y J.M. Román
Hospital Son Dureta, Palma de Mallorca, Hospital Materno-Infantil Vall d'Hebrón, Barcelona.
Antecedentes y objetivos: La Ataxia-Telangiectasia (AT) es una enfermedad autosómica recesiva, caracterizada por ataxia cerebelosa, telangiectasias, inmunodeficiencia, fragilidad cromosómica y predisposición a desarrollar neoplasias. Presentamos un paciente con AT, que desarrolló leucemia linfoblástica aguda (LLA).
Caso clínico: Niño de 3 años, remitido por dolores óseos de 3 semanas de evolución y fiebre de 10 días, sin otros síntomas. Antecedentes personales: desde los 6 meses de vida ataxia truncal estática y disartria. Estudiado por neuropediatria se realizó estudio constatando telangiectasias, hipogammmaglobulinemia y alfafetoproteina elevada, dianosticándole de AT. Antecedentes familiares: padres y hermana sanos, no consanguineidad. Exploración física al ingreso: regular estado general, irritabilidad, telangiectasias en pabellones auriculares y conjuntivas oculares, adenopatías laterocervicales (diámetro menor de 1 cm. ) y limitación en movilidad de ambas cinturas con dolor a la presión en metáfisis; resto sin hallazgos. Pruebas complementarias:Estudios microbiológicos negativos. Hemograma :Leucocitos 4,6 x 10 3/microL (48%N, 40%L), Hematíes 3,92 x 106/ microL, Plaquetas 215 x 103/microL. Estudio inmunológico en sangre periférica: 2% células mononucleadas CD 19+ CD10+ CD34+. Estudio inmunológico en médula ósea: 95% de células mononucleadas CD19+ CD34+ CD10+ DR+ IgM- Tdt-. Con el diagnóstico de LLA tipo B, el paciente se trasladó al Hospital Vall d´Hebrón en donde recibe tratamiento quimioterápico según protocolo Pethema (bajo riesgo). Actualmente se mantiene en remisión completa.
Conclusiones:1) El conocimiento de las variadas manifestaciones de la AT es fundamental para poderlas sospechar y diagnosticar, en especial las infecciosas y oncológicas, pues condicionan el pronóstico vital; 2) Debido la fragilidad cromosómica de la AT, las radiaciones ionizantes pueden aumentar su tendencia a desarrollar neoplasias, por lo que deben limitarse, tanto en el diagnóstico como en el tratamiento; 3) Destacamos la precocidad en el desarrollo de la LLA en este paciente y, 4) Es importante completar el diagnóstico con el estudio y consejo genético, pues los familiares heterocigotos también poseen mayor predisposición al cáncer.
Reumatología
Artritis psoriásica en la infancia. Revisión de nuestra casuística
A.I. Fernández Lorente, S.T. Jiménez Gil de Muro, J.J. Calvo Vera, C. Martín de Vicente, E. Gracia Cervera, R. García Romero,
D. Gros Esteban, T. Ureña Hornos, I. Pastor Mouron y S. Abio Albero
Hospital Materno Infantil Miguel Servet, Zaragoza.
Antecedentes: Se trata de una enfermedad de baja prevalencia. El 1-3% de la población padece psoriasis; de éstos un 2-8% en la edad pediátrica. La incidencia de artropatía psoriásica en la infancia no está bien establecida (1% de los casos de psoriasis según algunos estudios) y puede ser la única manifestación de la enfermedad antes de la 2ª década de la vida, cuando suele aparecer la lesión cutánea, con un intervalo de 8 años después del debut articular. Actualmente esta enfermedad se considera como una entidad separada de las espondiloartropatías seronegativas, por sus características peculiares al margen de éstas. Nuestro objetivo es presentar 3 casos que podrían corresponder a esta infrecuente enfermedad.
Métodos: Datos recogidos de las historias clínicas de los casos. Caso 1: Niña de 6 años que debutó en noviembre de 1993 con dolor e inflamación de tobillos de 2-3 semanas de evolución. Un familiar de 2° grado con psoriasis. ANA-, que tres años más tarde sepositivizó. HLA B27 +. Caso 2: Niña de 6 años y 6 meses que debutó en julio de 2000 con historia de 5 brotes de dactilitis en el mismo dedo de la mano en los 10 meses anteriores. Antecedentes familiares de psoriasis, ANA-, FR-. Caso 3: Niño de 7 años y 11 meses que debutó en noviembre de 2000 con dactilitis en ambas manos y artritis de muñecas y tobillos. Antecedentes de psoriasis en varios familiares de 2° grado, ANA- , FR-.
Criterios de Vancouver adaptados de Southwood: artritis psoriásica definida: artritis con rash psoriasiforme típico o artritis con tres de los cuatro criterios menores siguientes: dactilitis, cicatrices ungueales u onicolisis, rash psoriasiforme, historia familiar en 1° o 2° grado de psoriasis; artritis psoriásica probable: artritis con 2 de los 4 criterios menores.
Resultados: De los tres casos presentados ninguno presenta lesiones cutáneas típicas psoriásicas. Los tres casos han respondido bien al tratamiento con indometacina y habrá que seguir su evolución para confirmar o no el diagnóstico de artritis psoriásica.
Conclusiones: Además de ser una enfermedad infrecuente la artritis psoriásica, en la infancia nos encontramos con la dificultad añadida de que la lesión cutánea no aparece hasta una década después de la artritis con lo que el diagnóstico cumpliendo los criterios de Vancouver se realiza a posteriori.
LES con afectación glomerulonefrítica difusa que debuta con una sepsis neumocócica
S. Viver Gómez, E. Miravet Fuster, C. Fresneda Machado y M.T. Lacasa Pérez
Hospital Clínico Universitario San Carlos, Madrid.
Introducción: Presentamos el caso clínico de una adolescente que presenta una sepsis neumocócica diagnosticándose durante el ingreso de lupus eritematoso sistémico con afectación glomerular difusa. Esta asociación se ha visto en otras ocasiones en la literatura y plantea la relación entre los estados autoinmunes y una posible disfunción esplénica.
Caso clínico: Adolescente de 17 años sin antecedentes de interés que ingresa con cuadro de cinco días de evolución consistente en fiebre alta, malestar general y dolor precordial que aumenta con la inspiración. Desde hace tres meses presenta adenopatías, fiebre ocasional, artralgias en manos, lesiones cutáneas, úlceras orales y fotosensibilidad. En la exploración al ingreso presenta fiebre, mal estado general y solo destaca una adenopatía laterocervical derecha de 4 x 4 cm. En analítica presenta linfopenia, anemia normocítica con aumento de VSG y PCR. Bioquímica normal. S. orina: aislados cilindros granulosos finos. Orina de 24 h: proteinuria 0,56 g/dl. Rx. de tórax: ensanchamiento mediastínico. Ecocardiografía: Derrame pericárdico leve. Hemocultivos (2): positivos a Streptococo pneumoniae. Estudio inmunológico: ANA 1/1.280, anti-Sm+, antihistona, anticardiolipina y anti-DNA en títulos altos. CH 50, C3, C4, Cid disminuidos. Biopsia renal: glomerulonefritis difusa. Nefritis lúpica clase IV A. Se trata con Penicilina G i.v, con buena respuesta del cuadro séptico. Se inicia tratamiento con corticoides añadiéndose ciclofosfamida i.v en dosis mensual. Actualmente la paciente se encuentra asintomática con disminución significativa de los niveles de autoanticuerpos y de complemento.
Conclusiones: La sepsis neumocócica es una infección que amenaza la vida y se ha relacionado con estados de asplenia. Esta caso plantea la posible existencia de una disfunción esplénica en el lupus sistémico. Otro punto de discusión se plantearía en cuanto a la relación de causalidad entre el neumococo y la nefropatía.
Hiperferritinemia como marcador de la enfermedad de Still
C. Trillo Belizón, E. Ortega Páez, S. Broncano Lupiañez, A. Rodríguez Leal, J.M. Gómez Vida, J.A. Martín García, A. Valenzuela Soria, E. Fernández Segura, R. Nasrawin Lapaz y A. Cabrera Peña
Hospital General Básico de Motril, Motril.
Antecedentes: Aunque la elevación de la ferritina en el suero tiene lugar en múltiples situaciones patológicas (infecciones, enteritis, parasitosis, enfermedades hepáticas, insuficiencia renal crónica, linfomas, histiocitosis maligna, síndrome hemofagocítico y enfermedades inflamatorias crónicas), hay autores que consideran hiperferritinemias superiores a 1000 ng/ml muy sugestivas de enfermedad de Still y elevaciones superiores a 4000 ng/ml como específicas de esta enfermedad en presencia de un cuadro clínico compatible.
Caso clínico: Niña de 9 años que presenta picos febriles (39-40ºC) que se acompañan de erupción maculo-papulosa generalizada evanescente, artralgias y mialgias generalizadas y escalofríos. Asintomática en los períodos apiréticos. Entre los datos analíticos destacan: 29800 leucocitos, VSG 98 mm 1ª hora, PCR 41,3 mg/dl y ferritina 8005 ng/ml. Factor reumatoide y anticuerpos antinucleares negativos.
Otros datos analíticos: Haptoglobina, orosomucoide, fibrinógeno, dímero D y PDF ligeramente elevados. Bioquímica básica, transaminasas, inmunoglobulinas, complemento, hemocultivos, urocultivo, coprocultivo, serologías TABM, CMV, VEB, toxoplasma y borrelias negativas. Médula ósea con aumento de celularidad mieloide/eritroide de aspecto normal. Radiografía de tórax, ecografía abdominal y cardíaca y examen ocular normales.
Conclusiones: Ante una clínica compatible, como la de nuestra paciente, la elevación desmesurada ( miles de ng/ml) de la ferritina en el suero puede ayudar a un diagnóstico e inicio de tratamiento precoces de esta enfermedad. La hiperferritinemia será más especifica cuanto más elevada se encuentre.
Lupus Eritematoso Sistémico en paciente varón de 14 años
J. Pineda Sánchez, C. Muñoz Gil, Ll. Galcerán Jove, J.M. Muixi Rosset, M.D. Vila Serra y S. Nevot Falcó
Hospital General de Manresa, Manresa.
El lupus eritematoso sistémico (LES) es una enfermedad inflamatoria crónica del tejido conjuntivo de naturaleza autoinmune que afecta a múltiples órganos y sistemas cuyo diagnóstico se basa en criterios clínicos y de laboratorio.
Caso clínico: Paciente varón de 14 años que presenta astenia y sensación distérmica variable de 2 meses de evolución, iniciándose esta sintomatología tras una apendicectomía. Dos semanas antes de su ingreso hospitalario aparece fiebre, anorexia y aftas bucales de intensidad progresiva con dificultad para la alimentación y pérdida de peso significativa.
Antecedentes: Apendicectomía: apendicitis flemonosa según resultado anatomopatológico.
Exploración física al ingreso.
Febril (40 ºC). Regular estado general. Eritema del puente nasal, pabellones auriculares y del tercio distal de los dedos de manos y pies. Afectación de mucosa oral con enantema y aftosis dolorosa que impide la apertura de la boca. Resto exploración por aparatos normal.
Exámenes complementarios
- Analítica. Hemograma: Pancitopenia: Leucocitos 2,5 x 109/L (fórmula normal). Hematíes: 3,9 x 1012/L, Hb 10,8 g/dL, Hcto 31 % VCM 80 fL, plaquetas 124 x 109. Bioquímica: ALT 0,99 mkat/L AST 1,40 µkat/L , LDH 13,4 µkat/L , VSG 70 mm/h. Resto bioquímica normal. Hemocultivo negativo. PPD negativo. Serologías víricas: VHB, VHC, VIH, CMV: IgG, IgM negativas; E. Barr, rubeola, varicela, toxoplasma: IgG positivas, IgM negativas. ANA y anti DNA positivos. Ac antifosfolípidos y anticardiolipinas negativos. Crioaglutininas negativas. HLA B27 negativo. Complemento C3 y C4 disminuídos.
- Punción de médula ósea. Bloqueo medular no sugestivo de hemopatía.
- Sedimento de orina: hematuria microscópica con proteinuria.
- Función renal: proteinuria de rango no nefrótico (66 mg/dl).
- Rx tórax normal, ecografía abdominal normal.
- Biopsia de mucosa oral y cutánea: inespecífica.
Evolución y tratamiento: Ingreso con medidas de soporte y sintomáticas. Una vez finalizado el estudio y dado el empeoramiento progresivo del paciente y ante la sospecha de LES, se inicia tratamiento con corticoterapia e.v. con mejoría clínica de las lesiones mucocutáneas y del estado general del paciente. Se confirma el diagnostico de LES según los criterios diagnósticos de la American Rheumatism Association. Durante su estancia se detecta afectación renal (proteinuria significativa > 0,5 mg/dl en orina de 24 horas) y afectación articular interfalángica por lo que se decide su traslado a centro de tercer nivel para tratamiento y posterior control.
Conclusiones:
- La presencia de aftas bucales de evolución tórpida acompañada o no de otra sintomatología requieren al LES como uno de sus diagnósticos diferenciales.
- Aunque menos frecuente, el LES es una enfermedad que también afecta a niños varones.
- El diagnóstico del LES se basa en criterios clínicos y de laboratorio, sin embargo, los criterios clínicos son orientativos del diagnóstico en espera de los resultados inmunológicos.
- La afectación renal es un signo de mal pronóstico, que requiere estudio de la función renal desde un primer momento.
Fiebre persistente de origen desconocido como forma de presentación de lupus eritematoso sistémico
M. Los Arcos Solas, M. Fernández Fernández, B. Curros Novo, A. Moráis López, S. Campuzano Martín, I. Málaga, C. Polo Mellado, C. Bousoño García, E. Ramos Polo y N. García López
Hospital Central de Asturias, Oviedo.
Introducción: El lupus eritematoso sistémico (LES) es una enfermedad multisistémica, inflamatoria, de origen autoinmune, siendo la presentación clínica muy variable. La pubertad es la edad de presentación más frecuente, con predominio por el sexo femenino, aunque en menor proporción que en la edad adulta.
Caso clínico: Varón de 14 años que presenta fiebre persistente de origen desconocido de 3 semanas de evolución, acompañada de astenia y anorexia. No antecedentes personales de interés.
En exploración física al ingreso presenta buen estado general, destacando la presencia de eritema malar y pequeñas adenopatías generalizadas, no dolorosas ni adheridas y esplenomegalia de 3 cm.
En analítica sanguínea realizada al inicio del cuadro se observa pancitopenia progresiva y aumento de reactantes de fase aguda, sin hallarse atipias o displasias. En bioquímica realizada a las 24 horas del ingreso presenta elevación importante de ácido úrico y LDH, por lo que se inicia pauta de rehidratación y alcalinización, ante sospecha de lisis tumoral.
Se realiza TAC tóraco abdominal, con presencia de adenopatías tóraco-abdominales asociadas a esplenomegalia > 15 cm.
Se plantea el diagnóstico diferencial entre LES y proceso linfoproliferativo, confirmándose el primero con serología de auto-anticuerpos, que muestra positividad de ANA (1/320), con patrón moteado, anti-SM y anti-DNA.
Serología viral, detecta infección pasada por virus de Epstein -Barr.
Inicio de corticoterapia oral, con mejoría clínica, añadiéndose antipalúdicos ante la sospecha de afectación renal, no confirmada.
Presentó pequeño derrame pericárdico, con buena evolución.
Conclusiones: Ante un niño con fiebre persistente sin foco, el LES es una de las posibilidades diagnósticas, pudiendo confundirse con procesos linfoproliferativos si la afectación ganglionar es muy importante.
Síndrome S.E.A.: manifestaciones y evolución clínica en 11 pacientes
I. Calvo Penadés y L. Lacruz Pérez
Hospital Universitario Dr. Peset., Valencia.
Introducción: El síndrome de artropatía y entesopatía seronegativo, conocido como S.E.A, fue descrito por Rosenberg y Petty en 1982 para identificar a aquellos pacientes que probablemente podían desarrollar una de las enfermedades del grupo de las espondiloartroparias juveniles sin la necesidad de contar con la tipificación del antígeno de histocompatibilidad HLA-B27 y evitar que quedaran incorrectamente incluidos en la ACJ/ARJ. Los pacientes con SEA pueden evolucionar hacia una espondiloartritis juvenil u otra EAP, o hacia la remisión sin dejar de ser SEA, según los resultados de seguimientos de las series publicadas.
Objetivo: Es analizar las manifestaciones clínicas y evolutivas del Síndrome SEA.
Pacientes y métodos: Presentamos 11 pacientes, 8 niños y 3 niñas con Síndrome SEA que cumplían criterios de Rosenberg y Petty (Artritis Rheum 1982,25:1041-1047).
Valoramos los síntomas durante el primer años de enfermedad y su posterior evolución, determinando el grado de afección articular axial y periférica, así como control radiológico de las articulaciones afectas y sacroiliacas.
En todos ellos se realizó determinación del Factor Reumatoide (FR), Anticuerpos Antinucleares (ANA) y Antígeno de Histocompatibilidad HLA-B27.
Resultados: La edad media de inició fue de 11 años (rango 6-15) con un tiempo de seguimiento de 4 años.
Cuatro pacientes tenían antecedentes de Espondilitis Anquilosante y uno de Psoriasis.
Los 11 pacientes presentaron artralgias y 9 de ellos oligoartritis, predominante en rodillas seguido de tobillos y muñecas. Durante el primer año, 7 cursaron con entensopatia principalmente aquilea, plantar y suprarotuliana y en los tres restantes se presentó más tardíamente. Tres pacientes presentaron dolor lumbar inflamatorio sin signos radiológicos y dos presentaron sacroilitis con cambios radiológicos. Ningún paciente desarrolló uveitis.
El FR fue negativo en todos los casos, dos pacientes presentaron ANA positivos y el HLA-B27 fue positivo en 9 pacientes.
Los 11 pacientes recibieron AINES, asociado a Salazopirina en 4 casos y a Metotrexato en 3 pacientes. El grado funcional de Steinbroker fue en 10 pacientes de I y solo un caso presentó grado II.
Conclusiones: En nuestra serie el síndrome SEA presenta una buena evolución clínica ya que solo el 18% de los pacientes desarrollaron sacroilitis. Faltan series españolas para poder comparar resultados y un mayor número de años de seguimiento.
Piomiositis
S. Fernández Fernández, A. González Vergaz, T. Bracamonte Bermejo, J.M. de Cea Crespo, L. Echeverría Zudaire, B. García Cuartero, M. Llanos Carrasco y A. Arregui Sierra
Hospital Severo Ochoa, Leganés.
Introducción: La infección piógena del músculo esquelético (piomiositis), es una entidad poco frecuente en pacientes pediátricos inmunocompetentes. La inespecificidad de los síntomas y las pruebas de laboratorio, hacen que el diagnóstico y el tratamiento se demoren.
Material y métodos: Paciente varón de 14 años de edad que consulta por fiebre (38-39 ºC) de ocho días de evolución y dolor en flanco izquierdo. No refiere traumatismo previo, pero si haber realizado ejercicio físico intenso.
A.P.: Reflujo G-E con realización de funduplicatura de Nissen dos años antes de acudir a consulta.
E.F.: Afectación moderada del estado general; dolor a la palpación en F.I.I., fosa renal izqda. y región inguinal homolateral, con dolor a la marcha sin limitación articular ni signos inflamatoros.
P. Comp.: Seriadas cifras de leucocitos normales con neutrofilia discreta. V.S.G. y P.C.R. elevados (cifras máximas de 54 mm y 143,9 mg/dl respectivamente).Bacteriología: Hemocultivos, urocultivo, frotis nasofaringeo, L.C.R.: negativos. Serologías, incluyendo H.I.V., negativas. Rx. cadera y ecografía abdominal y de cadera sin hallazgos patológicos. Gammagrafía ósea con H.D.P.-99- Tc y 67-Ga.: hipercaptación a nivel de cresta iliaca izqda. y porción superior del hueso iliaco adyacente. R.M.N.: Afectación difusa de la musculatura del cuadrado lumbar , iliaco y porción profunda del psoas izqdo., presentando este último aumento de tamaño y área nodular sugerente de absceso de 1x 2 cm. compatible con piomiosistis en fase flemonosa. P.A.A.F. con control mediante T.A.C. aislándose Staphilococo coagulasa negativo.
Iniciándose tratamiento con Imipenem parenteral durante 15 días, con excelente evolución clínica, normalizándose los parámetros analíticos a la semana de iniciado el mismo.
R.M.N. de control a los dos meses : cambios residuales en músculo psoas iliaco sin evidencia de piomiositis.
Comentarios: 1) La triada clínica de fiebre, dolor y sintomatología de la cadera (articular y/o muscular), debe hacernos incluir en el dgco. diferencial la piomiositis del ms. Psoas. 2) El ejercicio físico intenso, aún sin antecedentes de traumatismo evidente, puede ser el desencadenante del proceso. 3) Las pruebas de imagen, st. la R.N.M. facilitan el diagnóstico del proceso. 4) La buena evolución clínica y analítica permite acortar la duración del tratamiento.
Enfermedad de Kawasaki: forma de presentación completa en un lactante
A. Peña Valenceja, F. Centeno Malfaz, J. Rodríguez Calleja, M.F. Omaña Alonso y J.M. Muro Tudelilla
Hospital Universitario del Río Ortega, Valladolid.
Introducción: La enfermedad de Kawasaki es una arteritis sistémica de origen desconocido. El diagnóstico se basa en la asociación de signos y síntomas clínicos, que por otro lado no son exclusivos de la enfermedad. Inicialmente descrita por Kawasaki y colaboradores en 1974, fue definida como fiebre de al menos 5 días de evolución, más la presencia de al menos 4 de 5 de los siguientes criterios clínicos: inyección conjuntival, alteraciones de la boca (labios enrojecidos y agrietados, lengua aframbuesada y enantema), alteraciones de pies y manos (eritema palmo-plantar, edema duro de pies y manos y descamación subungueal de dedos), exantemas polimorfos y adenopatías superiores a 1,5 cm. En el momento actual, la mayoría de los autores prefieren una definición modificada, en la que la fiebre se considera un síntoma más, no obligado, y sin exigirse una duración concreta para la misma.
El diagnóstico diferencial de esta enfermedad debe realizarse con un amplio número de entidades, siendo muy importante por la posibilidad de complicaciones, algunas de ellas graves. Esto es especialmente importante en las formas incompletas, en las que el cuadro no cumple los criterios clínicos, y que son frecuentes en menores de 6 meses.
La enfermedad cursa en tres fases: aguda (clínica); subaguda (descamativa); convalecencia (secuelas). El tratamiento consiste en la administración de gammaglobulina intravenosa y ácido acetilsalicílico. La instauración precoz del mismo disminuye la incidencia de complicaciones, especialmente de aneurismas coronarios.
Presentamos el cuadro de un lactante de 3 meses y medio de edad compatible con una forma típica de enfermedad de Kawasaki. Incidimos en la importancia del diagnóstico precoz para la instauración del tratamiento. Ilustramos el caso con abundante iconografía.
Caso clínico: Paciente varón de 3 meses y medio de edad que ingresa en nuestro servicio por presentar fiebre elevada de 24 horas de evolución y decaimiento con rechazo de tomas. A la exploración física se aprecia una ligera hiperemia faríngea, con un signo del trago en el oído izquierdo, y una adenopatía laterocervical derecha de 2x3 cm de diámetro, llamando la atención el decaimiento del niño, con quejido y aspecto de gravedad. Se realizó analítica de sangre que mostró una leucocitosis de 19.300, con ligera desviación izquierda, con una proteína C reactiva (PCR) de 38 mg/l. El sistemático de orina fue normal. Tras obtener muestras para cultivos, incluyendo LCR cuya citoquímica fue normal, se inició tratamiento con cefotaxima IV. Al tercer día de ingreso se aprecian labios secos agrietados, hiperemia conjuntival bilateral, y enrojecimiento llamativo de palmas y plantas, apareciendo de forma fugaz un exantema maculoso en tronco, etiquetándose de probable enfermedad de Kawasaki e iniciando tratamiento con gammaglobulina intravenosa a dosis de 2 g/kg en dosis única en perfusión lenta, y ácido acetilsalicílico a dosis de 100 mg/kg/día durante 5 días, pasando después a dosis antiagregantes de mantenimiento. Se mantuvo afebril desde el 3º día de tratamiento específico. La bacteriología fue negativa, retirándose la antibioterapia. Elevación de reactantes de fase aguda hasta 247 mg/l de PCR. Electrocardiograma y ecocardiografía normal. Serología para virus de Epstein Barr, Ricketsias, y citomegalovirus negativa. Trombocitosis progresiva alcanzando en el control prealta, realizado a los 12 días del ingreso, una cifra de 923.000 plaquetas, con normalización de reactantes de fase aguda. Evolución posterior favorable, con control ecocardiográfico normal.
Discusión: Aunque la enfermedad es de carácter autolimitado, y en la mayoría de las ocasiones de buen pronóstico, es muy importante su conocimiento por su frecuente asociación con coronariopatías de graves consecuencias. Al no existir un marcador biológico, el diagnóstico se basa en criterios clínicos, que son inespecíficos y fácilmente atribuibles a otras enfermedades. En muchas ocasiones la enfermedad no cumple todos los criterios, etiquetándose de formas incompletas. Estas formas son más frecuentes en lactantes, y se relacionan con una mayor incidencia de complicaciones.
La administración de gammaglobulina IV a dosis de 2 g/kg ha demostrado su eficacia en la reducción de la incidencia de aneurismas coronarios, cuando se administra antes de la fase descamativa. El ácido acetil salicílico se utiliza a altas dosis hasta la remisión de la fiebre, manteniéndose después a dosis antiagregantes durante un tiempo variable según la evolución analítica y ecocardiográfica.
Conclusiones: Queremos destacar la importancia del diagnóstico precoz de la enfermedad de Kawasaki. Ante la sospecha fundada del mismo, debe instaurarse el tratamiento específico lo más precozmente posible, debido a las importantes complicaciones, sobre todo cardíacas, que se pueden presentar a lo largo de la fase de convalecencia de la enfermedad.
Prevalencia y significado clínico de los anticuerpos anticardiolipina en la artritis idiopática juvenil
I. Calvo Penadés, L. Lacruz Pérez y E. Andreu Alapont
Hospital Universitario Dr. Peset, Valencia.
Los anticuerpos anticardiolipina (aCL) han sido referidos en una gran variedad de enfermedades autoinmunes y no autoinmunes. La presencia en niños con artritis idiopática juvenil (AIJ) no esta bien establecida.
Objetivo: Determinar la prevalencia de los aCL en pacientes con AIJ y establecer si existe relación con las manifestaciones clínicas.
Métodos: Se determinan por ELISA los aCL (isotipos IgG e IgM), anticuerpos antinucleares (ANA) y factor reumatoide (FR) en 74 niños con AIJ (35 oligoarticulares, 24 poliarticulares y 15 sistémicas). Los aCL también son realizados en 50 niños de grupo control. Se valoran los parámetro de actividad ( nº de articulaciones afectas, la VSG y la Hb) y situación funcional de los pacientes.
Resultados: Los aCL son detectados en 26/74 pacientes con AIJ (35,2%) y en 3/50 niños control (6%). Las cifras máximas de IgG se presentan por debajo de 46 y de 20 IgM. En un 69% los aCL se asocian a la presencia de ANA pero no al FR. La positividad alcanzó cifras máximas al inicio de la enfermedad (77%), 8 pacientes presentaron cambios en los niveles según la actividad de la enfermedad.
Conclusión: La prevalencia de aCL es similar a otras series. No parece guardar relación con los parámetros clínicos biológicos, aunque no podemos de la implicación con la actividad de la enfermedad.
Púrpura de Schönlein-Henoch: revisión de los últimos años
E.M. Camero Muñiz, A. Fernández Romero, E.M. Saavedra Chaves, C. Zamorano Lacalle y J. González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Introducción: La púrpura anafilactoide es un trastorno de origen inmunológico del endotelio vascular. Afecta a vasos de pequeño calibre, principalmente de la piel, articulaciones, tracto gastrointestinal y riñones.
Objetivo: Conocer las características epidemiológicas, clínicas, terapéuticas y evolutivas de esta enfermedad en nuestro medio.
Material y método: Revisión retrospectiva de historias clínicas con diagnóstico de Púrpura de Schönlein-Henoch en niños de 0 a 14 años, desde enero de 1995 a mayo de 2000. Se analiza: edad, sexo, antecedentes (infecciones en el mes previo y fármacos en la semana anterior), forma de comienzo, clínica, complicaciones, pruebas complementarias, evolución y tratamiento.
Resultados: Se revisan 53 historias, con diagnóstico de púrpura de Schönlein-Henoch, de los cuales 28 son mujeres (52,8%) y 25 son varones (47,2%); de edades comprendidas entre 1 y 9 años, con una media de edad de 4,8 años. El mayor número de casos se registraron en enero y octubre. En el 79,2% de las historias se encuentra antecedente de infección reciente, fundamentalmente de vías respiratorias altas (52,8%). El 60,3% del total de casos había recibido tratamiento farmacológico, la mitad de ellos antibióticos. En 34 casos (64,1%) la púrpura fue la forma de comienzo, apareciendo de manera exclusiva en el 55,8% y, en el resto, acompañada de otros síntomas, destacando las manifestaciones articulares (artralgias y/o artritis) en el 46%, seguido de dolor abdominal en el 20%. Como segunda forma de inicio más frecuente encontramos el dolor abdominal (20%). En cuanto a la clínica, la púrpura fue de características típicas en el 100%, el resto de síntomas encontrados por orden de frecuencia fueron: dolor abdominal (64%), manifestaciones articulares (60,3%, más frecuentes en tobillos), edemas (39,6%, más de la mitad localizados en miembros inferiores), vómitos (35,8%), hemorragia digestiva (22,6%), cefalea y/o trastornos del comportamiento (9,4%), epistaxis (9,4%), disfonía (7,5%), hematuria macroscópica (5,3%), parestesias (3,7%) y orquiepididimitis (1,9%). Respecto a las pruebas complementarias, el estudio de coagulación fue normal en todos los casos; en el sedimento de orina apareció hematuria aislada en el 20,7% y hematuria con proteinuria en el 15%, aislándose proteinuria sola en el 3,8%; ASLO y PCR se elevaron en menos de la mitad de los casos. Inmunidad humoral: Ig A normal en 42, elevada en 6 y disminuida en 1 caso; Ig G normal en 42 y elevada en 7; Ig M normal en 44 y elevada en 3; en 2 casos se elevaron ambas. Se solicitaron inmunocomplejos circulantes en 33 casos y en más de la mitad se encontraban elevados. Se realizó tratamiento sintomático en el 100 % de los casos, se administraron corticoides en el 30,2%, la mayoría de ellos durante menos de 2 semanas y a una dosis inferior a 2 mg/kg/día; un solo caso precisó tratamiento con ciclosporina por presentar síndrome nefrótico corticorresistente. Aparecieron las siguientes complicaciones: 3 casos de nefropatía (2 de síndrome nefrótico y 1 de microhematuria persistente) y 1 de invaginación intestinal. La media de estancia hospitalaria fue de 12,7 días, con un rango comprendido entre 3 y 52 días. El 75,4% requirió un solo ingreso.
Conclusiones:1) Encontramos un ligero predominio de mujeres sobre varones. 2) La media de edad de aparición de la enfermedad fue de 4,8 años. 3) Hubo antecedentes de infección en el 79,2% de los niños, la mayoría de vías respiratorias altas. 4) La forma de comienzo más frecuente fue la púrpura. 5) Los síntomas más frecuentes fueron la púrpura, el dolor abdominal, las manifestaciones articulares y los edemas. 6) El 35,7% de los niños presentó alteraciones en el sedimento de orina (hematuria y/o proteinuria). 7) La incidencia de complicaciones renales es tres veces más alta que la señalada en otros estudios, aunque la muestra revisada es insuficiente. 8) Aparecieron complicaciones en el 7,5% de los casos, siendo las más frecuentes las renales (5,6%). 9) El 30,2% de los niños precisó tratamiento con corticoides y el 1,9% ciclosporina. 10) La Ig A sólo se encuentra descendida en un caso, frente al 50% que refleja la bibliografía.
Incidencia de la Enfermedad de Kawasaki en nuestro medio
C. González Espinosa, B. González Álvarez, J. Feo González, R. Pérez Hernández, P. Lupiani Castellanos y M. Domínguez Suárez
Hospital Universitario de Canarias, Santa Cruz de Tenerife.
En la década comprendida entre 1990 y 2000, se diagnosticaron en nuestro Servicio un total de 23 casos de Enfermedad de Kawasaki, cuyas edades oscilaban entre los 2 y los 14 años, correspondiendo 14 a varones y 9 a mujeres.
Al ingreso se sospechó el diagnóstico en un total de 10 pacientes y el resto se confirmó durante su hospitalización. Todos ellos presentaron fiebre, adenopatías, eritema, hipertermia, lengua afresada e hiperemia conjuntival, apareciendo soplo cardíaco en 14 casos y dolores en extremidades inferiores en 15. En los exámenes complementarios se detectó leucocitosis con desviación a la izquierda, Proteína C Reactiva +, disminución de la albúmina en 11 casos y arritmia y prolongación del intervalo PR-QT en 13 casos. En dos de los pacientes se apreció miocarditis. Se instauró tratamiento con Ácido Acetilo Salicílico a altas dosis e inmunoglobulinas ev. a razón de 2 gr/kg . Todos los pacientes evolucionaron favorablemente y sólo uno de ellos hizo una insuficiencia cardíaca, de la que se recuperó favorablemente.
Artitris crónica juvenil de inicio sistémico: a propósito de un caso grave
M.C. Moliner Robredo, J. Blázquez Fernández, I. Cuadrado Pérez, M. Escudero Bergua, M. Garzón Gómez, M. Fernández Ibieta, A.P. Galera Peinado, M.T. Cortes Coto, J. García-Arroba Muñoz y J. Herrera Montes
Hospital Universitario de Getafe, Getafe.
Antecedentes y objetivo: La artritis crónica o idiopática juvenil (ACJ) es un grupo de enfermedades caracterizadas por sinovitis crónica y asociadas a varias manifestaciones inflamatorias extraarticulares. En la ACJ de inicio sistémico la clínica puede ser muy variada e inespecífica, pudiendo pasar desapercibida la artritis por la espectacularidad de los síntomas sistémicos. El diagnóstico es clínico por la persistencia de las manifestaciones sistémicas típicas a lo largo de 3 meses consecutivos, y exclusión de otras enfermedades. Presentamos un caso de una niña de 14 años con ACJ de inicio sistémico, por la gravedad y rápida progresión del deterioro clínico.
Caso clínico: Niña de 14 años con síndrome febril de 9 días de evolución, malestar general, exantema maculopapular (extremidades y cara), astenia y anorexia (pérdida del 3% de su peso en 3 días), y dolor articular en tobillo derecho, muñecas y codos, con aspecto tumefacto y pálido de los dedos de la mano, con morfología fusiforme. Pruebas complementarias: Aumento importante de reactantes de fase aguda (PCR; VSG, *1AT, Ferritina), anemización y trombopenia progresivas, severa vacuolización de neutrófilos, aumento moderado de enzimas hepáticos, e hipoalbuminemia. M.O.: severa hiperplasia granulocítica e hipoplasia eritroide llamativa. Gammagrafía con Ga: depósito patológico del radiotrazador a nivel laterocervical bilateral, más intenso en lado derecho, así como en área teórica pectoral y/o axilar derecha. Hepatoesplenomegalia. ECO axilar derecha: adenopatías axilares derechas, la mayor de ellas de unos 2 cm, de localización profunda. Se descarta etiología infecciosa por VEB, CMV, C. Burnetti, R. Conorii, Borrelia, Brucella, Hepatitis A, B y C, Chlamydia Pneumoniae y Mycoplasma. Hemocultivo, urocultivo y cultivo de M.O. (parásitos, hongos, micobacterias típicas y atípicas): negativos. Marcadores reumáticos negativos (ASLO, Inmunoglobulinas, Complemento, ANA y F.R.). Resto de exploraciones complementarias sin hallazgos patológicos.
Resultados: Tras tratamiento antibiótico empírico de amplio espectro durante 10 días sin respuesta favorable y, ante el deterioro clínico y analítico rápidamente progresivo, se instaura tratamiento con Prednisona oral (1,5 mg/kg/día, en 3 dosis) + profilaxis de osteoporosis con Calcio y vitamina D (500 mg Ca y 400 UI de vit D3/día) + protección gástrica (Magaldrato). Evolución clínica muy favorable, disminuyendo los picos febriles, así como la astenia y anorexia, y desapareciendo el exantema y la fiebre definitivamente tras 6 días de tratamiento, presentando ocasionalmente artralgias erráticas sin signos inflamatorios aparentes. Alta definitiva tras 8 meses de tratamiento corticoideo, con disminución lenta progresiva, sin recaídas.
Conclusiones: Imposibilidad de predecir la evolución en un paciente determinado, pudiendo presentar exacerbaciones y remisiones o persistencia de los síntomas durante años (artritis leve, artritis grave), remitiendo al llegar a la pubertad o dando artritis activa en la edad adulta, así como exarcebaciones después de años de aparente remisión completa, que pueden asociarse a enfermedades intercurrentes. La ACJ tiene baja mortalidad pero puede ocurrir en casos graves de afectación sistémica. El tratamiento corticoideo está indicado en la enfermedad sistémica grave que no responde a salicilatos.
Pediatría Extrahospitalaria
Enfermedad tuberculosa infantil ¿es posible su control y eliminación?
A. Bercedo Sanz, C. Borragán Torre, A. Valdés Joglar y M. Serrano Acebo
Centro de Salud Meruelo, Santander, Centro de Salud Gama, Santander.
Antecedentes: La enfermedad tuberculosa (TBC) es uno de los problemas de salud pública más importantes que quedan pendientes de resolver como se demuestra por el aumento de su incidencia en los últimos años (57/100.000 habitantes/año). En Cantabria, al igual que en el resto de España, la prevalencia de la infección tuberculosa a los 6 y 14 años es de 1,32% y 4,21%, respectivamente. La TBC infantil suele ser un reflejo de la TBC del adulto conviviente por lo que el estudio familiar debe ser obligado siempre. Presentamos dos casos de TBC infantil diagnosticados recientemente, con el objetivo de potenciar el interés sanitario para conseguir su control y eliminación en los próximos años.
Caso 1: Varón de 9 años y 2 meses que presenta fiebre prolongada de 8 días de evolución en tratamiento con eritromicina oral. Exploración dentro de la normalidad salvo secreciones de vías altas. Exámenes complementarios: Hemograma, bioquímica y orina normales. VSG: 56 mm/hora. PCR: 7,4 mg/dl. Rx Tórax: adenopatías hiliares izquierdas e infiltrado perihiliar asociado. Mantoux: 14 mm. Tratamiento: Isoniacida, rifampicina y pirazinamida a las dosis establecidas. Estudio familiar: Abuelo y tía maternos afectos de TBC pulmonar.
Caso 2: Varón de 2 años y 10 meses afecto de síndrome febril de 7 días de evolución asociado a grillera y tos productiva en tratamiento con amoxicilina-clavulánico oral. A. personales: mantoux negativo a los 18 meses de edad. Asma bronquial leve intermitente con bronquitis de repetición desde los 23 meses. Exploración: Buen estado general. Cardiopulmonar: roncus dispersos en ambos hemitórax y subcrepitantes medios e inferiores derechos. Resto sin hallazgos. Exámenes complementarios: Hemograma (Hb: 10,4; VCM: 76,8), VSG: 130 mm/hora. PCR:2,6 mg/dl. Bioquímica normal. Mantoux: 14 mm. Rx Tórax: Adenopatías hiliares izquierdas. Neumonía en lóbulo medio derecho. Cultivo de Löwenstein-Jensen negativo. Tratamiento: Isoniacida, rifampicina y pirazinamida a las dosis habituales. Estudio familiar: padres y hermano de 2 meses mantoux y Rx tórax normales. Tío y primo maternos: mantoux + con Rx Tórax normal.
Conclusiones: La alta incidencia de la TBC aconseja una mayor implicación de los profesionales sanitarios en su prevención y control. La prueba tuberculínica debe seguir realizándose de forma periódica al año, a los 4-6 años y a los 11-16 años si queremos controlar la endemia existente en nuestro país, siendo obligado el estudio familiar ante un caso de TBC en la edad pediátrica.
Planteamiento de mejora de la calidad en la asistencia neonatal en un centro de atención primaria
J. Romero Sánchez, A.M. León Ruiz y M.I. Romero Sánchez
Centro de Salud Las Albarizas, Marbella.
Antecedentes y objetivo: En la asistencia programada al recién nacido en nuestro centro de salud de titularidad pública se implementaron unas actividades para favorecer la captación activa de los neonatos y su incorporación al programa de seguimiento de salud infantil vigente en la comunidad andaluza. Se había detectado de forma cualitativa en los años previos un frecuente abandono de dicho programa en etapas precoces (período neonatal) incluso en recién nacidos de riesgo social. Nuestra población corresponde a un nucleo costero en importante crecimiento demográfico debido a inmigración intra e internacional, con tasas de crecimiento vegetativo intercensal de un 20 %.
Métodos: Se centraron en un grupo focal que agrupó a los pediatras del centro de salud, las matronas, las enfermeras con dedicación preferente al área infantil y representantes de los administrativos. Como método de identificación de problemas se utilizó el "brainstorming" y la técnica del grupo nominal y para priorización el método Hanlon. Como herramientas de análisis de la oportunidad de mejora se emplearon el diagrama de Isihawa y el gráfico de Pareto. Con estos métodos se acordaron criterios de mejora y se implementaron las medidas correctoras.
Resultados: Los criterios de mejora fueron la captación activa del recién nacido en el acto de la toma de muestras de las pruebas endocrino-metabólicas por la matrona y la citación en dicho momento mediante el sistema informático TASS hacia los 15 días de vida, incluyéndolo en el programa de seguimiento de salud infantil. En dicha revisión de los 15 días, efectuada por el pediatra, se citaba de igual modo para los 2 meses de vida. De este modo no se constataron abandonos en el programa de seguimiento de salud infantil en su fase neonatal en los niños de riesgo social, tan solo en un porcentaje (alrededor del 20%) de los niños que eligieron para su seguimiento a centros no públicos.
Gestión interniveles asistenciales del proceso otitis media en la edad pediátrica. Nuestra experiencia
J. Romero Sánchez, A.M. León Ruiz y M.I. Romero Sánchez
Centro de Salud Las Albarizas, Marbella.
Antecedentes y objetivo: En la asistencia al niño con otitis media subsidiaria de valoración evolutiva tanto por el pediatra de asistencia primaria como por el especialista hospitalario de otorrinolaringología (ORL) se habían detectado de forma cualitativa en los años previos frecuentes problemas en la coordinación entre dichos niveles, que sobre todo repercutían en la lentitud del proceso con el consiguiente incremento de la lista de espera (situándose ésta en más de 2 meses). Nuestro centro de salud tiene de referencia en ORL a un hospital de segundo nivel situado a 5 km de distancia y a un centro periférico de especialidades situado en el mismo centro de salud.
Métodos: Se centraron en un grupo focal que agrupó a los pediatras del centro de salud, los otorrinolaringólogos del hospital de referencia y del centro de especialidades y representantes de los administrativos. Como método de identificación de problemas se utilizó el "brainstorming" y la técnica del grupo nominal y para priorización el método Hanlon. Como herramientas de análisis de la oportunidad de mejora se emplearon el diagrama de Isihawa y el gráfico de Pareto. Con estos métodos se acordaron criterios de mejora y se implementaron las medidas correctoras.
Resultados: Los criterios de mejora se centraron en la elección entre los niños evaluados por el pediatra del centro de salud y subsidiarios de examen ORL aquellos con alta probabilidad de precisar cirugía (adenoidectomía y/o colocación de tubos de drenaje timpánicos) de aquellos otros con baja probabilidad de necesitar dichos procedimientos. Estos criterios fueron cualitativos y se basaron en la edad del niño, el número de episodios de otitis media, tiempo promedio de curación clínica, calidad de la audición detectada por la familia y número de tratamientos antibióticos que habían precisado. Las medidas correctoras fueron el envío preferente de los niños con alta probabilidad de cirugía y el envio de los demás niños a una consulta especial de audiometría cuyo resultado podía ser valorado por el pediatra del centro de salud. Este manejo del proceso otitis media ha redundado en una disminución significativa de la lista de espera, situándose ésta en 2 - 3 semanas de forma mantenida en el último año.
Asma pediátrico e intervenciones educativas
R. Peiró, C. Casanova, J. Colomer Revuelta, A. Martorrell, C. Colomer, R. Ferrero, J. Latour y O. Valls
Centro de Salud Pública de Alzira, Alzira, Universidad de Valencia. Facultad de Medicina., Valencia, Escuela Valenciana de Estudios en Salud, Valencia.
Introducción: El ingreso por asma es una de las hospitalizaciones pediátricas consideradas evitables puesto que es una enfermedad que se puede manejar ambulatoriamente tanto por la posibilidad de mejorar y controlar un episodio agudo como por el adecuado seguimiento de la enfermedad crónica en los períodos intercrisis. Esto permite mejoras en los resultados en salud tanto por la reducción de las hospitalizaciones como la reducción de la frecuentación a los servicios de urgencias y la mejora de la calidad de vida de los niños asmáticos. En la Comunidad Valenciana se producen aproximadamente 400 hospitalizaciones anuales por esta causa y no existe información sobre la frecuentación por asma agudo en niños de los servicios de urgencia aunque se sospecha que es mucho mayor. Por otra parte la incidencia de asma infantil se ha incrementado durante los últimos años, siendo una de las enfermedades pulmonares crónicas más comunes entre los niños. Aunque actualmente no existe tratamiento para su curación, esta puede ser controlada con tratamiento y seguimiento adecuado
El objetivo de este estudio es identificar las intervenciones realizadas por los profesionales sanitarios de Atención Primaria del Servicio Valenciano de Salud para el control y seguimiento del asma infantil.
Material y métodos: Estudio de las intervenciones realizadas a niños asmáticos para seguimiento de su enfermedad. La información se obtuvo mediante cuestionario autoadministrado a los profesionales que trabajan en pediatría: médicos y enfermería. Se envió una carta explicativa con los objetivos y un grupo de cuestionarios para que el coordinador del centro los distribuyera y se realizó un reenvio de recordatorio.
El cuestionario fue diseñado para el estudio a partir de la guía americana de manejo del asma de 1997 y consta de cinco partes: Datos socioeconómicos (6 ítems), realización de actividades educativas (14 ítems), información sobre control ambiental (10 ítems), terapia farmacológica (22 ítems), y medidas objetivas para valorar la gravedad (18 ítems), Las preguntas tenían 5 opciones de respuesta: si, no lo hago por falta de tiempo, no lo considero necesario, no pertinente, y no. Se mandaron encuestas a 151 centros de AP. Se realiza un análisis descriptivo.
Resultados: Se recibieron encuestas desde 113 centros de los 151 centros a los que se envió. Siendo la tasa de respuesta por centros de un 74,8%. El total de cuestionarios recibidos fue de 361 siendo respondidos por personal de enfermería 116 (32%) y por personal médico el resto (68%), siendo 208 pediatras. Con respecto a las actividades educativas y la valoración de la gravedad mayoritariamente son los pediatras los encargados de transmitir esa información al niño y la familia. La información sobre control ambiental y terapia farmacológica es realizada también por enfermería (60-70%). Mayoritariamente no se da información por escrito (60-70%) y no tienen actividades establecidas con ellos ni con los familiares (75-95%)
Conclusiones: No existen programas educativos de seguimiento y control para niños asmáticos en AP. Existe mucha variabilidad en cuanto a pautas de actuaciones entre centro y entre diferentes tipos de profesionales.
Las hospitalizaciones por AMBULATORY CARE SENSITIVE CONDITIONS (ACSC): Selección de los códigos diagnósticos válidos para la edad pediátrica en España
J. Colomer Revuelta, R. Peiró, C. Casanova, J. Caminal, C. Fernández y C. Ramírez
Universidad de Valencia. Facultad de Medicina., Valencia, Centro de Salud Pública de Alzira, Alzira, Hospital de Sagunto, Sagunto.
Antecedentes: Las hospitalizaciones por "ambulatory care sensitive conditions" (ACSC) han sido utilizadas en diversos estudios como un indicador del funcionamiento de la atención primaria.
Los ACSC son un grupo de diagnósticos de las hospitalizaciones potencialmente evitables, es decir de problemas de salud que podrían haber sido prevenidos o evitados mediante una intervención efectiva en atención primaria. El objetivo de este trabajo es identificar el listado de códigos de diagnósticos de ACSC útiles como instrumento de medida para la población de 0 a 14 años de nuestro entorno sanitario.
Método: Se realiza una selección inicial a partir de todos los códigos de diagnósticos de ACSC identificados en la literatura, aplicándoles los criterios que definen el indicador como apropiado, según Solberg y Weissman. Se utiliza un método de consenso para recabar información sobre el cumplimiento de dos criterios esenciales: 1-¿Los servicios sanitarios de Atención Primaria pediátrica pueden evitar la hospitalización por este problema de salud mediante prevención primaria, diagnóstico precoz y tratamiento del problema de salud y/o control y seguimiento adecuados?; 2- ¿En la mayoría de los casos, cuando el problema de salud se ha producido, es necesaria la hospitalización?. En él participan pediatras con experiencia clínica en atención primaria y en atención especializada.
Resultados: Se consideró necesario el acuerdo del 75% de los participantes para establecer consenso en cada cuestión planteada. Los códigos de diagnósticos que no obtuvieron consenso fueron excluidos, así como los que no cumplieron los otros criterios establecidos. Se obtuvo un listado definitivo de códigos de diagnósticos de problemas de salud que se proponen como válidos para analizar la capacidad de resolución de la atención primaria pediátrica en nuestro entorno sanitario.
Conclusión: Se propone un listado de códigos de diagnósticos de ACSC adaptado a nuestro entorno sanitario, válido para analizar la capacidad de resolución de la atención primaria pediátrica en España, pues limita la influencia de la variabilidad de la práctica clínica hospitalaria y de las políticas de admisión de los centros.
La Traducción directa de terminología biomédica pediátrica y sus posibles equívocos
J. Romero Sánchez, A.M. León Ruiz y M.I. Romero Sánchez
Centro de Salud Las Albarizas, Marbella.
Antecedentes: La mayor proporción de información escrita en publicaciones biomédicas en la actualidad lo está en idioma inglés y procede en su mayor parte de Estados Unidos, Japón y la Unión Europea. A menudo, muchos lectores con mínimos conocimientos de esta lengua creen estar capacitados para su lectura (e incluso la redacción) de los artículos biomédicos en idioma inglés. Es obvio que la sintaxis y el léxico son más complejos y ricos en castellano, pero es un hecho incestionable que el idioma inglés se ha situado como líder en la comunicación biomédica a escala mundial. Con frecuencia pues hay que traducirlo. Los términos parecidos en ambos idiomas pueden llevar al equívoco si no se tiene la precaución siempre de revisar su significado.
Métodos: Se revisaron cualitativamente en bases de datos biomédicas de acceso electrónico (http://gateway.ovid.com) los primeros diez abstracts disponibles de seis publicaciones pediátricas de alto factor de impacto (Pediatrics, Arch Dis Child, Am J Dis Child, J Pediatr, Eur J Pediatr, Clin Pediatr). Se detectaron las palabras de traducción posiblemente equívoca y se revisó su significado en diccionarios (Collins, http://www.britannica.com ).
Resultados: Se detectaron 20 palabras con posibles equívocos en su traducción. Se presentan a continuación: actual, America, billion, to collect, compliance, condition, constipation, crude, definite, discoloration, dramatic, emergency, eventual, evidence, infant, node, ointment, portentous, rate, siblings.
Conclusiones: Se presentan con su traducción correcta los términos referidos que a menudo pueden producir equívoco en caso de efectuar la traducción directa sin asegurarse su significado, lo cual puede tener repercusiones importantes en el manejo adecuado de la documentación pediátrica.
Síntomas de inicio del neuroblastoma y su relación con el tiempo tardado en diagnosticar. Revisión de 151 casos
M.T. Alonso Salas y A. Álvarez Silván
Hospital Universitario Virgen del Rocío, Sevilla.
El cáncer es la primera causa de muerte en la infancia por enfermedad, a pesar del enorme avance experimentado en los últimos años, consiguiéndose en la actualidad una supervivencia a los cinco años entre el 56-70%. En los países subdesarrollados esta mejoría no ha sido similar, en parte debido a la limitación de medios diagnósticos y terapéuticos, y en parte también al estado avanzado que presenta la enfermedad en el momento del diagnóstico. Los pediatras nos enfrentamos ahora a nuevos retos, de una parte si el cáncer se cura es nuestro deber diagnosticarlo precozmente, y por otro lado nos encontramos ante una patología nueva: las secuelas con la que muchos de estos niños se incorporan a la sociedad tras su curación. El tumor sólido extracraneal más frecuente en nuestro medio es el neuroblastoma, que supone en nuestra serie el 34% de la totalidad.
Hemos estudiado los síntomas ó signos de inicio que presentaron estos niños (151 casos a lo largo de 18 años), y que fueron descubiertos por sus padres, ellos mismos o el pediatra; el tiempo transcurrido desde el inicio de los síntomas que puedan ser atribuidos al tumor hasta el diagnóstico y el estadío que presentaban en ese momento. Cuando asociamos el tiempo de retraso diagnóstico y el estadio encontramos cómo evidentemente descienden los estadios iniciales cuando el retraso diagnóstico es mayor y aumentan los estadios avanzados, de manera significativa. Encontramos que los síntomas con los que debuta el neuroblastoma son extraordinariamente diversos, de hecho es el tumor infantil que en más ocasiones se presenta con un síndrome paraneoplásico, y el segundo tumor, después de la leucemia, que lo hace con síntomas neurológicos, además de síntomas comunes en la infancia que pueden pasar desapercibidos durante un tiempo y confundirse con otros procesos. El signo más frecuente sin embargo es la palpación de una masa abdominal, seguido por síntomas generales anodinos y por dolor abdominal. Es este síntoma, el dolor abdominal el que más veces puede pasar desapercibido y el que se asocia con un retraso diagnóstico mayor, seguido de los síntomas generales y las adenomegalias.
Concluimos en este estudio que el diagnóstico precoz del cáncer es esencial para aumentar las posibilidades de curación, que aún está en duda la posibilidad de un screening a la población sana que ya se hace en Japón desde los años 80, y que aún no se ha iniciado en nuestro país; en un futuro podremos diagnosticar antes el neuroblastoma congénito con la generalización de la ecografía prenatal, y en general con una mayor facilidad para la realización de una ecografía abdominal en los niños con dolor abdominal en los que queramos descartar organicidad. Proponemos fomentar los recursos diagnósticos en asistencia primaria, potenciar la sensibilización de este tema a todos los profesionales, y facilitar las interconsultas con los servicios de cirugía, radiología y oncología hospitalarios.
Conocimientos, actitudes y creencias de los padres de la Región de Murcia sobre las vacunaciones de sus hijos
J.C. Ñiguez Carbonell, P.J. Bernal González, J.A. Navarro Alonso, J.F. Amoraga Bernal, L. Fernández Sáez y T. Saura Robles
Servicio de Promoción de Salud. Dirección General de Salud. Consejería de Sanidad. Murcia.
Objetivos: Hemos pretendido analizar el nivel de conocimientos, actitudes y creencias que los padres de la Región de Murcia poseen sobre las vacunaciones de sus hijos, siguiendo las directrices de la OMS en su Asamblea Mundial de 1982.
Material y métodos: Estudio descriptivo transversal, a través de una encuesta postal autoadministrada, previamente validada. La población diana fueron una muestra representativa de padres de RN antes de recibir la documentación e información del Programa de Vacunas. Dicha encuesta consta de 25 preguntas, donde además de los datos sociométricos se recogen preguntas sobre enfermedades vacunables que conocen y gravedad de las mismas; reacciones de las vacunas; nivel de ansiedad ante la vacunación; nivel de información recibida y sobre los mitos de vacunar resfriado o en verano.
Resultados: Se enviaron 746 encuestas, obteniéndose un nivel de respuesta del 68,9%. Fue contestada en su mayoría por las madres (54,9%), con una edad media de 30,1 (29,69-30,48) años y de las que eran primíparas el 50,4%. El 67,5% tenían estudios primarios o medios y solamente el 20,4% estudios universitarios medios o superiores. El 57,6% de los padres, considera que el grado de información que poseen sobre las vacunas de sus hijos es insuficiente, habiéndola recibido fundamentalmente del pediatra (56,6%), de libros o revistas (39,3%) y de otros padres/familiares en el 34,4%; solamente el 22,6% refirieron como fuente de información otro personal sanitario. De las nueve enfermedades incluidas en el calendario de vacunaciones, recordaban una media de 4,0 (2,8-5,1), siendo las tres más referidas la rubéola (64,0%), el tétanos (61,7%) y la polio (53,3%). El 77,3% creen que las enfermedades contra las que se vacunan son graves o muy graves. Un alto porcentaje (43,6%) desconocen que para que una vacuna sea eficaz hay que administrarla varias veces. El 86,4% consideran que las vacunas ocasionan reacciones leves y sólo el 7,8% señalan que pueden producir reacciones importantes. El 57,0% refieren sentirse tranquilos y sin miedo, el 37,4% algo nerviosos y sólo el 3,1% muy nerviosos al ir a vacunar por primera vez a sus hijos. Los padres vacunarían en verano en el 85,2%, pero no los vacunarian estando resfriado en el 72,5% de los casos o con fiebre de más de 38ºC en el 96%. El 82,5% consideraron que las vacunas deberían ser obligatorias.
El hecho de tener más de un hijo fue estadisticamente significativo con tener mas grado de información (p < 0,0001); saber que las vacunas han de ser administratadas más de una vez (p < 0,01); considerar que las vacunas producen reacciones leves (p < 0,05); estar más tranquilas a la hora de vacunar (p < 0,05); conocer que se puede vacunar en verano (p < 0,001) o estando resfriado (p < 0,0001).
Conclusiones: Los problemas más importantes detectados han sido: La baja incidencia como fuente informante del personal sanitario (salvo el pediatra) y los medios de comunicación audiovisual. Como hechos que pueden llevar a retrasar o incumplir el calendario vacunal: 1) El alto porcentaje de padres que refieren tener poca información sobre las vacunas que se les van a administrar a sus hijos; 2) el desconocimiento de la administración secuencial de una mayoría de vacunas y 3) la persistencia del mito de no vacunar cuando el niño está acatarrado. Todos estos hechos se ven agravados si la madre tiene un sólo hijo.
Nefrología
Pielonefritis focal: una entidad infradiagnosticada en la infancia
P. González Santiago, A. Lassaletta Atienza, E. Vegas Muñoz, P. Sevilla Ramos, A.M. Carrasco Torres, M. Alonso Cristobo,
C. Torrijos Román y E. García Frías
Hospital Universitario Príncipe de Asturias, Alcalá de Henares.
Antecedentes: La pielonefritis focal es una forma evolutiva de la pielonefritis bacteriana aguda. El objetivo del estudio es describir el diagnóstico, tratamiento y evolución de nuestros casos para una mejor caracterización de la enfermedad.
Métodos: Estudio retrospectivo, mediante revisión de historias clínicas, desde enero de 1995 a diciembre de 2000. Los parámetros valorados fueron edad, sexo, signos y síntomas de presentación, pruebas de imagen al diagnóstico y durante la evolución (ecografia renal, TC abdominal, gammagrafía con DMSA, cistografía), resultados de analítica sanguínea y microbiología al ingreso, tratamiento antibiótico administrado y respuesta al mismo y estancia media de hospitalización.
Resultados: El número de casos fue de 4, todas niñas, de edades comprendidas entre 3,2 y 12,08 años (media 6,7). Al ingreso todos los casos presentaban fiebre, 2 asociaban dolor abdominal, solo una niña refería síndrome miccional. El sedimento urinario en todas mostraba piuria. La fórmula sanguínea revelaba leucocitosis con neutrofilia, con aumento significativo de la proteína C reactiva en todos los casos. Todos los urocultivos fueron positivos para E. coli. La función renal estaba conservada en todos los casos. La ecografía renal al ingreso evidenció en tres de los casos masa de ecogenicidad heterogénea en riñón derecho En el cuarto, la ecografía al ingreso fue normal, al evolucionar torpidamente, se realizó TC abdominal donde se objetivó en polo de riñón derecho disminución de captación de contraste e imagen hipodensa en ese área, la ecografia mostraba en ese momento lesión anecoica. Se administró tratamiento antibiótico parenteral durante una media de 11,5 días (rango 5-20 días).La fiebre cedio al 6º día y el urocultivo se negativizó al 5º día de media (rangos 2-15 y 2-9 respectivamente). La estancia media fue de 12,5 días (rango 9-20). Todas las ecografías de control se normalizaron durante la evolución, todas las cistografías fueron normales y el DMSA mostró defecto de captación en 2 de las pacientes.
Conclusiones:1) La pielonefritis focal es una entidad todavía infradiagnosticada en la infancia. Debe considerarse en pacientes con infección urinaria que presenten una masa identificada radiológicamente y/o evolucionen de forma tórpida. 2) La ecografía es el método de elección para el diagnóstico y el seguimiento. Otras pruebas de imagen como la TC, se deben considerar cuando la ecografía no es concluyente y la clínica y la evolución sugieran el diagnóstico. 3) El tratamiento se basa en la antibioterapia parenteral prolongada. El tratamiento insuficiente puede conducir a la formación de abscesos y a largo plazo a la formación de cicatrices renales En nuestros casos, la evolución fue favorable y no se requirió tratamiento quirúrgico en ninguna de las pacientes.
Fístula arterivenosa renal postraumática de resolución espontánea
C. Chaves González, A. Colino García, M.C. Llopis Garrido, J.I. Muñoz Bonet, F. Núñez Gómez, C. Benlloch Sánchez, A. López Sáiz y J. Esteban
Hospital Clínico Universitario, Valencia.
Introducción: La aparición de una fístula arteriovenosa (FAV) como complicación tras un traumatismo renal, es un hecho infrecuente. Generalmente aparecen asociadas a traumatismos renales penetrantes, siendo muy pocos los casos descritos tras traumatismos cerrados. Se presenta un caso de traumatismo renal cerrado con grave lesión del parénquima, complicado con el desarrollo de una FAV, en el que una actitud conservadora fue suficiente para resolución del mismo.
Caso clínico: Adolescente varón de 13 años, sin antecedentes de interés, que consulta por dolor abdominal tras caída casual con traumatismo directo sobre región lumbar derecha. Exploración clínica: constantes normales, hematoma sobre cresta iliaca derecha y defensa selectiva a la palpación en hipocondrio y fosa iliaca del mismo lado. Asocia hematuria macroscópica. Se realiza Ecografía abdominal: colección líquida perirrenal derecha. TAC
Abdominal (con contraste): hematoma perirrenal derecho con foco de contusión en polo inferior y laceración profunda del parénquima a ese nivel. No afectación del pedículo vascular.Hemograma y bioquímica sanguínea normales. Queda ingresado en unidad de cuidados intensivos adoptando actitud conservadora ante la estabilidad clínica y hematológica. Evolución favorable inicial persistiendo unicamente hematuria microscópica a partir del tercer día. El 7ª día de ingreso reaparece hematuria macroscópica acompañada de coágulos junto con incremento de la clínica abdominal. Se realiza Ecografía donde aparece una lesión quística (15 mm de diámetro) en polo inferior de riñón derecho, adyacente a hematoma, con movimiento en su interior. Con la exploración en modo Doppler pulsado se detectan flujos de tipo arterial y venoso en esta lesión; el flujo venoso de salida en riñón derecho esta muy elevado (43 cm/seg.) en relación con el de la vena renal izquierda (17 cm/seg.). Todos estos hallazgos son compatibles con FAV postraumática. Se solicita arteriografía renal, permaneciendo en Unidad de cuidados intensivos con estrecho control clínico y analítico hasta la realización de la misma. En controles ecográficos se aprecia a partir de las 48 horas contenido hiperecogénico en el interior de esta malformación, con trombosis completa y desaparición de los flujos el 4º día. Coincidiendo con cierre espontáneo de la fístula a nivel ecográfico, se objetiva mejoría clínica desapareciendo dolor abdominal y hematuria macroscópica el 5º día, persistiendo únicamente microhematuria al alta ( al 21º día del ingreso).
Discusión: Las FAV renales son raras, sobre todo en la infancia. Pueden ser congénitas o adquiridas, siendo estas últimas generalmente secundarias a lesiones penetrantes, sobre todo biopsias renales, y excepcionalmente a traumatismos cerrados. Clásicamente el diagnóstico se ha realizado mediante Angiografía, permitiendo el desarrollo de la Ecografía Doppler realizar el diagnóstico de forma no invasiva y el seguimiento evolutivo. La embolización transcatéter con control arteriográfico es el tratamiento de elección. Debe mantenerse un estrecho control clínico (UCIP) y ecográfico hasta la realización de la misma. Debe tenerse en cuenta, la posibilidad de una resolución espontánea en aquellas FAV de pequeño tamaño, hecho descrito en la literatura aunque de forma inusual.
Detección mediante gammagrafía renal 99m Tc-DMSA del daño renal precoz asociado a reflujo vésico-ureteral en lactantes
B. Amil, R. Pardo de la Vega, I. Málaga, F. Angel Ordóñez, F. Santos, S. Málaga, N. Zeidan, J.P. Suárez y C. Roig
Hospital Central de Asturias, Oviedo.
Antecedentes y objetivo: Las lesiones gammagráficas compatibles con nefropatía por reflujo (NR) se detectan a partir de los 6-12 meses siguientes a una infección urinaria (IU). Sin embargo, existe evidencia de daño renal asociado a reflujo vésico-ureteral (RVU) antes de que se hayan constatado episodios de IU o con IU pero sin que haya transcurrido el tiempo suficiente para generar las lesiones de NR. El propósito del estudio fue caracterizar mediante la realización de gammagrafía renal (GR) la presencia de daño renal en lactantes con RVU estéril o detectado tras el primer episodio de IU.
Métodos: Se estudiaron de forma prospectiva 13 niños (8 varones) menores de 2 años diagnosticados de RVU, distribuídos de acuerdo con el motivo de petición de la GR: Grupo I (hidronefrosis no obstructiva de diagnóstico prenatal, n = 5) y Grupo II (primer episodio de IU, n = 8). Excepto en un caso, la GR (realizada con 99mTcc-DMSA) fue realizada en los 3 primeros meses de vida. Tres especialistas informaron de forma independiente como función renal relativa disminuída cuando era inferior al 40%, moderadamente disminuída entre 39-20% y severamente disminuída por debajo del 20%, identificando cuatro patrones gammagráficos diferentes, según presentasen disminución de la captación relativa con y sin defectos focales (A y B, respectivamente), defectos focales sin disminución de la captación (C) y defectos focales bilaterales (D).
Resultados: Cuatro de los cinco pacientes del Grupo I presentaron captación renal moderada-severamente disminuida, con preferencia del patrón B (3/5) y del riñón derecho (3 vs 1). Seis de los 8 casos del Grupo II mostraron captación renal modera-severamente disminuida, con la misma preferencia por el riñón derecho (5 vs 3), presentando 3 de ellos patrón A, 3 patrón B y 2 patrón C (tabla):
GrupoF. RenalF. RenalF. RenalPatrónPatrónPatrónPatrón > 40%39-20%< 20%ABCD
I (n = 5)122131
II (n = 8)242332
Conclusión: La GR realizada en los primeros meses de vida es una exploración altamente sensible para la detección precoz de lesiones renales. En base a la ausencia de antecedentes de IU y/o un intervalo suficiente entre el episodio de la IU y la afectación renal específica de NR, los hallazgos deben ser atribuidos a un daño renal congénito.
Historia natural de la displasia renal multiquística: seguimiento durante la infancia
B. Amil, R. Pardo de la Vega, I. Málaga, F. Ángel Ordóñez, F. Santos y S. Málaga
Hospital Central de Asturias, Oviedo.
Antecedentes y objetivo: La displasia renal multiquística (DRM) continúa generando interrogantes por su relación potencial con complicaciones evolutivas. El objetivo del estudio fue conocer la evolución natural de la DRM en los casos tratados de forma conservadora, incidiendo de forma especial en la cronología de la involución renal.
Métodos: Se siguieron durante un período medio de 9 años y 7 meses (rango: 1 mes-14 años) a 35 pacientes (13 mujeres) diagnosticados de DRM entre los años 1974 y 2000. El protocolo de trabajo consistió en controles clínico-analíticos y realización de una ecografía renal cada seis meses durante los dos primeros años de vida y anualmente a partir de entonces hasta la completa desaparición del riñón displásico.
Resultados: La sospecha de malformación renal mediante ecografía prenatal fue el motivo de diagnóstico de la DRM en el 69% de los casos (24/35). Doce pacientes (34%) presentaron anomalías urológicas asociadas, siendo la ureterohidronefrosis y el reflujo vésicoureteral las malformaciones más frecuentes. Mientras que la nefrectomía precoz de la DRM en el período neonatal o la lactancia fue la decisión terapéutica en 9 casos, el resto se siguió clínica y ecográficamente de forma conservadora:
SeguimientoTamaño renalTamaño renalInvolución (años)sin cambios (%)más pequeño (%)acumulada (%)
< 111/22 (50)10/22 (45)1/22 (5)
1-24/22 (18)13/22 (59)5/22 (23)
2-44/22 (18)9/22 (41)9/22 (41)
4-63/17 (18)2/17 (12)12/17 (79)
> 63/17 (18)----14/17 (82)
Durante el seguimiento se realizaron 4 nefrectomías por persistencia del riñón displásico no involucionado. Exceptuando los pacientes con uropatías asociadas que presentaron infecciones urinarias (5 casos) o insuficiencia renal crónica (3 pacientes), no se registraron otros con hipertensión arterial o malignización atribuibles a la DRM.
Conclusiones: El seguimiento de la DRM indica una alta tasa de involución espontánea, por lo que el manejo conservador debe ser, en principio, la modalidad terapéutica de elección en estos pacientes. El riesgo de complicaciones de la DRM es bajo, en especial si no se asocia a otras malformaciones urológicas.
Técnicas de recogida de orina en el lactante
L. Sanguino López, C. Moscardó Guilleme, M. Belda Anaya, O. Manrique Moral, M.C. Vicent Castelló y J. Flores Serrano
Hospital General Universitario, Alicante.

Objetivos: Evaluar nuestro nuevo protocolo de diagnostico de i.t.u.:
- Valorar los falsos positivos de la bolsa perineal en nuestro medio.
- Confirmar la rentabilidad de la punción y el sondaje vesical .
- Identificar las complicaciones de la punción.
- Comprobar la utilidad de la ecografía previa a p. suprapúbica y sus parámetros.
Material y métodos: Estudio prospectivo en lactantes remitidos con sospecha de ITU desde sept/1999 a en/2001. Se analiza los resultados de la recogida de orina por punción, sondaje, mitad de chorro o bolsa según protocolo de diagnóstico de ITU aprobado en nuestra Unidad. Se evalua los datos de la ecografía previa a la punción vesical.
Resultados: El número total de casos fue 54 (29 V, 25M). Edad media: 5,9 m.
En 20 (37%) se confirmó ITU, 7 (13%) resultaron dudosos .
38 cultivos por bolsa: 34 (89%) positivos y de estos, 10 (29%) resultaron falsos positivos y en 4(12%) no se obtuvo orina por otro método.
En 24 niños se intentó el sondaje obteniéndose orina en 20 (83%), siendo 15 (75%)positivos.
En 40 niños se intentó punción, obteniéndose orina en 23 (57,5%), siendo 10 (43%) positivos;
A 38 de los 40 se les realizaron un total de 61 ecografías (1,6 por paciente). El tiempo de anuria y diámetro vesical medios fueron de 42 min y 3,6 cm. respectivamente. Cuando, según protocolo, se realizó punción con diámetro vesical >= 3,5 cm el índice de aciertos fue de 19/25 (76%).
En solo 2/40 (5%) niños hubo complicaciones (1 hematuria microscópica y 1 macroscópica).
Conclusiones:
- El diagnóstico de ITU por bolsa supone al menos un 29% de falsos positivos
- Sobre una población similar el método que porcentualmente ha dado menos resultados positivos ha sido la punción vesical.
- La única complicación observada en la punciones ha sido el sangrado (en el 5%) y siempre se resolvió de forma espontánea.
- Realizando una ecografía vesical previa y puncionando solo con diámetro vesical >= 3,5 cm, el rendimiento de la técnica mejora ostensiblemente.
Síndrome nefrótico y elevación de anticuerpos anti-citomegalovirus en un lactante de 5 meses
M. González Santacruz, C. Pancho Flores, P. González Delgado, M.C. Vicent Castelló, O.Manrique Moral, M.J. Martín Aguado y J. Flores Serrano
Hospital General Universitario, Alicante.
Introducción: La aparición de un síndrome nefrótico en el primer año de vida es infrecuente y, cuando ocurre, se produce por un grupo heterogéneo de afecciones con diferentes etiología, histología, evolución y pronóstico. Presentamos el caso de un lactante de 5 meses en el que se asoció el desarrollo de un síndrome nefrótico por una glomerulonefritis mesangial IgM, con una titulación elevada de anticuerpos IgG e IgM anti-citomegalovirus (anti-CMV).
Caso clínico: Varón de 5 meses, que un mes después de presentar lesiones petequiales en mano y muñeca derechas, en el contexto de un catarro de vías altas, ingresó por vómitos, anorexia y pérdida ponderal. De entre sus antecedentes familiares, resalta abuela materna con hipertensión arterial y nefropatía no filiada. En la exploración al ingreso se apreció ligero edema palpebral y de miembros inferiores (con discreta fóvea), TA: 114/67 mmHg (p95/p90) y peso de 7050 gr (p25). En las pruebas complementarias en ese momento destacaban la presencia de proteinuria de rango nefrótico (150 mg/Kg/24hrs), con microalbuminuria (713,7 mg/24hrs, menor del 80%), oliguria (diuresis: 130 ml/24hrs; Excreción Fraccional de Na: 0,04) y disminución del aclaramiento de creatinina (59 ml/min/1,73m2). Las proteínas plasmáticas totales estaban disminuídas (3,5 gr/dL) destacando la presencia de hipoalbuminemia (629 mg/dL) e hipogammaglobulinemia (89,6 mg/dL). Del mismo modo, existía hipercolesterolemia (341 mg/dL), hipertrigliceridemia (412 mg/dL), e hiperaldosteronismo (1200 mg/dL). La bioquímica plasmática demostró, además, hiponatremia (130 mEq/L), hipopotasemia (3 mEq/L, con signos electrocardiográficos compatibles), e hipocalcemia (7,6 mEq/L). Existía igualmente disminución de las concentraciones urinarias de Na (3 mEq/L), K (3 mEq/L), urea (268 mg/dL) y creatinina (10,6 mg/dL). Una ecografía abdominal demostró un incremento del tamaño de ambos riñones, sin alteraciones en su ecogenicidad. En una serología frente a microorganismos del grupo TORCH y VIH, destacaron concentraciones elevadas de anticuerpos IgG (345) e IgM (480) frente a CMV. La cuantificación de las fracciones C3 y C4 del complemento y de anticuerpos Antiestreptolisina-0 fue normal, y el análisis del frotis faríngeo fue negativo. El paciente fue remitido a la unidad de referencia para realización de biopsia renal, cuyo resultado demostró la presencia de una glomerulonefritis mesangial IgM.
Con la sospecha clínica y analítica de síndrome nefrótico se inició tratamiento con prednisona oral (60 mg/m2/día), apareciendo la crisis diurética al cuarto día del inicio del tratamiento. La normalización de los parámetros analíticos se alcanzó a las 3 semanas, por lo que el paciente fue dado de alta.
Durante el seguimiento posterior presentó diversas complicaciones infecciosas. En una nueva determinación de anticuerpos, realizada 6 semanas después de la anterior, se habían elevado aún más los niveles de IgG (3635) e IgM (553) anti-CMV.
Discusión: En el caso expuesto, la asociación entre los niveles elevados de anticuerpos frente al CMV y la glomerulonefritis mesangial IgM, parece sugerir que ésta es secundaria a una primoinfección por dicho virus. Aunque el pronóstico teórico es desfavorable (edad menor de 1 año, proliferación mesangial), ha respondido bien al tratamiento con corticoides. A pesar de su corta edad y su relación con el CMV nuestro caso no parece tratarse de un síndrome nefrótico e infección congénitos por CMV.
El aumento de la reabsorción ósea es un fenómeno universal en la hipercalciuria idiopática
M.I. Luis Yanes, V. García Nieto, A. Ruiz Rabaza, M. Monge, C. Vázquez, A. Callejón Callejón y J.S. León González
Hospital Nuestra Señora de la Candelaria, Santa Cruz de Tenerife.
Introducción: En estudios previos se ha comunicado que alrededor del 40% de los pacientes con hipercalciuria idiopática (HI) tienen reducción de la densidad mineral ósea ( DMO). En HI se ha descrito la existencia de un aumento de la producción de IL-1, PGE, o calcitriol que puede producir un incremento de la reabsorción ósea. No obstante, en algunas series de pacientes adultos con HI se ha descrito una reducción de la actividad osteoblástica en las biopsias óseas.
Material y métodos: Se estudiaron 74 pacientes con HI (25V, 49M) (9,45 ± 2,87 años) y 105 controles (C) (44V, 61 M) (9,09 ± 2,91 años). Se determinaron los niveles de PTHi, calcitriol, osteocalcina, fosfatasa ácida resistente al tartato (FART) y los cocientes urinarios con respecto a la creatinina de deoxypyridioline (DPyr/Cr) y del telopeptide C-terminal del colágeno tipo I (Crosslaps) (TLC/ Cr). Se midió la DMO mediante un densitómetro Hologic QDR-4500SL (DEXA). Se definió la osteopenia cuando Z-DMO fue inferior a -1 DS.
Resultados: El valor de Z-DMO del grupo con HI fue inferior al de C (-0,56 ± 0,75 vs 0,03 ± 0,93, p < 0,001 ). No hubo diferencias entre grupos en los niveles de PTHi, calcitriol o FART . Los niños con HI tuvieron valores significativamente más elevados de TCCI/Cr con respecto a C (909,1 ± 375 mcg/mmol) tanto en aquellos con osteopenia (1824,8 ± 7229; p < 0,001 ) como en los que tenían DMO normal (2184,7 ± 1317; p < 0,001). Similares resultados se observaron con el cociente DPry/Cr. Los niveles de osteocalcina en niños con HI y DMO normal fueron significativamente mas elevados que los de C (338,4 ± 21,3 vs 24,8 ± 11,9 ng/ml; p = 0,01) sin observarse diferencias en aquellos con osteopenia.
Conclusiones: La reducción de la DMO observada en los niños con HI parece estar relacionada con un incremento de la resorción ósea. Los niños con DMO normal tienen una respuesta osteoblástica superior a la de aquellos con osteopenia.
Sensibilidad gustativa a la sal y Tensión Arterial en la edad pediátrica: ¿una relación esperada?
S. Málaga, J. Argüelles, C. Perillán, J.J. Díaz, C. Rey, F. Díaz, A. Diéguez y M. Vijande
Universidad de Oviedo, Oviedo.

Un reciente metaanálisis realizado en población infanto-juvenil ha confirmado una significativa relación entre tensión arterial (TA) e ingesta de sodio, que pudiera estar condicionada por la sensibilidad gustativa a la sal.
Objetivo: Estudiar la sensibilidad gustativa a la sal en una población pediátrica y su relación con la distribución de la TA.
Sujetos y métodos: 44 jóvenes sanos, de ellos 25 varones, con una edad media de 15 2 ± 0,4 años (rango 13,1 ± 20,5). Para su inclusión en el estudio se obtuvo autorización personal y/o paterna, previa explicación detallada del protocolo a los sujetos experimentales y sus familias. Fueron sometidos a un test de sensibilidad (Técnica "staircase", adaptada de Cornsweet), con vistas a reconocer la presencia de sal cuando se les ofrecía simultáneamente agua destilada-desionizada y una solución acuosa de baja concentración sódica. La TA estimada fue la media de dos determinaciones realizadas con el sujeto sentado y en reposo durante 15 minutos, mediante un esfigmomanómetro de mercurio Erkameter, por personal certificado para este cometido. Procedimiento estadístico: descriptivo convencional, análisis de regresión y correlación. Valores expresados como media ± ESM.
Resultados: Los valores medios de TA sistólica (TAS) fueron 110,7 ± 2,0 mmHg en varones y 111,2 ± 2,7 mmHg en mujeres, que fueron procesados en conjunto al no detectarse diferencias entre sexos. El umbral medio de sensibilidad al NaCl en la totalidad de la muestra estudiada fue 5,1 ± 0,7 mM. Como se observa en la figura, la TAS y la sensibilidad al NaCl presentan una asociación lineal que es estadísticamente significativa (rx,y = -0,40; p < 0,01), mostrando los sujetos con mayor sensibilidad gustativa, los valores de TAS más elevados.
Conclusión: La sensibilidad gustativa a la sal muestra una correlación positiva con la TAS en población pediátrica, por lo que no puede excluirse que exista una relación funcional entre ambas variables.
Financiada por el FIS. Proyecto n°.00/0401
Perfil ingestivo de sal en población infanto-juvenil: aplicación de tests de sensibilidad y consistencia
S. Málaga, J. Argüelles, C. Perillán, J.J. Díaz, C. Rey, F. Díaz, A. Diéguez y M. Vijande
Universidad de Oviedo, Oviedo.

Antecedentes y objetivo: La relación entre el consumo de sal y la hipertensión arterial (HTA) ha sido suficientemente documentada. Los estudios en población pediátrica requieren instrumentos adecuados a sus características etáreas. El papel de la sensibilidad gustativa a la sal sobre la HTA no ha recibido suficiente atención por los investigadores. El objetivo del presente trabajo ha sido diseñar y poner en práctica test objetivos que permitan delimitar el perfil ingestivo del niño y adolescente, basándonos en su capacidad para detectar diferentes cantidades de sal (Test de sensibilidad - TS), así como para diferenciar entre soluciones salinas de distinta concentración (Test de consistencia o capacidad de discriminación - TC).
Métodos: Mediante experimentos piloto se ha podido determinar el rango de solu-ciones salinas a utilizar en el TS (10 soluciones de 0,2 a 13,7 mM, en incrementos de 1,5mM) y el TC (6 soluciones de 0 a 50 mM, en incrementos de 10 mM). El TS se desarrolló sobre el esquema clásico "staircase", ofreciendo simultáneamente dos vasos (agua destilada-desionizada y solución salina), comenzando por la concentración más alta. Cada concentración se probaba dos veces y un solo fallo se consideró resultado negativo, obligando a volver a la concentración inmediatamente superior. Resultados negativos en tres concentraciones detenían el test, considerándose como umbral la media geométrica de los tres valores. El TC consistía en la ordenación, respecto a su sabor salado, de 6 vasos conteniendo otras tantas soluciones salinas variando entre 0 y 50 mM. La ordenación perfecta arroja un valor "0" en la prueba. Cada fallo en la ordenación se puntuó con el valor absoluto de la diferencia de colocación respecto al valor correcto. Los test se han aplicado a una población pediátrica de 44 individuos sanos (25 varones) de 15,2 ± 0,4 años (rango 13,1 a 20,5). Al no detectarse diferencias entre sexos, los datos se procesaron en conjunto. Procedimiento estadístico: descriptivo convencional y test de correlación y regresión. La inclusión en el estudio exigió la autorización personal y/o paterna previa explicación detallada del protocolo.
Resultados: El umbral de sensibilidad hallado en el TS fue de 5,1 ± 0,7 mM NaCl, y la valoración media en el TC resultó ser de 1,77 ± 0,36 puntos. Hemos hallado una correlación positiva estadísticamente significativa entre la sensibilidad y la consistencia rxy = 0,37 (p < 0,05), de forma que los individuos con una mayor sensibilidad son los que mostraron una mayor capacidad de discriminar entre dos concentraciones de sal próximas (Figura).
Conclusión: La aplicación conjunta de los test de sensibilidad y consistencia constituye una herramienta eficaz y coherente para la determinación del perfil ingestivo de sal de la población infantojuvenil, pudiendo ser de utilidad para los estudios de relación entre tensión arterial e ingesta salina.
Financiado con la ayuda FIS.: 00/401
Tema libre
Embolización no selectiva de arterias bronquiales como tratamiento de una hemoptisis masiva en un niño con fibrosis quística. A propósito de un caso
T. Pérez Arvelo, F. Díaz Romero, H. Valles, J.R. Torino Casasnovas y F. Toledo Trujillo
Hospital Universitario de Canarias, Santa Cruz de Tenerife.
Objetivo: La hemoptisis es frecuente en pacientes afectos de fibrosis quística y enfermedad pulmonar, pero la hemorragia masiva es una rara complicación,representando una emergencia médica.
El tratamiento clásico para la hemoptisis masiva ha sido: la angiografía, la embolización selectiva y en ocasiones la cirugía. La embolización no selectiva de las arterias bronquiales es una de las opciones terapéuticas para la hemoptisis en aquellos pacientes con sangrados masivos debidos a la proliferación de arterias bronquiales y vasos colaterales sistémicos.
Material y métodos: Describimos el caso de un niño de 9 años de edad con un episodio de hemoptisis masiva.
Nuestro paciente presentaba una severa y avanzada enfermedad pulmonar, y fue hospitalizado al presentar un sangrado pulmonar masivo. No fue posible realizar broncoscopia, y por lo tanto saber el origen del sangrado. Se decidió entonces, ante la extrema gravedad, la práctica de una angiografía, descubriéndose un gran número de arterias bronquiales anormales y vasos colaterales sistémicos. En la misma sesión se intentó embolizar para controlar la hemoptisis. Se realizó embolización no selectiva de las arterias bronquiales y el sangrado cedió. Fueron utilizados microcatéteres y microalambres (coils).
Resultados: Fue posible parar el sangrado al primer intento y salvar la vida al niño.
Conclusión: Pensamos que la embolización no selectiva de las arterias bronquiales puede ser una buena alternativa para el control inicial de una hemoptisis masiva en la fibrosis quística.
Pioderma gangrenoso en un niño de 4 meses de edad
R. Ruiz-Cano, L. Pérez-García, C. Gutiérrez-Junquera, M.C. Escudero Cantó, M. Lillo-Lillo y E. Escario-Travesedo
Complejo Hospitalario de Albacete-Hospital General, Albacete.
Introducción: El pioderma gangrenoso (PG) es una dermatosis neutrofílica que se caracteriza por la aparición de lesiones pustulosas con cráter central y base eritematosa e histológicamente por un infiltrado estéril de neutrófilos. La edad media de afectación es de 40 años. Se ve raramente en la infancia y se asocia con frecuencia a enfermedades tales como leucemias e inmunodeficiencias. El tratamiento de elección en los niños son los corticoides sistémicos, siendo la evolución de los casos publicados, tórpida a pesar del tratamiento, presentando lesiones nuevas y necesitando varios meses para su curación. Presentamos el caso de un niño de 4 meses cuya lesión inicial se desarrolló en la zona de inyección de la 2ª dosis de vacuna y cuya evolución fue favorable sin tratamiento corticoideo.
Caso clínico: Varón de 6 meses de edad sin antecedentes personales ni familiares de interés, que tras la vacunación a los 4 meses de edad de la pentavalente bacteriana y polio presenta lesiones vesiculo-eritematosas, primeramente en el sitio de la inyección para extenderse en pocos días, a varias localizaciones de la piel y cuero cabelludo. Las más importantes están en la cara lateral de ambos muslos, de 10 - 12 cm con sobreelevación de los bordes y necrosis central. Resto de exploración física normal. Exploraciones complementarias: Hemograma: 30.970 leucocitos (60,5% neutrófilos, 27,5% linfocitos, 7,3% monocitos, 2,3% eosinófilos), hemoglobina 11,7; plaquetas 777.000, en la extensión al MO no se observan formas anormales. Bioquímica sanguínea y sistemático de orina normales. PCR: 59. Inmunoglobulinas A, G, M, E, C3 y C4: normales. Parásitos en heces: negativos. Rx tórax: normal. Biopsia de piel: cultivo aerobio y anaerobio negativos, Histología: sugestiva de pioderma gangrenoso. Estudio inmunológico completo: normal. Tratamiento: en su ingreso se administró cloxacilina IV durante 7 días y oral 4 días más y antisépticos tópicos (sulfato de cobre y de zinc) durante 3 semanas. Previamente a su ingreso, había recibido tratamiento con cefaclor sin mejoría. Evolución: la fiebre desapareció al quinto día del ingreso y las lesiones fueron regresando progresivamente hasta su cicatrización.
Discusión: El PG es una enfermedad muy infrecuente en la infancia, siendo éste el segundo caso más joven recogido en la literatura. Otros autores también han relacionado, el inicio de las lesiones, con la administración de la vacuna bacteriana inyectable. Presentó patergia, observada con más frecuencia en los niños que en los adultos (20%) con PG. Otras lesiones bacterianas, que producen lesiones parecidas, fueron descartadas mediante cultivo lesional e histología. En nuestro caso no encontramos ninguna enfermedad ni alteración de base de las asociadas con esta enfermedad. Es de destacar la mejoría llamativamente rápida, para lo esperable, aun sin tratamiento específico.
Secuestro pulmonar extralobar en mediastino superior. Una localización infrecuente
M.L. Cid Vila, R. Quintás Martínez, M. Carames Varela, N. Martínez Ortega y R. Marini Rodríguez
Hospital Comarcal Valdeorras, O Barco de Valdeorras.
Presentamos un caso de secuestro pulmonar en una localización muy poco frecuente que puede confundirse en las exploraciones radiológicas habituales (Rx. tórax. TAC pulmonar, con procesos de origen tumoral.
Se trata de un paciente de 5 años que acude a la consulta por adenopatías de localización cervical. La exploración física es normal, salvo adenopatías cervicales de pequeño tamaño (la mayor de 1,5 cm por 0,8 cm región submandibular izda.). Entre las exploraciones complementarias se efectúa hemograma y bioquímica habituales que son normales; en Rx de tórax se evidencia ensanchamiento mediastínico superior con probables adenopatías a ese nivel que comprimen traquea. Con este resultado se decide efectuar TAC torácico con contraste: en cortes de 5 y 10 mm se aprecia masa en mediastino anterosuperior de consistencia blanda. En ecografía existe la misma masa con movilidad con los grandes vasos y una pequeña formación quística de 3mm. El diagnóstico probable es de Timoma.
Se envía al Hospital Vall d'Hebron para diagnóstico definitivo y tratamiento. Las exploraciones practicadas son coincidentes con las nuestras. Los marcadores Alfafetoproteína y B-HCG son negativos así como las serologías a VIH, rubeola y toxoplasma.
El 22-1-99 se interviene quirúrgicamente realizándose timectomía y exerisis de masa penducular con vasos aberrante de subclavia izda. aorta y pulmonar adherido a hilio pulmonar y localizacion extrapericárdica. En la invención se secciona nervio frénico izdo que ocasiona una parálisis diafragmática izda. sin repercusión clínica.
Diagnóstico definitivo y anatomía patológica: Secuestro pulmonar extralobar con quistes broncogénicos displásicos, mucocele y vasculopatía secundaria a hiperaflujo pulmonar y sistémico.
En el momento actual el paciente está asintomático con un desarrollo pondoestatural normal y sin problemas respiratorios ni cardíacos. Se hace así mismo una revisión de la bibliografía, sobre secuestros extrapulmonares.
El hábito del consumo televisivo en adolescentes en una zona básica de salud
J. Fernández Villar y M.M. Díez López
Centro de Salud Vidriales, Zamora.
Antecedentes: Es importante conocer el hábito televisivo en los adolescentes por la posible repercusión físico-psíquica.
Objetivos: Conocer los hábitos de consumo de televisión (TV) en nuestros adolescentes.
Material y métodos: Estudio descriptivo transversal. Se estudió la población adolescente (9-14 años) de los colegios de nuestra zona básica (n = 210). Valoramos mediante cuestionario autoadministrado la programación preferida, horas de exposición, consumo previo a la jornada escolar, distancia de visionado, hora límite nocturna y el lugar de la TV como actividad de tiempo libre.
Resultados: Varones 56,2%, mujeres 43,8%. La programación preferida en el 26,4% incluía series o videos de acción-violenta o programas para adultos, el 35,3% prefería programas o series infantiles juveniles, sólo un 2% incluía documentales y un 2,8% películas. El 46,4% refería una exposición > a 2 horas. El 41,6% ve TV antes de ir a clase. El 14,4% ve TV hasta las 21 horas, el 64,5% hasta las 23,30 horas y sobrepasa esta hora el 21,1%. La distancia al TV era ¾ 2 metros en el 39,2%. La TV ocupa el tercer lugar entre las preferencias de tiempo libre.
Conclusiones: Alta presencia de contenidos de acción-violencia y programación para adultos. Un alto porcentaje ve TV un tiempo excesivo, en una franja horaria más propia de adultos y a corta distancia, lo que podría tener consecuencias en la salud de nuestros adolescentes. La TV aparece entre sus principales preferencias de tiempo libre.
Dermatitis de contacto por mercurio. Síndrome de Baboon
M. Vázquez Olivares, R. de Fátima Lafuente Urrez y J. Piñol Montserrat
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Antecedentes y objetivo: El síndrome de Baboon se define como una dermatitis alérgica de contacto sistémica con un inicio agudo en personas sensibilizadas caracterizada por un exantema que adopta una distribución peculiar afectando nalgas, región anogenital y flexuras. El estudio histopatológico es inespecífico, observándose leve infiltrado linfohistiocitario perivascular en dermis superficial, acompañado de cambios espongióticos focales en la epidermis.
Caso clínico: Se presenta el caso de una niña de 5 años de edad, sin antecedentes familiares de interés y con antecedentes personales de asma bronquial, sinusitis maxilar, adenoidectomía, otitis serosa y celulitis periorbitaria del ojo derecho. Acude al Servicio de Urgencias de nuestro hospital por presentar erupción cutánea, ligeramente pruriginosa, de 12 horas de evolución en muslo y pierna derecha consistente en una placa eritematosa, caliente, infiltrada, de límites difusos en cuya perifería se aprecian lesiones papulosas, eritematosas, aisladas de aproximadamente 2-3 mm de diámetro. Asimismo se aprecian lesiones papuloeritematosas confluentes a nivel de la espalda. No dolor a la palpación. No adenopatías inguinales. Buen estado general. Afebril. Bioquímica normal. La paciente es diagnosticada de celulitis y se instaura tratamiento con antibióticos y antiinflamatorios vía oral y corticoides y antibióticos tópicos. A la exploración física se aprecia una placa ritemato-violácea en muslo derecho y hiperpigmentación residual a nivel de los espacios interdigitales de los dedos de las manos y de los pies. En la anamnesis realizada cabe destacar que 2-3 días antes de la aparición de la erupción cutánea la paciente ha tocado mercurio tras romperse un termómetro en su domicilio. Asimismo refiere intolerancia a la mercromina.
Resultados: Las pruebas epicutáneas resultaron positivas al mercurio. Se llega al diagnóstico de S. de Baboon por sensibilización al mercurio. El exantema desaparece después de 12 días con tratamiento corticoideo tópico.
Conclusiones: Es importante la realización de una correcta anamnesis para el diagnóstico precoz de esta patología. El mercurio puede producir severos cuadros de reacciones alérgicas por contacto o inhalación en pacientes previamente sensibilizados por mercuriales tópicos. Se ha especulado que la sensibilización al mercurio puede ser de dos tipos: una reacción exclusiva al mercurio ionizado, y otra, al mercurio ionizado y no ionizado. Este fenómeno puede ser debido a diferencias en la biodisponibilidad o en la penetración percutánea. Para la evaluación de la hipersensibilidad al mercurio, es más seguro emplear mercurio ionizado y no ionizado, antes que sólo el mercurio clorado o el mercurio amoniacal. Una reacción alérgica tipo IV frente a alergenos administrados sistémicamente probablemente produzca reacciones de este tipo, por lo que el Síndrome de Baboon puede ser inducido por níquel, ampicilina y heparina.
¿Debemos y podemos mejorar nuestra actitud médica y humana frente al niño enfermo crónico grave? Autocrítica constructiva de una madre y pediatra
S. Bello Miró
Consorcio Sanitario de la Selva, Blanes.
Antecedentes: Conocemos la dificultad que entraña la atención del niño enfermo crónico grave, aumentada si su enfermedad es poco conocida y el pronóstico es poco esperanzador, y a todo esto hay que sumar la variabilidad de la reacción de los padres que conviven con esta problemática y la diversidad de las cualidades personales de los facultativos. Por otro lado con el avance de la Pediatría el numero de pacientes de este tipo va en aumento progresivo, por tanto más de nosotros entraremos en contacto con algunos de ellos y es muy importante que estemos preparados para su atención tanto en sus aspectos médicos como en los humanos. El objetivo de esta comunicación es aunar el punto de vista como madre y el ojo crítico como pediatra para analizar los problemas detectados y con ello invitarnos a una reflexión. También proponemos algunas posibles soluciones o sugerencias.
Material y métodos: La base de esta comunicación es el análisis de la experiencia real, a lo largo de 9 años, de una pediatra y madre de un niño afecto de una enfermedad crónica grave, evolutiva, de mal pronóstico, poco conocida, de diagnóstico difícil, con múltiples descompensaciones y que precisó atención en varios centros sanitarios de distintas provincias españolas, por diversos médicos y pediatras de todas las categorías profesionales y de casi todas las especialidades. También hemos contado con la colaboración de otros compañeros que han vivido situaciones similares o que trabajan en contacto con estos niños y sus familias. Referente a la metodología, se ha intentado mantener la máxima objetividad eliminando cualquier sentimiento de animadversión generado por la situación. Evidentemente no se hace referencia a ninguna entidad ni a ningún profesional en particular.
Resultados: Se han detectado múltiples problemáticas en diversos terrenos, diagnóstico, diálogo con los padres, toma de decisiones relevantes, coordinación entre especialistas, animadversiones personales entre facultativos, información al pediatra de cabecera, delegación de responsabilidades, atención en los períodos vacacionales, atención en los servicios de urgencias, falta de personal y sobrecarga del mismo, falta de humanidad y actitud ante y tras el fallecimiento del niño. Se plantean soluciones o sugerencias abarcando la actitud del profesional y su relación con los padres y el niño, la coordinación entre facultativos, la supervisión del paciente y su tratamiento, la atención urgente (presentamos la Tarjeta de Atención al Niño), el pediatra en formación y la necesidad de la epicrítica y cita con la familia tras el fallecimiento del niño.
Conclusiones: Esta comunicación puede aportar mejoras en la atención del niño enfermo crónico grave tanto en el aspecto de la praxis médica como en el humano. No creemos estar en posesión de la verdad absoluta pero sí de poder aportar una visión diferente desde "el otro lado de la mesa". Dada nuestra calidad de pediatras conocemos las dificultades de la falta de personal, la problemática laboral, sobrecarga de trabajo, falta de tiempo, remuneración económica, limitaciones humanas y otras tantas, pero debemos y podemos contribuir a mejorar algunas de las situaciones descritas. Es necesario que nos replanteemos nuestra actitud del pediatra frente al niño enfermo crónico grave.
Mastocitosis multinodular globulosa: a propósito de un caso
A.E. Jerez Calero, F. Contreras Chova, R. Ruiz Villaverde y R. Bayes García
Hospital Universitario San Cecilio, Granada.
Introducción: La mastocitosis multinodular globulosa es una mastocitosis inicialmente cutánea que se caracteriza por la presencia de lesiones nodulares globulosas abundantes en número y que en algunos casos pueden llegar a presentar importantes fenómenos congestivos. Algunos autores la incluyen como una variante de la mastocitosis xantelasmoidea descrita por Tilbury Fox en 875.
Caso Clínico: Antecedes familiares: Padre afecto de Dermatitis seborreica y Madre con Xerosis cutánea.
Antecedentes obstétricos: Embarazo controlado, sin incidencias. Parto: Cesárea por SFA. RNAT de sexo femenino. Peso al nacimiento 3.200 gr. Lactancia materna exclusiva 1 mes.
A la edad de 1 mes acude a Consulta de Dermatología por la presencia de lesiones eritemato-descamativas en cara, línea de implantacion del pelo y área del pañal, diagnosticandose de D. Seborreica. Se prescribe cambio de leche materna a fórmula láctea hipoalergénica. Debido a la progresión y cambio de aspecto de las lesiones dermatológicas a pesar de tratamiento vuelve a consultar. En esta ocasión presenta a los 2 meses de edad nódulos globulosos abundantes de tamaño guisante o algo mayores, de color orsado o marillento, salientes, firmes o hemisféricos que suelen acompañarse en ocasiones de fenómenos congestivos. Signo de Darier +.
Cursa con anemia propia del lactante. Sideremia baja. Ganancia pondero-estatural normal. Desde los 2 meses de edad hasta la edad actual (3 años y 3 meses) ha sido ingresada en varias ocasiones por fenómenos congestivos asociados a su clínica cutánea: La anamnesis por órganos y aparatos es normal, así como la exploración de los diferentes sistemas (cardiorespiratorio y digestivo). No adenopatías ni visceromegalias. En los exámenes complementarios se observa un predominio linfomonocitario en ausencia de leucocitosis. No existe eosinofilia. La bioquímica sanguínea es normal, salvo sideremis 41 microgr/ml y discreta elevación de la VSG 11/33 mm/h. Las radiografías del huesos largos a los 14 meses son normales. Ha estado a tratamiento periódica con Ciproheptadina 1 cc/8 h y Cromoglicato disódico 200 mg/6 h entre brotes. Con 2 años y 9 meses se ha realizado una biopsia de médula ósea por punción esternal y se ha objetivado Elementos linfoides moderadamente elevados en número, aunque bien diferenciados. "-3/mil de estos elementos nucleados son mastocitos. Todas las series son normales.
En las ultimas radiografías de huesos largos, realizadas a los 3 años de edad aparecen signos sugestivos de osteopenia que recomiendan la práctica de densitometria ósea.
Comentario: Los fenómenos congestivos de la Mastocitosis Multinodular suelen ser la principal causa de ingreso y están producidos por la liberación paroxística de histamina. Entre ellos se incluyen Signo de Darier; Dermografismo en piel no afecta, Manifestaciones ampollosas y Flush.
En todos los casos es pertinente solicitar pruebas complementarias: Hemograma, Bioquímica, Estudio histológico, Aspirado de MO y Rx de huesos largos. La evolución depende del tipo clínico, de la edad, afectación visceral y gravedad de los síntomas ocasionados por los mediadores de los mastocitos. Las formas generalizadas, sobre todo las de comienzo precoz pueden desaparecer sin dejar señal en la pubertad.
Cuidados del lactante y prevención del Síndrome de Muerte Súbita del lactante en un área rural, 1999-2000
M.A. Zafra Anta, C. Camara, A. Nuñez, M.A. Amor, C. Blanco y A. Pelayo
Hospital Pozoblanco, Pozoblanco, Centro de Salud ASNC, Pozoblanco.
Objetivo: El objetivo general es conocer algunos aspectos sobre los cuidados del lactante en la actualidad en un Área Rural, el Área Sanitaria Norte de Córdoba (ASNC). En concreto se estudian algunas variables sociobiológicas del entorno del lactante, y se pretende determinar la prevalencia de la postura para dormir "boca-arriba" o "de lado" en los primeros meses de vida, la frecuencia de lactancia materna, así como el evitar el consumo de tabaco en el entorno, el compartir la cama con el bebé y el exceso de abrigo. Estos aspectos epidemiológicos han demostrado, en diversos estudios, colaborar muy positivamente en la prevención del síndrome de la muerte súbita del lactante (SMSL).
Este estudio se efectuó antes del inicio de la campaña "Ponle a dormir Boca Arriba" de la AEP.
Población y métodos: Se diseñó un estudio epidemiológico observacional, descriptivo. La población incluye todos los lactantes de 3-5 meses, nacidos entre el 15-8-1999 y 15-2-2000, residentes en el ASNC. La recogida de datos se realizó mediante un cuestionario descriptivo y basado en hechos, diseñado por los propios autores, durante la revisión del Niño Sano efectuada en la consulta de Pediatría de los Centros de Salud o, en caso contrario, en su domicilio o en el hospital. Se recogieron datos de clasificación, sociobiológicos y variables de cuidados del lactante, descritas de interés en la bibliografía médica.
El estudio estadístico descriptivo de las variables se efectuó mediante el programa SX 4.0.
Resultados: En 293 cuestionarios recogidos (sobre un total de 354 recién nacidos en dicho período de 6 meses):
- Prevalencia de decúbito prono para dormir fue 46,8%. De ellos, 31,696 recordaban dicha postura en maternidad, un 19,8% la aducían por experiencia con hijos previos y el 16,9% señalaban no haber recibido información. Recibían lactancia artificial exclusiva un 24,9% al primer mes, 35,8% el 2° y 46,8% el 3° mes.
- Afirmaban fumar el 26,3 % de las madres y el 49,5% de los padres. Un 19,5% de las madres fumaban durante el embarazo.
- Compartían ocasional o frecuentemente la cama con el bebé, el 7,5% y el 1,5% respectivamente. Riesgo de exceso de abrigo ("estrés térmico"), en un 28,3% por uso de calefacción nocturna y 68,3% por "sobrecobertura".
Conclusiones: Alta prevalencia de la postura prono para dormir y de lactancia artificial al 2° y Ser mes de vida. Necesidad de adoptar un mensaje explícito y reiterado sobre los factores de prevención del SMSL por parte de todo el personal sanitario implicado en la educación sobre las prácticas de cuidados del lactante en nuestro Área y nuestro entorno.
Hipoglucemia hipocetosica neonatal en un caso de deficiencia periférica 6-Peruvoil-Tetrahidrobiopterina Sintasa (PTPS)
A. Belanger-Quintana, M. Martínez-Pardo, M.A. Mena, M.J. García, M. Castro y M. Ugarte
Hospital Ramón y Cajal, Madrid, Universidad Autónoma de Madrid. Facultad de Ciencias (Dpto. Biología Molecular), Madrid.
La tetrahidrobiopterina (BH4) es el cofactor de las hidroxilasas de aminoácidos aromáticos, enzimas que controlan la degradación de fenilalanina (Fen) y la síntesis de los neurotransmisores dopamina y serotonina. El defecto más común en la biosíntesis de BH4, es la deficiencia en 6-piruvoil tetrahidrobiopterina sintasa (PTS, OMIM 261640). Existen dos formas de la enfermedad, la forma clásica o típica, que se caracteriza por tener niveles muy deficientes de biopterina y neurotransmisores en LCR y la forma periférica o atípica con niveles normales de biopterina y neurotransmisores en LCR.
Presentamos un caso de deficiencia en PTPS, forma periférica o atípica, que tuvo desde el nacimiento varias crisis de hipoglucemia hipocetósica. Primer hijo de padres no consanguíneos, nacido a término por cesárea, con peso en p10, talla en p50 y perímetro craneal en p10 (microcefalia respecto a talla). Ingresa a los 26 días de vida, remitido por el Centro de Detección Precoz de Talavera, por presentar niveles de Fen en sangre de 600 µM/l (N < 120 µM/l). En los antecedentes personales destacan crisis de hipoglucemia (22-33 mg/dl) desde las 13 horas hasta los 10 días de vida, en que fue dado de alta. La analítica al ingreso, con 3 horas de ayuno, mostraba una glucemia de 39 mg/dl con ácidos grasos libres (FFA) y cuerpos cetónicos disminuidos y una relación FFA-3/OH-Butirato de 10 y niveles de insulina de 13 µU/ml (insulinemia/glucemia = 0,33; N < 0,34) con un cortisol de 14mg/ml.
En el diagnóstico diferencial de hiperfenilalaninemias, presentaba niveles de Fen en sangre de 900 µM/l, fuerte excreción de neopterina (10,8 mM/Mcreat; N = 1,1-4) con niveles bajos de biopterina (0,48 mM/Mcreat; N = 0,6-3) y un porcentaje de biopterina del 4,5% (N = 44-77), compatible con una deficiencia de PTPS. Los niveles de biopterina en LCR eran normales y los neurotransmisores en el límite inferior de la normalidad. La sobrecarga de BH4 (20 mg/Kg) normalizó los niveles de Fen plasmáticos, así como los de neurotransmisores en LCR.

