

Introducción
Como señalaba Holland, hace mas de 35 años1, la vida nació en el seno de los mares primitivos, y la individualidad se consiguió internalizando el océano. Los líquidos orgánicos, el "medio interno", de Claud Bernard, reproducía el medio ambiente. Cuando la vida dejó el agua de los mares y se internó en la tierra los seres vivos para evitar la desecación, tuvieron que defenderse mediante gruesas envoltura quitinosas (ricas en calcio), como la de los quelonios o los reptiles. El perfeccionamiento de los mecanismos "homeostáticos", mediante sistemas de canales iónicos y sistemas de bombeo que, con consumo de energía, permitían mantener concentraciones distintas a un lado y otro de la membrana, posibilitó independizarse de las variaciones que a lo largo de los siglos se producían en el medio ambiente, como ocurrió con el calcio. Finalmente, para levantarse sobre la corteza terrestre, permitiendo correr o volar sobre ella, como los mamíferos o las aves, se necesitó un endoesqueleto que mantuviera la forma constante, y al mismo tiempo, sirviera de anclaje para los tendones de los músculos. En todo lo expuesto el calcio ha jugado un papel un papel fundamental.
a) En primer lugar, en lo referente al "medio interno", los iónes cálcicos son esenciales para una amplia variedad de funciones biológicas que incluyen procesos extracelulares vitales, como la formación del hueso, ser cofactor de los factores II, VII, IX y X de la coagulación, e intervenir en la adhesión intercelular. Igualmente, procesos fundamentales intracelulares, como son la regulación del crecimiento y división celular, el acoplamiento hormonarespuesta, acoplamiento del estímulo eléctrico-respuesta en la contracción muscular, y en la liberación de neurotrasmisores. Por ello, los organismos terrestres de vida libre deben mantener la concentración del pool extracelular de calcio "casi-constante", con mínimas variaciones, en la especie humana alrededor de la media, los 10 mg/dL (2,5 mM/L). En el hombre un 40% del Ca plasmático está unido a proteínas, fundamentalmente a la albúmina (el 80-90% del ligado), y el 60% restante es ultrafiltrable, (por ello la disminución de 1 g/dL en la concentración de albúmina disminuye 1 mg/dL, el calcio sérico total). De este 60% del calcio ultrafiltrable, un 46% está ionizado, difusible, (4,8 mg/dL) y el 14% restante forma complejos con el fosfato o el citrato. El calcio ionizado (Ca2+) es el elemento biológicamente activo y está en equilibrio con el Ca unido a proteínas, unión que varía con la concentración de hidrogeniones: la variación de 0,1 unidad en el pH supone un cambio del 10% en el Ca ionizado. La acidosis la eleva y la alcalosis lo disminuye.
De forma sintética, el Ca se absorbe en el intestino y se elimina por el riñón Lo cual supone que ambas magnitudes son equivalentes. Para mantener esta homeostasis existen diferentes y sutiles mecanismos cibernéticos, el conocimiento de alguno de los cuales es de reciente adquisición.
b) Por otra parte, el endoesqueleto de los mamíferos o las aves, está, en su casi totalidad, formado por sales fosfato-cálcicas, (cristales de hidroxiapatita y fosfato octacálcico, junto a complejos amorfos, como el fosfato cálcico, carbonato cálcico, etc). En contra de lo que cabría suponer el hueso es un órgano dinámico, capaz de un rápido turn-over, y con un constante modelado y remodelado. El hueso representa un gran reservorio de Ca, (y de P y Mg), en contínuo equilibrio con el pool extra-celular de Ca, actuando de voluminoso tampón. Los procesos que afectan a este órgano y a la mineralización, constituyen las "enfermedades metabólicas óseas". De los 1.000 g de Ca que aproximadamente tiene un adulto el 99% está en el hueso y solo el 1% (unos 10 g), en los fluidos orgánicos. Esta proporción es la misma en todas las edades, pero, obviamente, son distintos el contenidos total de Ca y la mineralización del hueso, (en el lactante unos 400 mEq /kg peso corporal frente a los 950 mEq/Kg del adulto).
Regulación del Ca corporal
Vamos a pasar breve revista a estos mecanismos, deteniéndonos en los aspectos mas actuales, aceptando que no es este el lugar para comentar las múltiples facetas de la patología, formas clínicas, diagnóstico o tratamiento (hipo-hiperparatiroidismos, raquitismos, osteoporosis, etc.,) que, por otra parte, se encuentran excelentemente recogidos en las últimas ediciones de los tratados de endocrinología, que acaban de aparecer en España2,3, y otras de inmediata aparición4. Nos dedicaremos a comentar en este apartado algunos aspectos puntuales, cuya adquisición es mas reciente, y que, en algún caso, han marcado un hito en nuestros conocimientos, sobre la fisiología y fisiopatología del metabolismo de este metal.
Los principales reguladores de la homeostasis cálcica son la parathormona (PTH) y la vitamina D. El PTHrP ("PTH-related-peptide") y la calcitonina parecen ser primariamente importantes en la vida fetal. Veremos sucesivamente:
- La absorción del Ca, la importancia de la vitamina D y sus metabolitos polares, y, como tópico reciente, motivo todavía de controversias, los polimorfismos del receptor de la vitamina D.
- El sistema cibernético que mantiene "casi-constante" el nivel sérico de Ca2+l, regulado por el CARS ("Calcium-ion-sensing cell-surface receptor"), y su patología. A su estímulo responde la PTH actuando, a través de su receptor, sobre los riñones y otros tejidos.
- Finalmente, el "parathyroid hormone-related peptide" (PTHrP) y la calcitonina, que, como hemos adelantado, es de menor trascendencia en el control de la calcemia, en la vida postnatal.
Vitamina D
La cantidad de Ca en el organismo está regulada, fundamentalmente, por la absorción intestinal. La mayor parte (90%), se realiza a través del intestino delgado (íleon, yeyuno y duodeno, respectivamente 60, 20 y 10%), el resto por el cólon (8%) y el estómago (2%). La ingesta diaria recomendada es de 360 mg, en el primer semestre, 540 mg, el segundo, 800 mg durante la infancia, y de 1.200 mg durante la pubertad5.
El calcio de la dieta se absorbe gracias a un mecanismo de transporte activo, mediado por un transportador (calbindinas fundamentalmente), jugando en todo ello un papel central la vitamina D, la cual comprende un grupo de esteroles con actividad fisiológica similar. No se trata de un nutriente estricto, dado su origen mixto, (endógeno y exógeno), y su metabolización a componentes activos. Por su origen y mecanismo de acción, se comportan como una hormona:
- El 7-Dehidrocolesterol presente en el citoplasma de células de dermis y epidermis, y que, mediante la radiación ultravioleta (290-320 nm), se convierte en vitamina D3 (colecalciferol).
- La alimentación es también fuente de vitamina D3 de origen animal y de vitamina D2 (ergosterol) de origen vegetal, que acaba también, en vitamina D3. No existe control limitante a la absorción intestinal de estas vitaminas. Para la absorción intestinal son necesarias las sales biliares.
La vitamina D3 sérica se transporta, ligada a una proteína, DBP ("vitamina D-binding-protein"), llegando al hígado, donde sufre una primera hidroxilación: 25-hidroxi-colecalciferol, o calcidiol (25-OHD). En ello intervienen dos hidroxilasas, una microsomial y otra mitocondrial, ambas dependientes del citocromo p450 monooxigenasa. La segunda no es específica de la vitamina D y tiene menos afinidad que la microsomial, pero, a diferencia de esta, no es retroinhibida por el producto resultante (lo cual representa un cierto mecanismo de control en la primera vía), por lo que ante grandes dosis de vitamina D (intoxicación), continúa hidroxilándose.
La forma hormonal de la vitamina D3 es la 1-25-dihidroxi-colecalciferol o calcitriol (1,25(OH)2D), a la que se llega tras sufrir otra hidroxilación en el riñón. Es el metabolito mas potente, hallándose en la circulación a nivel de pg./mL La 1-a-hidroxilasa renal, es el paso limitante de toda la vía metabólica, se activa por los niveles séricos disminuidos de Ca2+ y fosfato, o elevados de la PTH, (en respuesta a la hipocalcemia). La enzima se encuentra en el interior de la membrana mitocondrial de las células del túbulo proximal. El papel de otros metabolitos como el 24,25 D, no está claro, pero ciertamente es el camino para la degradación metabólica en forma de 1,24,25,D (ác. calcitrioico), hidrosoluble, y eliminable por orina y heces. Todos los metabolitos de la vitamina D se trasportan ligados a la DBP.
El metabolito más abundante de vitmina D, y más útil para informarnos acerca del estado de replección de vitamina D, es el 25OHD (< de 12 ng/mL, puede diagnosticar un "raquitismo sutil", el mas común en nuestro ambiente, V.N 20-80 ng/mL), siendo también útil en el diagnóstico de la hipervitaminosis. Por su parte las concentraciones plasmáticas de 1,25(OH)2D, dan una información mas precisa de la replección o necesidades del Ca y P en relación con la ingesta. Su elevación (> de 70 pg/mL), se presenta en casos de raquitismo carencial inveterado6.
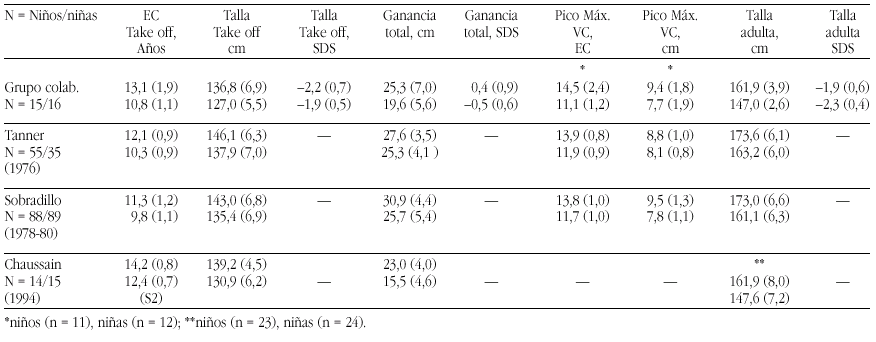
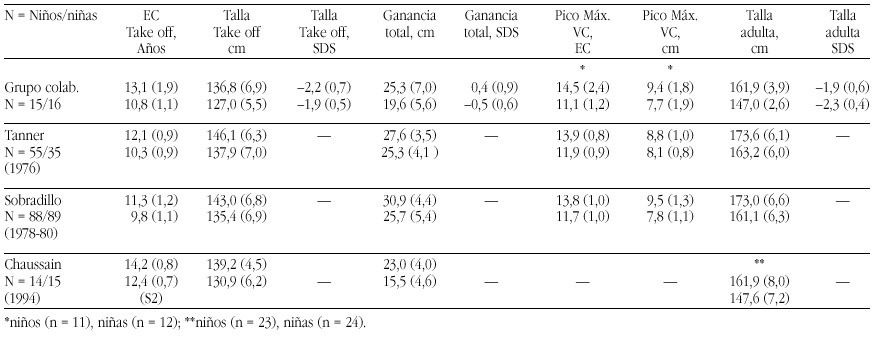
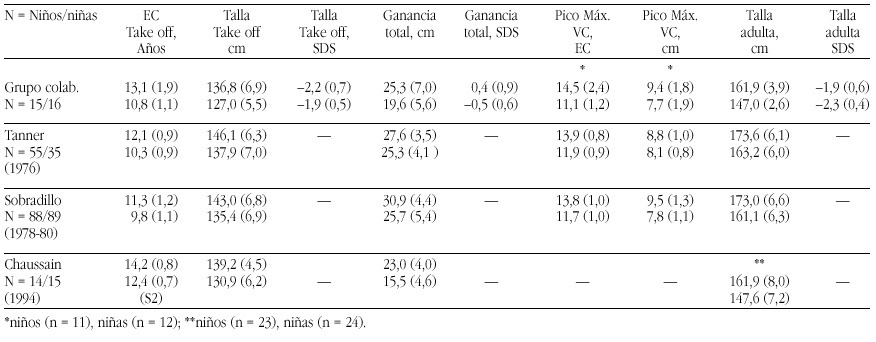
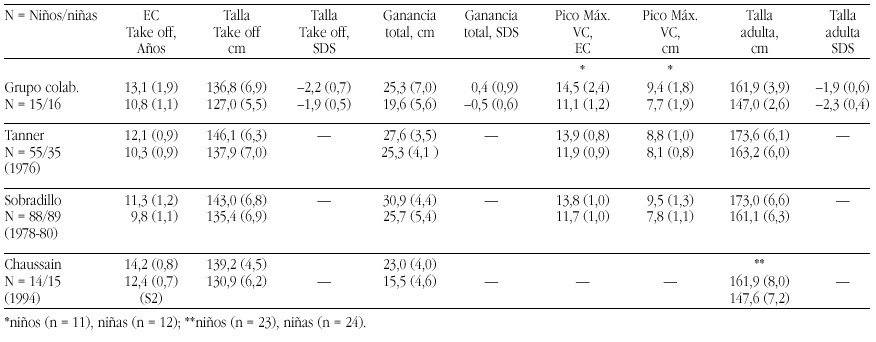
Las tasas de los valores plasmáticos normales de los metabolitos de la Vitamina D figuran en la tabla 17.

Funciones endocrinas clásicas de la vitamina D
El papel fundamental del metabolito activo de la vitamina D es mantener el ambiente calcio-fosfórico adecuado en el líquido extracelular para promover la calcificación del cartílago epifisario de los condrocitos neoformados, para ello facilita la absorción intestinal de Ca y, hasta cierto punto de P, regulando, asimismo, el nivel de fosfatasa alcalina necesario para esta aposición. Sus órganos diana son por ello el intestino y el hueso.
Receptores de la vitamina D. El 1,25(OH)2 D, se comporta como una hormona esteroidea. Tras su síntesis renal se trasporta unido a la DBP, hasta las células diana (fundamentalmente hueso e intestino), donde se une a un receptor específico (VDR) de alta afinidad, miembro de los receptores nucleres, con cierta homología con los receptores de estrógenos y progestrerona. El complejo VDR-1,25D, forma un heterodímero, con el receptor para el retinoide X (XRX), que se une al DNA específico, gen que, mediante la acción de una serie de coactivadores, regulará la síntesis de ADN, la traducción de mARN específicos (activando la polimerasa II) y, en última instancia la síntesis de una serie de proteínas, de entre las cuales las mas importantes son la calbindinas (varias, de diferentes p.m.)que permiten la absorción del Ca a través del enterocito. En otras células diana como las paratiroidea o la renales, disminuye respectivamente la síntesis de PTH y de 1-a-hidroxilasa renal (y con ello se autoregula la síntesis de 1,25D) (figura 1)8.

Existen otros mecanismos rápidos estimulantes de la absorción de calcio, presumiblemente por mecanismos no genómicos, quizá a través de un modificación directa de los fosfolípidos de las membranas de los enterocitos, con flujo de iones de Ca, vía trans e intracelular9.
El efecto predominante sobre el hueso es aumentar la resorción ósea, probablemente independientemente de los efectos directos sobre el osteoclasto. El efecto neto de estas acciones es, como hemos dicho, elevar el Ca sérico y hasta cierto punto el P.
Acciones "no clásicas" de la vitamina D8
Dado que los receptores para la vitamina D se han identificado en múltiples tejidos, se le presupone otras muchas acciones, algunas ya mencionadas, como regular la síntesis de PTH, y de 1-a-hidroxilasa renal, junto a otras como intervenir directamente sobre los osteoblastos (facilitando la síntesis de proteínas como la osteocalcina y osteopontina), estimular la diferenciación de los osteoblastos, y facilitar, probablemente, la reabsoción de Ca y P a nivel renal etc. Al mismo tiempo de inhibir la 1-a-hidroxilación, estimula la 24-hidroxilación, con lo cual se inhibe su propia síntesis, y se facilita su metabolización inactivante.
Pero su acción mas característica es, repetimos, la mineralización del hueso en crecimiento, una alteración de esta función conduce al raquitismo, resultante de una defectuosa mineralización de la matriz del cartílago epifisario. El osteoide no calcificado da lugar a la característica metáfisis raquítica. Remitimos al lector interesado a alguna de las revisiones citadas, que sobre el tema se han realizado recientemente2-4.
Solamente sintetizaremos que a estos procesos se puede llegar por interferencia de cualquiera de los mecanismos que hemos mencionado (figura 28 y tabla 2).

Polimorfismos en el receptor de la vitamina D
VDRGP, ("VDR gen polymorphism"). El gen del receptor de la vitamina D (VDR), está situado en el cromosoma 12 (q13-14), posee 11 exones10, los tres primeros (IA, IB, IC) codifican la región no traducida 5'UTR, los 8 restantes van ordenados del II al IX. Se han descrito diversos polimorfismos en la longitud de los fragmentos de restricción (RFLP), en los exones II y IX y en el intrón VIII.
Para el estudio del exón II se ha utilizado el enzima de restricción Fok I. Al estar situados en este exón los codones que inician la traducción del código, se originan dos variantes polimórficas del receptor, una corta de 424 aa (F o M4), y otra larga de 427 aa (f o M1). En el intrón VIII, se encuentran los sitios para dos enzimas de restricción la Bsm I (genotipos BB, Bb y bb), y Apa I (genotipos AA, Aa, y aa). Recientemente se ha descrito un nuevo polimorfismo en este mismo intrón, el Tru9 I, un alelo que ha podido pasar desapercibido, al estar situado en el primer punto de unión de los "primers" utilizados originalmente para genotipar los fragmentos de restricción Bsm I. Está situado a + 443 pb del final del exón VIII (12). Su importancia está por definir. Finalmente, en el exón IX actúa la enzima Taq I, detectandose en el codón 352 un cambio silente (genotipos TT,Tt y tt). Un resumen de ello se recoge en la tabla 3.

La importancia y el significado de estos polimorfismos en el VDR, es motivo de investigación en el momento actual. En primer lugar, existen bastantes trabajos en los que se demuestra que la relación entre los VDRGP, y la densidad mineral ósea es, por lo menos, débil13-15. Sin embargo, y a pesar de estas opiniones, el meta-análisis de 75 artículos aparecidos antes de 1997, demuestra una asociación con alto nivel de confianza entre VDRGP, y la densidad mineral ósea, efecto que no se interfiere por la heterogenicidad genética ni factores no-genéticos, cuando se analizan los factores concurrentes en los resultados "positivos"16. Estos factores concurrentes obligan a estudiar grupos, según consideremos el sexo, la iniciación o no de la menopausia, la inclusión o no de individuos osteoporóticos, el lugar donde se realiza la densitometría, y, sobre todo, porque no todos los polimorfismos descritos tienen el mismo significado. Por ejemplo, los distintos genotipos de los fragmentos Bsm I del intrón VIII, no parecen guardar correlación con la densitometría ósea en distintos estudios13-15. Sin embargo los polimorfismos Fok I, del exón II, que, como hemos señalado, determina receptores con distinto nº de aminoácidos, si que parecen modificar la afinidad del receptor por el 1,25 (OH)2 D17, y su reflejo en la osificación.
Se ha demostrado la interacción con otros factores que regulan el metabolismo del Ca. Por ejemplo, existe una interacción entre la acción de la vitamina D y la respuesta del metabolismo óseo al ejercicio, que varía con los polimorfismos Fok I, incrementándose la "up-regulation" en los portadores del alelo f y no los del F18. Otro ejemplo, en relación con la predisposición genética a la osteoporosis: la ausencia del sitio de restricción para el Fok I (F o M4), en la que faltan los 3 primeros aminoácidos del VDR, hace que el receptor interactúe mas eficientemente con el factor de transcripción IIB, o, en otras palabras, el genotipo fVDR si que está asociado a menor actividad y, con ello menor densidad ósea19. Esta interacción entre distintos factores, puede explicar los resultados aparentemente contradictorios, que estamos comentando. Un ejemplo final muy claro, e interesante, es la interacción gen con gen. Hemos mencionado varias veces que las distintas isoformas de los fragmentos de restricción VDR Bsm I, no están asociadas a diferencias en la densidad mineral ósea; pero ello, si se evalúan aisladamente. Se han demostrado influencias entre los distintos genotipos de los receptores de vitamina D (VDR Bsm I) y genotipos del receptor de estrógenos (ER): las mujeres con la combinación (-/-) PvII ER y bb VDR presentan una media muy alta de densidad mineral ósea, mientras la combinación (-/-)PvII ER con BBVDR, tienen niveles significativamente mas bajos20. Estos datos sugieren que variaciones genéticas del locus ER en relación con el VDR, si que tienen influencia en la obtención y mantenimiento de la masa ósea en mujeres jóvenes20.
El papel de los polimorfismos en la masa ósea se pone mas de manifiesto cuando existen otros factores desfavorables que pueden interferir la aposición cálcica, por ejemplo el genotipo BB en pacientes hemodializados parece contribuir a la mayor o menor severidad del hiperparatiroidismo secundario, por un mecanismo relacionado con la fosfatemia21. En Nigeria el déficit de vitamina D es raro, sin embargo hay raquitismo por déficit alimentario de calcio. Es frecuente demostrar una asociación familiar. Estudiando los distintos genotipos determinados por la presencia o ausencia de puntos de actuación de los enzimas de restricción conocidos, Bsm I, Apa I, Taq I y Fok I, se demuestra que la presencia del alelo F (y ninguno de los demas, ni sus combinaciones), está asociado a la mayor presencia de raquitismo22.
Los receptores para la vitamina D, como hemos señalado, se han descrito en múltiples tejidos, por lo cual sus funciones deben suponerse múltiples y no todas conocidas, de ahí que se haya sugerido su participación en procesos cuya contribución patogénica no es fácil de explicar. Citaremos, como ejemplo, la diabetes. En un reciente estudio23, se han analizado 157 diabéticos tipo 1, frente a 248 controles, determinando anticuerpos ICA y tasas de ác. glutámico decarboxilasa (GAD), correlacionádolo con los polimorfismos de tres enzimas de restricción para el VDR (Bsm I, Apa I y Taq I). Las diferentes frecuencias de todos los alelos de los polimorfismos están asociados a diabetes, excepto los de Taq I, pero solo en los polimorfismos Bsm I, fue significativa esta asociación. Hacen falta futuros estudios funcionales para establecer su papel en la patogenia de la enfermedad.
Polimorfismos de vitamina D y talla corta
Para terminar este apartado, comentaremos uno de los aspectos mas interesantes y de gran interés pediátrico, cual es la posible relación entre los polimorfismos del VDR y el crecimiento, mas concretamente la talla corta. Es sabido que la vitamina D deficiencia o vitamina D resistencia, está asociado a talla extremadamente corta, lo cual se corrige con el tratamiento de vitamina D, excepto cuando hay severa resistencia. También hemos mencionado que uno de los papeles de la 1,25 D3 en el cartílago de crecimiento es regular la diferenciación y proliferación de los condrocitos. No es de extrañar por ello que diferentes investigaciones se hayan polarizado en el estudio de los polimorfismos del VDR, en relación con este problema.
Los polimorfismos de exón 2 del gen VDR, estudiado con Fok I predicen en el niño en crecimiento la mayor o menor absorción de Ca, de tal forma que en un estudio en este sentido los homocigotos FF tenían un 45.5% mas de absorción que los ff y un 17% mas que los heterocigotos fF24. El genotipo tt del polimorfismo Taq I, se asocia con mayor peso al año de vida (25). Es sabido que el exón 2 del VDR, posee los codones de inicio de la traducción, habiendo ya comentado que existen dos polimorfismos para iniciar esta traducción. Los individuos con el polimorfismo de inicio ATG codifican una proteína VDR de 427-aa, mientras que el codón de inicio ACG codifica un VDR de 424-aa. La secuencia CGATG crea un sitio de restricción para la endonucleasa FokI (f), lo cual permite detectar polimorfismos en la longitud de los fragmentos de restricción. Este polimorfismo del exón 2 es uno de los determinantes mas importantes de la talla adulta, al menos en la mujer japonesa, donde se realizó este estudio26. La importancia del diferente tamaño de estos receptores no está clara, pero parece que, a pesar de que la afinidad de ambos por la 1,25 D3 es similar, su eficiencia no es la misma, siendo la variante mas larga hasta un 50% menos eficaz en la trans-activación de ciertos genes mediada por la vitamina D27.
Para terminar, en un estudio transversal y longitudinal Lorentzon M et al (2000)28, demuestran la asociación de determinados polimorfismos con el peso al nacimiento, el crecimiento hasta la adolescencia y la talla final Los individuos con genotipo BB (del Bsm I en el intrón 8), son mas pequeños al nacimiento, crecen menos hasta la pubertad y al final tienen menos talla que los genotipos bB y bb. Si incluimos la talla media parental, el peso y talla al nacimiento y los alelos del VDR, se podía predecir hasta un 39% de la talla final y donde las variantes alélicas del VDR, aisladas, contribuyen hasta el 8% de todas la variaciones. Podemos concluir señalando que quizá en el futuro estos estudios puedan permitir el sacar del "cajón de sastre" de las tallas cortas idiopáticas algunos casos para los que se pudiera encontrar una explicación.
El CARS ("Calcium-ion-sensing cell-surface receptor") y la PTH. Mecanismos reguladores
de la homeostasis del Ca2+l.
El CARS
Si el mecanismo fundamental que regula la absorción del Ca, está ligado a la vitamina D, el mecanismo que regula la concentración sérica de Ca2+ se sabe desde antiguo que está encomendado básicamente a la PTH, regulando su eliminación renal y el intercambio del calcio sérico con el gran "pool" cálcico representado por el hueso. Que la PTH era sensible, por lo tanto, a la concentración sérica de Ca2+, era evidente, pero hasta que en 1993 Brown EM et al. no clonaron y caracterizaron el receptor sensible al calcio, en paratiroides bovina, se desconocía su mecanismo de actuación29. El receptor clonado resultó también sensible a otros agonistas catiónicos, como el magnesio y el gadolinio, y otros nó iónicos, como la neomicina. La identificación del gen del receptor sensible al calcio (CARS), permitió localizar otros tejidos que expresaban esta proteína, aparte de las células paratiroideas: las células parafoliculares C-tiroideas, que secretan calcitonina, las células de la porción ascendente del asa de Henle, y otras células que no tienen nada que ver, al parecer, con la regulación cálcica, como células en el cerebro.
Mecanismo de acción. Se trata de un receptor de la numerosa familia de los receptores con siete dominios transmembrana, acoplados a una proteína G. El gen está situado en el brazo largo del cromosoma 3 (3q1.3), y codifica una proteína de 1.078 aa., la cual posee un gran dominio N-terminal extracelular de 600 aa, al que se une el Ca ionizado, con los 7 dominios transmembrana y un dominio intracelular carboxi-terminal. Es el primer receptor de este tipo cuyo ligando es un ion y no una molécula completa. La activación del receptor activa la proteína Gq lo cual provoca un aumento transitorio del segundo mensajero, el inositol-3-fosfato, liberando el Ca almacenado en el compartimento intracelular. El aumento de Ca citoplásmico frena la secreción de PTH en las células paratiroideas y bloquea la reabsoción renal de Ca.
En última instancia, y recopilando lo que llevamos dicho, del Ca2+ extracelular depende todo el manejo del Ca2+ por el hueso, riñones e intestino, como se aprecia en el esquema tomado de Brown EM et al. (1995)30 (figura 3)(ver también mas adelante "La PTH y su receptor)".Antes de describirse el CARS, toda la fisiología de estas interacciones, se suponían mediadas, directa o indirectamente por la PTH. Efectivamente, la PTH ocupa una posición central del proceso, pero la presencia de receptores en el riñón, y las enseñanzas que nos han proporcionado las enfermedades originadas por mutaciones del gen del CARS, sugieren que gran parte de los mecanismos reabsortivos renales del calcio se deben a un efecto directo del Ca2+, paratiroide-independiente, hecho que, si conocido desde hace algún tiempo31, es ahora cuando tiene su explicación. Aclarando también otros fenómenos, como la diabetes insípida vasopresin-resistente que acompaña a la hipercalcemia. Específicamente, la hipocalcemia es detectada por los CARS a nivel renal, lo cual incrementa la reabsorción del Ca, tendente a corregir el desequilibrio. Además, a nivel de la nefrona distal existen, junto a los receptores para el Ca2+, re ceptores para la PTH, lo cual permite un efecto sinérgico de ambos receptores, y de sus mecanismos de acción, en orden a un mejor control renal de este ion.


Por otra parte, este receptor, el CARS, es de una relativa baja afinidad, lo cual le permite seguir manteniendo sensibilidad, ante concentraciones de Ca2+ superiores al rango fisiológico.
Enfermedades producidas por mutación del CARS. Se han descrito mutaciones activantes e inactivantes que, como señalábamos mas arriba, nos han permitido un mejor conocimiento del funcionamiento de este receptor. Sus principales características las recogemos en la tabla 4.

Mutaciones con pérdida de función. La hipercalcemia familiar benigna(HFB), fue descrita en 1972 por Foley et al32; forma de hipercalcemia bien tolerada, con herencia autosómica dominante, y muy poco frecuente. Cursa con hipocalciuria e hipermagnesemia. La hipocalciuria persiste a pesar de la paratiroidectomía, lo que sugiere la autonomía renal del proceso, la PTH es normal. El análisis genético demuestra que el rasgo de la enfermedad está ligado al 3q13, pero no en todos los casos, lo cual supone la existencia de otros locus33. El hiperparatiroidismo neonatal severo (HPNS), es un trastorno a menudo fatal, los pacientes muestran signos bioquímicos y radiológicos de hiperparatiroidismo importante. Las dos enfermedades se dan en la misma familia, lo que condujo a Marx et al34, a pensar que la primera fuera la forma heterocigota, y la segunda la homocigótica de un rasgo común. Dadas las características de estos procesos, y una vez conocido el CARS, la investigación se dirigió a este receptor. Los estudios de Pollak et al, demostraron todas estas suposiciones, al describir las mutaciones de este gen en ambos procesos35, la forma leve heterocigota (HFB) y homocigota en la neonatal grave. Sin embargo, las investigaciones de Pearce SH et al, contradicen estos hechos36, al encontrar pacientes con hiperparatiroidismo neonatal severo, con mutaciones del CARS, tanto homo, como heterocigotas.
Mutaciones con ganancia de función. Son varias la enfermedades consecuencia de este tipo de mutaciones en receptortes acoplados a proteinas G (pubertad precoz gonadotropin-independiente en varones, el bocio nodular etc). Una mutación de este tipo conduce a la hipocalcemia con hipercalciuria autosómica dominante (HHAD). Pearce et al37 demostraron un cierto número de mutaciones de este receptor con un incremento de sensibilidad al Ca2+ y otros ligandos agonistas.
Por último el descubrimiento de este receptor ha permitido la búsqueda de agentes calciomiméticos con fines terapéuticos, cuya importancia en el futuro para el tratamiento de los hiperparatiroidismos (síndromes MEN, niños dializados etc.) está por definir. Recientemente Coburn et al38 ha recogido los ensayos clínicos con un ligando agonista artificial (NPS-R-658), que demuestra su actividad, a dosis en rango nanomolares, con resultados prometedores.
La PTH y su receptor
Como acabamos de ver (figura 2) la parathormona es el elemento central que el organismo posee para mantener la concentración extracelular de Ca2+, dentro de los estrictos límites de normalidad. Sus órganos diana son el riñón y el hueso e, indirectamente (a través de la vitamina D), el intestino. La hipocalcemina determina un estímulo del CARS, y la secreción inmediata de PTH, por parte de las células de las glándulas paratiroideas. La PTH tiene una acción inmediata sobre el riñón estimulando la reabsorción de calcio en la porción gruesa ascendente del asa de Henle y el túbulo distal. Al propio tiempo se estimula la 1a-hidroxilasa renal convirtiendo el 25 (OH)D en 1,25 (OH)2D, activando con ello la absorción intestinal de Ca. Un mecanismo mas lento es la movilización del calcio óseo, aumentando el nº y la actividad de los osteoclastos, células en las que no se han demostrado receptores de la PTH, lo que hace suponer que su mecanismo de acción no es directo sino mediada por substancia liberadas por los osteoblastos, cuando la PTH actúa sobre ellos.
El receptor de la PTH es, como el CARS, otro prototípico receptor ligado a la membrana celular, dependiente de una proteína G y que, tras la unión con su ligando activa el AMP-c como segundo mensajero, pertenece por ello, también, al amplio grupo de receptores con siete dominios hidrófobos transmembrana y bucles de unión intra y extra celulares. Ha sido clonado y su gen asignado al cromosoma 3p22-p21.1. El PTHrP es también ligando de este receptor, para alguna o todas sus funciones. Los receptores de PTH de hueso y riñón no son idénticos, con 591 y 585 aa. respectivamente, tienen 78% de identidad.
El sistema de transducción de la señal es el de la adelinato-ciclasa (receptor hormonal-proteínas G de acoplamiento-adenilatociclasa). El mecanismo de activación de la proteína G heterotrimérica inactiva (aGDPbg), se inicia con la activación del receptor por el ligando que interacciona con la subunidad a de la proteína G, induciendo cambios conformocionales y liberando el nucleótido GDP, lo que permite la unión del GTP. La unión del GTP al trímero produce la disociación del receptor y la liberación del nuevo heterodímero bga GtP. Con la hidrólisis del GTP unido a la subunidad a, debido a la actividad GTP-ásica intrínseca de la subunidad a, acaba la activación del efector. Las subunidades a activadas, pueden ser activadoras (as), o inhibidoras (ai), de la adenilato ciclasa. La activación de la adenilato ciclasa, lleva consigo un incremento del AMP-c, y éste segunado mensajero regula la permeabilidad de los canales del Ca y, finalmente el incremento del Ca2+ citosólico. Otro mecanismo es la vía del fosfo-inositol (IP3), activado por la proteína Gq que estimula la liberación del Ca de las organelas al citosol.
El término "pseudohipoparatiroidismo" recoge diversos procesos en los que los tejidos muestran un grado variable de resistencia a la PTH. Aparte del clásico test de Ellsworth-Howard (respuesta a la PTH en términos de fosfaturia o cambios en la relación AMPc / creatinina), lo definitorio es la presencia de un hipoparatiroidismo clínico con PTH sérica alta. Son diferentes las alteraciones genéticas del sistema adenilato-ciclasa del receptor de la PTH:
- Tipo IA. Defecto genético, por una mutación39, de la subunidad a de la proteína estimuladora de la unión del nucleótido guanina (Gsa), cuyo gen se sitúa en el cromosoma 20q13.2. Se trata de un defecto celular generalizado, por lo que además de la resistencia a la PTH, hay resistencia de otros receptores (TSH, gonadotropinas, glucagón), acoplados a la proteína G. Presenta un fenotipo conocido, característico: talla corta, braquidactilia del 4º metacarpiano, facies redonda. etc. (osteodistrofia hereditaria de Albright). Se ha descrito asociada a pubertad precoz independiente de las gonadotropinas: a la temperatura corporal (37ºc), la proteína G mutada se degrada, pero a la temperatura de los testículos (33ºc), la mutación de G produce una activación del receptor de la LH y pubertad precoz).
- Tipo IB. Fenotipo normal, resistencia a la PTH, pero no a otras hormonas y con proteína G normal. La causa no está clara (¿anomalía del receptor?, ¿de la adenilciclasa?, ¿PTH bioinactiva?), posiblemente sea heterogénea.
- Tipo II. Se trata de un cuadro muy raro, que cursa con hipocalcemia, pero que difiere del tipo I en que el AMP-c urinario está elevado, tras el estímulo con PTH la fosfaturia no aumenta. Fenotipo normal. El defecto parece ser del AMP-c distal, pues generalmente está activado pero la célula no responde a la señal.
La calcitonina y el "parathyroid hormone-related peptide" (PTHrP)
La calcitonina es una hormona peptídica de 32 aa. sintetizada en las células C parafoliculares del tiroides (derivadas de la última bolsa branquial, la 5ª), desarrolladas al comienzo de la gestación. Pero también por otros tejidos (SNC, hipófisis, timo, pulmón etc.). Su gen se ha situado en el cromosoma 11p, muy cercano al de la PTH. Codifica tres péptidos: la CT, la catacalcina y otro péptido de 37 aa, el CGRP ("péptido relacionado genéticamente con la CT"). La expresión de cada mRNA, se debe al procesamiento alternativo del gen y depende de cada tejido: en las células parafoliculares predomina la CT y en el SNC el CGRP.
Su receptor, como el CARS o el de la PTH, pertenece a la superfamilia de receptores con 7 dominios transmembrana, ligado a proteína G y sistema adenilato-ciclasa, presenta 482 aa. De forma aguda, la calcitonina, disminuye el Ca2+ ionizado extracelular, mediante una inhibición de la resorción ósea, debido a una disminución del número y la actividad de los osteoclastos. Su papel en la especie humana, y, en general en los mamíferos, parece poco relevante, desde el momento en que la calcemia se mantiene en rangos de normalidad, tanto en su ausencia (atireosis), como cuando se segrega en cantidades excesivas (carcinoma medular de tiroides). Su papel en la vida prenatal y neonatal no está claro, habiéndose sugerido que la hipercalcitoninemia puede ser responsable de hipercalcemina neonatal.
El "parathyroid hormone-related peptide" (PTHrP). Buscando el factor responable de la hipercalcemia, con que cursan algunos tumores, (particularmente de mama, y de epitelio escamoso)40, se aisló una proteína que estimulaba los receptores de PTH, produciendo AMP-c, pero en estos tejidos no se expresaba el gen de la PTH. Se trataba de una subtancia similar, pero que no era PTH. Efectivamente su gen se ha clonado y se sitúa en el cromosoma 12p en situación análoga al de la PTH en el cromosoma 11. Los 13 aa. del extremo aminoterminal de la proteína son idénticos a la PTH, siendo los dos primeros claves para la activación de la adenilatociclasa. La porción 14-34, aunque distinta a la de la PTH, es fundamental para su unión al receptor.
Activa los receptores de PTH en las células renales y óseas, con acciones similares sobre el AMP-c urinario y sobre la 1a-hidroxilasa renal, sintetizando 1,25 (OH)2D. Se produce en casi todas las células del organismo, y en todos los tejidos embrionarios, pareciendo ser necesaria para un normal desarrollo fetal. Las mutaciones inactivadoras del receptor PTH/PTHrP conduce a un trastorno óseo letal, la condrodisplasia de Blomstrad. El PTHrP parece ser fundamental para la transferencia materno fetal del calcio, requiriéndose unos 30 g, para la maduración normal del feto y su correcta osificación. Los altos niveles encontrados también en la leche, hacen suponer alguna acción en el lactante41.
Finalmente, el calcio, el crecimiento y la evolución
El crecimiento en longitud de los huesos largos, es el determinante último de la talla adulta, y crecer, en última instancia, es aponer sales cálcicas a la zona de osificación encondral del cartílago epifisario de crecimiento. La gran cantidad de calcio que se deposita en el esqueleto, -por término medio 1 kg. en el individuo adulto-, nos obliga a pensar si en alguna circunstancia la disponibilidad de este elemento pudiera ser un factor limitante del crecimiento. Tanto a nivel individual, (el hecho de que la juventud bebiera mas leche, a lo que parece se nos incita desde la pantalla en algunas películas americanas, ¿puede tener influencia en la talla adulta?), como colectivo, (a lo largo de la evolución, - y no hablamos de siglos, sino de eras,- la talla de la especie humana ha sufrido variaciones, ¿ha podido tener alguna influencia en ello, aparte de otros nutrientes, la biodisponibilidad del calcio?), ha sido motivo de estudio.
Aunque el raquitismo está ligado al déficit de vitamina D, el ingreso inadecuado de Ca en la dieta puede ser también un factor desencadenante: Niveles habitualmente bajos de ingesta de Ca del orden de 150 a 250 mg/día, está demostrado que produce alteraciones de la masa ósea en niños, y cuadros de raquitismo, tanto sutil bioquímico-, como clínico. A nivel de bibliografía es sobradamente conocida la situación en Nigeria, donde a la falta de consumo lácteo, se unen la dietas ricas en maíz, con alto contenido en fitatos42), pero que también se ha descrito en países desarrollados (dietas macrobióticas, etc.)43. Ya hemos mencionado, que hay factores genéticos predisponentes, lo que explica el acúmulo familiar de casos en estos países. Los recientes estudios de polimorfismos en el VDR, son parte de la explicación genética del problema22,24.
Pero, dejando a un lado estos casos de claro déficit de Ca en la dieta, que induce patología evidente, se han acumulado en las dos últimas décadas mucho estudios y experiencia sobre contenido mineral óseo (CMO) en la infancia. El reciente desarrollo de la absorciometría con doble haz de RX (DXA), ha hecho que se conozcan mejor los factores genéticos y ambientales que afectan a la adquisición de masa ósea en lactantes y niños, lo cual ha sido motivo de recientes revisiones44. Entre los factores genéticos que pueden afectar a la masa ósea y el crecimiento en longitud, figuran los que hacen referencia a la efectividad en el funcionamiento de diferentes receptores. Los principales genes que pueden jugar un papel son el VDG, el gen del receptor de estrógenos y el gen para el factor de crecimiento transformación b1, TGFb1, ("transforming growth factorb1"). Para no ser reiterativos remitimos al lector a los comentarios hechos mas arriba, de aspectos como la influencia de los polimorfismos del gen VDR en la absorción intestinal de Ca y en la masa ósea en niños24, en el crecimiento del lactante y en la masa ósea adulta25, o su asociación con el peso al nacimiento, el crecimiento en la adolescencia y talla adulta26,28. También hemos mencionado la demostrada interacción entre genes, que puede modificar la expresión clínica, dependiendo de las diferentes coincidencias20. Todos estos aspectos pueden dar explicación a la documentada observación de las diferencias raciales45. Si la adquisición de masa ósea tiene una base genética, la interacción con los factores mutricionales ambientales y el estilo de vida, es importante.
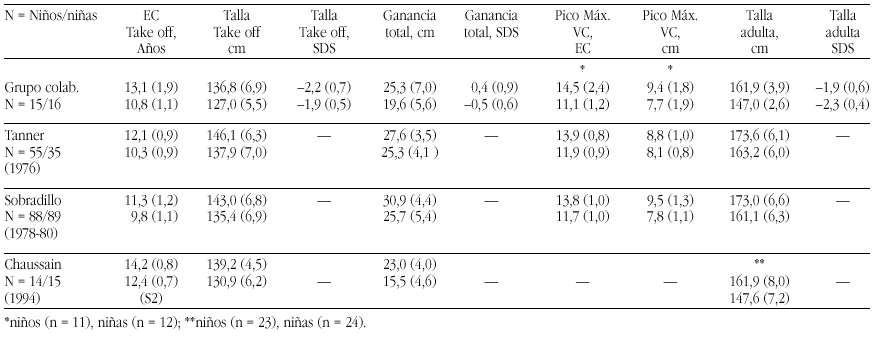
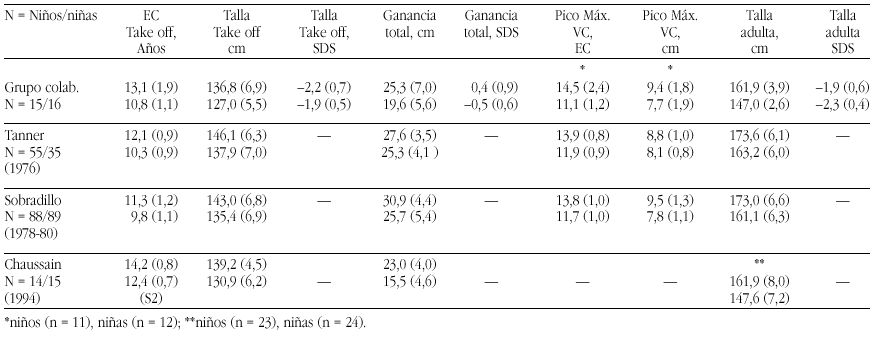
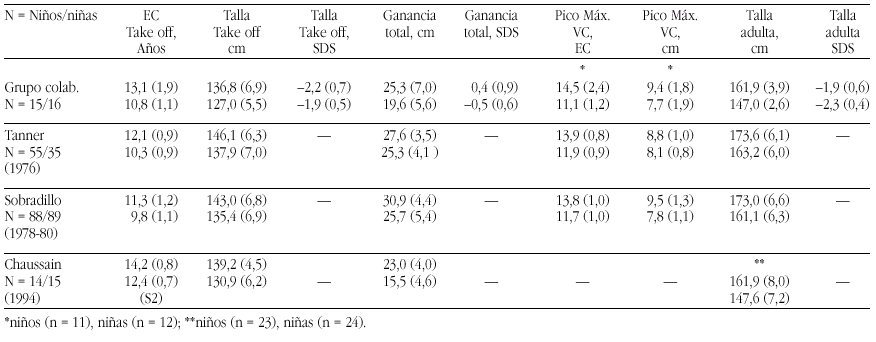
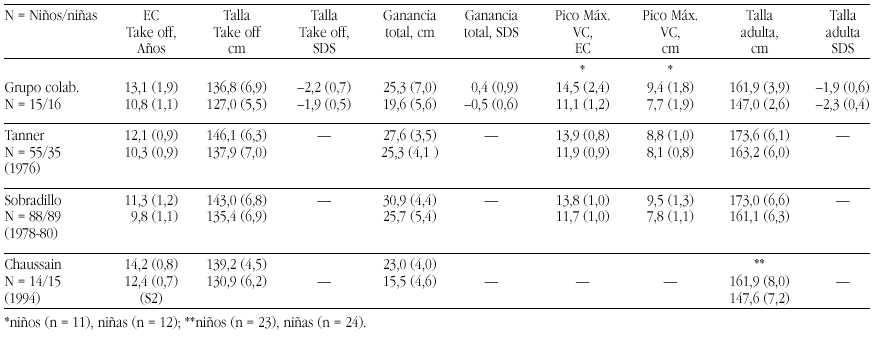
Contando con la genética y el ambiente, cabe preguntarse si puede ser un factor beneficioso la suplementación de calcio en la dieta. En el último decenio se ha recogido mucha información acerca de la relación existente entre ingesta de calcio, y la acumulación de masa ósea y el crecimiento durante la niñez y adolescencia. El tema también ha sido motivo de recientes revisiones46,47. Varios estudios observacionales, aunque no todos, sugieren que un aumento en la ingesta de Ca estimularía una mayor ganancia de masa ósea. Bonjour J-P et al 199947, hacen una revisión del tema y añaden dos estudios consecutivos de este tipo, realizados por ellos. Estudian el contenido mineral óseo (CMO), la densidad mineral ósea (DMO), y el crecimiento longitudinal, en un grupo de 200 niños y niñas, evaluando la ingesta diaria de nutrientes, (2 registros diarios durante 5 días). Como grupo completo no hubo diferencias significativas, ni tampoco en el grupo que incluía los estadios P2 a P4 de Tanner, y tampoco las hubo separados por sexo. Sin embargo, en el estadio P1 (prepuberales) hubo correlación significativa entre ingesta de Ca, la DMO, la CMO y el aumento de talla, tanto como grupo, como separado por sexos. Como resumen podemos decir que, estudios observacionales, concluyen que en estadío prepuberal, la suplementación de Ca estimula la ganancia de masa ósea y está asociado a una influencia positiva sobre la talla. Con los métodos de medición actuales debemos señalar que el efecto sobre la masa ósea podría estar mediado, al menos en parte, por el aumento del tamaño del hueso y el crecimiento longitudinal. Como hemos señalado no todos los estudios encuentran resultados positivos, por ejemplo, el grupo de Lee WT48, que lleva publicando anualmente sus resultados desde 1993, en un estudio observacional, no encuentra correlación entre la ingesta de calcio y la DMO de columna y radio. Tampoco con el ejercicio físico. Pero teniendo en cuenta las conclusiones del trabajo el trabajo anterior sobre la influencia de los estadios de Tanner, hemos de señalar que aquí se trataba de adolescentes.
De todas formas, en todos estos estudios no es fácil separar la influencia del mayor ingreso de proteínas y/o de energía, que suele acompañar al mayor ingreso de calcio. Para ello, se han llevado a cabo unos pocos estudios de intervención clínica, mediante la administración de suplementos de Ca. Estos mismos autores (Bonjour J-P et al 1999)47, realizaron un estudio a doble ciego, con placebo, en 149 niñas de 7 a 9 años, (de los cuales 108 terminaron el estudio). Idénticas proteínas y energía, con suplemento de 850 mg de Ca de la leche o placebo. Hubo un incremento de DMO y de la talla en el grupo suplementado con Ca. El incremento fue mas significativo en la niñas que consumían espontáneamente menos Ca (por bajo de 800mg/día), las cuales alcanzaron a las niñas consumidoras espontánea de niveles elevados de Ca. Los resultados obtenidos en columna lumbar y diáfisis femoral sugieren que el Ca podría estimular el crecimiento en longitud y el aumento de la sección transversal. Un año después del estudio todavía persistían los efectos. Las conclusiones de un estudio similar a largo plazo, realizado en China, efectuada por el otro grupo citado anteriormente (Lee WT et al)48, con aporte suplementario de 300 mg de calcio/día, en niños de 7 años, encuentran resultados similares, pero a diferencia del anterior, seguidos 30 meses después, el efecto había desaparecido49. En vista de ello, la ganancia mineral tras el aporte suplementario cálcico, lo interpretan como una reducción transitoria de la tasa de "turn-over" del hueso.
Parece que, efectivamente, los suplementos de Ca inhiben la remodelación. La osteocalcina sérica, marcador de remodelado óseo disminuye al administrar suplementos de calcio50, mecanismo que parece relacionado con inhibición de la PTH. Pero se sugiere otro efecto positivo sobre el modelado, ya que el Ca estimula in vitro la proliferación celular y la síntesis de ADN, acción mediada por los receptores celulares de Ca (CARS), sin excluir otros mecanismos, que expliquen el efecto estimulador de la proliferación osteoblástica, como podrían ser la síntesis de IGF-I51, y diferentes hormonas calciotrópicas, incluido el PTHrP, que juega un gran papel en la diferenciación del cartílago a nivel de placa epifisaria52.
Hay que tener en cuenta que la ingesta alimentaria de Ca se puede ver modificada por otros componentes de la alimentación, algunos conocidos desde antiguo, como los fitatos, en los que son ricos algunos cereales53, o el elevado contenido de las agua en fluoruros54, y otro de conocimiento mas reciente, como la demostración de que en niños, la mayor ingesta de Na incrementa significativamente la excreción urinaria de Ca, y que puede conducir a un menor depósito óseo55. En este sentido se conoce que la activación de la bomba del Na, con aumento de la actividad de la Na+/K+- ATP-asa, bloquea el incremento del calcio citosólico libre inducido por la GH en adipocito de rata, al interferir los canales del Ca voltaje-sensibles, bien por hiperpolarización de la membrana, o por interacciones desconocidas entre las bombas de Na y Ca56.
Aporte alimentario cálcico y la evolución de la talla en la especie humana
Al inicio de este trabajo, hacíamos referencia al origen de la vida y la evolución. Para terminar esta revisión, volveremos al principio haciendo unos breves comentarios sobre este tema. Es sobradamente conocido efecto que tiene sobre la talla la mejoría de los factores socioeconómicos. La mejora en la alimentación supone un mayor ingreso de calorías y de proteínas, pero estudios bien documentados, acerca de la relación entre la evolución de estos factores y la talla, demuestran que el incremento en la ingesta de Ca es mucho mayor que la de los otros factores, lo cual ha llevado a considerar que el calcio per se pudiera jugar un papel en este bien conocido efecto promotor del crecimiento, afectando positivamente el modelado óseo e inhibiendo el remodelado, como acabamos de ver. Todo ello ha conducido a la hipótesis, formulada por diferentes autores57-59 de que los ajustes en el tamaño de los huesos, que constatan los estudios paleontológicos, guardaría relación con la disponibilidad de la ingesta diaria de calcio, pudiendo representar un mecanismo adaptativo en el proceso de la evolución de la especie humana.
Los requerimientos de nutrientes en la especie humana fueron establecidos a lo largo de siglos de experiencia evolucionaria y la evidencia disponible indica que esta evolución se desarrolló, inicialmente, en un ambiente nutricional muy rico en calcio. El ejercicio y los patrones alimentarios de los humanos que vivieron hasta el final de la Edad de Piedra, como señalan Eaton y Nelson58 se puede considerar un paradigma: El ingreso cálcico era el doble que en el hombre actual, y el ejercicio físico, en pueblos nómadas de cazadores, también era, obviamente, mucho mayor que el del hombre moderno. El estudio de los huesos fósiles que nos han llegado de este período sugiere que desarrollaron una masa ósea mucho mayor y que experimentaban menos pérdida de masa ósea relacionada con la edad, que los huesos de los humanos del siglo XXI.
La primitivas especies de homínidos, eran de muy corta estatura (homo habilis 2-2,4 millones de años)60. Pero la talla de sus sucesores, el homo erectus, 1,5 millones de años, eran como los hombres actuales61 y durante todo el paleolítico permaneció constante: el hombre de Cro-Magnon medía, casi exactamente, la media actual58. Pero ya en el mesolítico la talla media empezó a declinar, llegando en el neolítico una disminución de entre 5,0-15,2 cm, por debajo de las tallas anteriores (tabla 5).

Se ha estudiado la alimentación de estos períodos. Hace 150 millones de años la mayor parte de los mamíferos que poblaban la tierra, nuestros antepasados, eran insectívoros, cuya riqueza en calcio es muy alta (124 mg/100 gr), localizado fundamentalmente en su exoesqueleto de quitina, y aunque en los grandes primates de hoy, la utilización de esta fuente es muy limitada, los actuales prosimios, como los lemúridos, aún se alimentan de insectos y pueden digerir quitina. Hace 50 millones de años los primates de aspecto moderno, nuestros antecesores más inmediatos, eran omnívoros, consumiendo insectos y otros invertebrados, pequeño número de vertebrados y, sobre todo, gran cantidad de fruta. Los monos actuales de gran peso, como los gorilas, con 150 kg de peso, deben consumir un 6-10% de su peso para satisfacer sus necesidades energéticas, puesto que siendo la dieta a base de vegetales fundamentalmente, con pocas calorías, necesitan ingerir grandes cantidades, lo que supone un gran aporte de calcio.
Referido ya a la especie humana, durante el alto paleolítico, en pueblos pre-agricultores y sin animales domésticos (que aparecieron hace ~10.000 a.), la base de la alimentación eran vegetales salvajes, pequeños animales, y ausencia de productos lácteos, (pasada la lactancia). En estas condiciones y remitiendo al lector interesado a artículos científicos especializadas58,62, sobre tipo de plantas que se podían consumir, animales, composición de los mismos, etc. se ha calculado la ingesta media diaria de calcio, en la Edad de Piedra, era ~1.800 mg/día. Incluso se han hecho estudios alternativos en diferentes situaciones de subsistencia. De todas formas, muy por encima de los 500-8000 mg/día del hombre actual.
El paleolítico tardío, entre 25.000 y 35.000 millones de años, puede considerase el último período en el que el colectivo humano interactúa con circunstancias bioambientales típicas de los períodos en los que originalmente se seleccionó la especie. A partir de entonces cambió la alimentación y elpatrón de ejercicio. De nómada y cazador pasó a ser cultivador y sedentario. La revolución del neolítico llevó consigo el pasar de las plantas silvestres a las cultivadas, lo que conlleva una serie de circunstancias: menos proteínas, mucho menos calcio, alimentación rica en fibras y muchos fitatos (maíz, trigo), lo cual disminuyó notablemente su biodisponibilidad de calcio. La investigaciones antropológicas citadas, en las que tampoco podemos extendernos, sugieren que las fluctuaciones en la disponibilidad diaria de calcio ha jugado un gran papel, como mecanismo de adaptación, en la evolución secular de la talla59,63, verificándose una disminución de la misma que, iniciada en el mesolítico, fue máxima como hemos visto en el período neolítico.

