


El fallo hepático agudo (FHA) secundario a enfermedades metabólicas hereditarias (EMH) es una enfermedad grave infrecuente de mal pronóstico. La intervención temprana puede salvar vidas.
ObjetivoDescribir la presentación clínica, la investigación y la evolución de FHA asociado a EMH en niños pequeños.
Material y métodosEstudio retrospectivo realizado en una unidad terciaria de cuidados intensivos neonatales y pediátricos, por un período de 27 años. Se analizaron los registros médicos de todos los niños hasta 2 años de edad, hospitalizados por FHA y con etiología metabólica documentada.
ResultadosDe los 34 casos de ALF, 18 se asociaban a EMH: galactosemia (4), síndrome de depleción del ADN mitocondrial (SDM) (3), deficiencia de ornitina transcarbamilasa (3), defectos congénitos de la glucosilación (2), tirosinemia tipo 1 (2), deficiencia de 3-hidroxi-acil-CoA deshidrogenasa de cadena larga (1), intolerancia hereditaria a la fructosa (1), aciduria metilmalónica clásica (1) y citrulinemia tipo 1 (1). La edad mediana fue de 1,3 meses. En el 67% de los casos se había observado al menos un síntoma o signo indicativo de EMH con anterioridad (vómitos, fallo de medro, hipotonía o retraso del desarrollo). Los signos físicos más frecuentes en el momento del ingreso fueron: hepatomegalia (72%), ictericia (67%) y encefalopatía (44%). Los niveles analíticos pico fueron: razón internacional normalizada media, 4,5; lactato mediano, 5mmol/L; bilirrubina media, 201μmol/L; alanina aminotransferasa (ALT) mediana, 137 UI/L; y amonio mediano, 177μmol/L. Un paciente fue remitido para trasplante de hígado en contexto de FHA (MSD). La tasa de mortalidad fue del 44%.
DiscusiónLa identificación de EMH como causa frecuente de FHA permitió el uso de medidas terapéuticas específicas y un asesoramiento familiar apropiado. Sus manifestaciones clínicas particulares y niveles moderados de ALT y bilirrubina pueden llevar a sospechar esta condición.
Pediatric acute liver failure (ALF) due to inherited metabolic diseases (IMD) is a rare life-threatening condition with a poor prognosis. Early intervention may be lifesaving.
ObjectiveTo describe clinical presentation, investigation and outcomes of ALF related to IMD in young children.
Material and methodsRetrospective review of the medical records of children aged up to 24 months, admitted to a tertiary pediatric and neonatal Intensive Care Unit during a 27-year period, fulfilling the ALF criteria, with documented metabolic etiology.
ResultsFrom 34 ALF cases, 18 were related to IMD: galactosemia (4), mitochondrial DNA depletion syndrome (MDS) (3), ornithine transcarbamilase deficiency (3), congenital defects of glycosylation (2), tyrosinemia type 1 (2), long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (1), hereditary fructose intolerance (1), classic methylmalonic aciduria (1) and citrulinemia type 1 (1). The median age was 1.3 months. At least one previous suggestive sign/symptom of IMD (vomiting, failure to thrive, hypotonia or developmental delay) was observed in 67% of the cases. The most common physical signs at admission included: hepatomegaly (72%), jaundice (67%) and encephalopathy (44%). The peak laboratorial findings were: mean international normalizad ratio 4.5, median lactate 5mmol/L, mean bilirubin 201μmol/L, median alanine aminotransferase (ALT) 137 UI/L and median ammonia 177μmol/L. One patient was submitted to liver transplant in ALF context (MSD). The mortality rate was 44%.
DiscussionThe identification of IMD as a frequent cause of ALF allowed specific therapeutic measures and adequate family counselling. Particular clinical features and moderated ALT and bilirubin levels can lead to its suspicion.
El fallo hepático agudo (FHA) es una enfermedad rara grave, con afectación severa de la función hepática, que puede progresar rápidamente. Puede reconocerse por la presencia de necrosis hepatocelular y coagulopatía refractaria, asociadas o no a encefalopatía. La etiología del FHA varía según la edad del paciente y la región geográfica. En neonatos y lactantes, las infecciones virales (por enterovirus, herpes simple y otras), la hemocromatosis neonatal, las enfermedades metabólicas hereditarias (EMH) y la linfohistiocitosis hemofagocítica son las causas principales. En niños mayores, las causas más comunes son la hepatotoxicidad por fármacos, la hepatitis autoinmune y las infecciones virales. Dentro de estas últimas, las causadas por el virus de la hepatitis A son las más comunes en países en desarrollo1. Las EMH merecen especial atención en el diagnóstico diferencial del FHA pediátrico, especialmente en lactantes y niños pequeños, en los que se asocian al 13-43% de los casos2. En los 3 primeros años de vida, las EMH asociadas a FHA más frecuentes son: galactosemia, tirosinemia tipo 1, citopatía mitocondrial, defecto de la oxidación de ácidos grasos, intolerancia hereditaria a la fructosa, trastorno del ciclo de la urea y defectos congénitos de la glucosilación2. En este grupo etario en particular es fundamental llevar a cabo una investigación exhaustiva, estructurada y expeditiva para minimizar el número de casos indeterminados que podrían deberse a una EMH no detectada.
El pronóstico del FHA asociado a EMH suele ser desfavorable y depende del diagnóstico concreto3. El reconocimiento temprano de estas etiologías es fundamental, ya que algunas requieren tratamiento específico o pueden contraindicar el trasplante de hígado (TH), como ocurre en casos con afectación multisistémica1,2,4. El TH puede mejorar el pronóstico en casos con EMH que no responden a tratamiento conservador2,5.
Para mejorar el diagnóstico y manejo de la EMH en casos de FHA es fundamental mejorar nuestro conocimiento de su presentación clínica y sus marcadores bioquímicos. El objetivo del estudio era caracterizar la presentación clínica, el perfil analítico y la evolución de niños pequeños con FHA asociado a EMH, ingresados en una unidad de cuidados intensivos (UCI) neonatal y pediátrica durante el período de estudio de 27 años.
MétodosEstudio descriptivo realizado en una UCI neonatal y pediátrica de un hospital universitario terciario, que es el centro nacional de referencia para el TH. Fueron revisados los registros médicos de los niños hasta 2 años de edad, ingresados en la UCI por FHA y con EMH documentada, durante el período de estudio de 27 años (entre enero de 1989 y diciembre del 2015).
El FHA se definió de acuerdo con los siguientes criterios establecidos por el grupo de trabajo sobre insuficiencia pediátrica aguda6: 1) niños sin manifestaciones conocidas de enfermedad hepática crónica; 2) evidencia bioquímica de daño hepático agudo, y 3) presencia de coagulopatía de origen hepático con una razón internacional normalizada (international normalizad ratio [INR]) ≥ 1,5 no resuelta con vitamina K en presencia de encefalopatía hepática (EH), o IRN ≥ 2 independientemente de la presencia de EH. La encefalopatía se evaluó por medio de electroencefalograma y/o manifestaciones clínicas y se clasificó en 4grados: grado 1: cambios de conducta, cambios mínimos en nivel de consciencia y alteraciones del sueño; grado 2: letargo, confusión, comportamiento inapropiado, desorientación, cambios de humor; grado 3: con somnolencia pero estimulable, desorientación profunda, comportamiento extraño, rigidez muscular, clonus e hiperreflexia, y grado 4: coma y descerebración1,7.
Se analizaron datos demográficos, los antecedentes familiares, los antecedentes médicos y síntomas y signos clínicos. Se registraron los valores pico del recuento leucocitario, IRN, lactato, amonio, bilirrubina total y alanina aminotransferasa (ALT), así como la glucosa basal y los niveles valle de albúmina. El estudio metabólico inicial incluyó: perfil de acilcarnitinas, perfiles de aminoácidos séricos y en orina y de ácidos orgánicos en orina, niveles séricos de transferrina deficiente en hidratos de carbono, y niveles en orina de sustancias reductoras y cetonas. Se solicitaron otras analíticas y estudios metabólicos de manera individualizada. Se llevó a cabo extracción de ADN para pruebas moleculares/genéticas específicas. También se recogieron datos sobre los resultados de pruebas moleculares, el momento de confirmación del diagnóstico etiológico (in vivo o post mortem), la decisión con respecto al TH, el tiempo transcurrido hasta el alta de la UCI, la mortalidad al alta y la mortalidad global.
El análisis estadístico se realizó con el programa Statistical Package for the Social Sciences® versión 20 (IBM Corp. Released 2011. IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp.). Las variables cuantitativas se evaluaron con medidas de tendencia central y dispersión, y las cualitativas mediante el cálculo de frecuencias.
ResultadosEn el período de estudio, del total de 34 niños de hasta 24 meses de edad ingresados en la UCI con FHA, 18 tenían una EMH confirmada. La edad mediana fue de 1,3 meses (p25: 0,2; p75: 5,5), 8/18 eran neonatos y 8/18 varones.
Las EMH etiológicas identificadas y los resultados del estudio genético se exponen en la tabla 1. En casos de intolerancia hereditaria a la fructosa, el diagnóstico se confirmó por estudio enzimático en biopsia hepática, objetivándose una reducción en la escisión de la fructosa-1-fosfato por la aldolasa y un cociente de fructosa-1,6-bifosfato/fructosa-1-fosfato de 38 (N ≈ 1). Los pacientes más jóvenes se diagnosticaron en el período neonatal de galactosemia (3), síndrome de depleción del ADN mitocondrial (SDM) (2), deficiencia de ornitina transcarbamilasa (OTC) (1), aciduria metilmalónica clásica (AMMc) (1) y citrulinemia tipo 1 (1).
Etiologías de FHA y estudios genéticos
| EMH | n | Estudio genético |
|---|---|---|
| Galactosemia | 4 | Gen GALT: Q188R (homocigosis) (2) Gen GALE: p.Val94Mel c.280G>A (homocigosis) (2) |
| SDM | 3 | Gen DGUOK: c.677A>G (homocigosis) (2) y c.749T>C (homocigosis) (1) |
| Deficiencia OTC | 3 | Gen OTC: Tyr143 stop exón 5 (1); R92Q CGA>CAA codón 92 (1) y R23X CGA>TGA codón 23, exón 1 (homocigosis) (1). |
| CDG | 2 | Gen CCDC115: p.leuc31Ser c.92T>C (1). |
| Tirosinemia tipo 1 | 2 | Gen FAH: IVS6-1(G>T)/IVS7+1(G>A) (1) y IVS6-1(G>T) (homocigosis) (1) |
| Deficiencia LCHAD | 1 | Gen HADHA: G1528C (homocigosis) |
| Intolerancia hered. fructosa | 1 | – |
| AMMc | 1 | Gen MUT: c.1172delA (p.D391Vfs*9) (homocigosis) |
| Citrulinemia tipo 1 | 1 | Gen ASS1: c.970G>A (p.G324S) (homocigosis) |
AMMc: aciduria metilmalónica clásica; CDG: defecto congénito de la glucosilación; EMH: enfermedad metabólica hereditaria; FHA: fallo hepático agudo; LCHAD: deficiencia de 3-hidroxi-acil-CoA deshidrogenasa de cadena larga; OTC: ornitina transcarbamilasa; SDM: síndrome de depleción de ADN mitocondrial.
Se documentaron datos sobre consanguinidad en 13/18 pacientes, con antecedentes positivos en 3. Los antecedentes familiares fueron anodinos en todos los casos, exceptuando los de 2 pacientes: un paciente con SDM cuyo hermano falleció en otro hospital con un diagnóstico neonatal de hemocromatosis, en quien también se confirmó con posterioridad el diagnóstico de SDM por deficiencia de deoxiguanosina cinasa (DGUOK), y una recién nacida, hermana de un paciente de mayor edad que ya estaba incluido en el estudio, que murió con FHA y cuyo estudio post mortem permitió diagnosticar galactosemia por deficiencia de galactosa-4-epimerasa (GALE) en ambos hermanos.
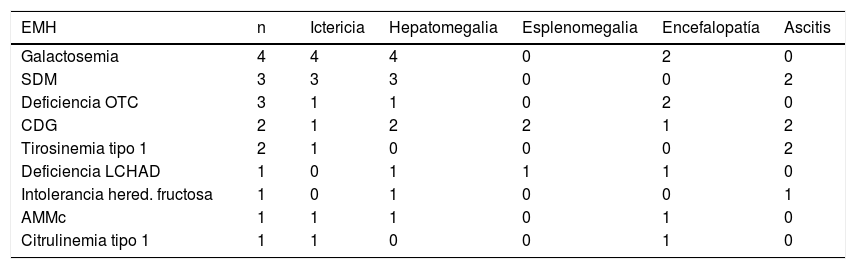
Con anterioridad al ingreso, el 67% (12/18) de los casos habían presentado al menos uno de los siguientes síntomas/signos: vómitos, fallo de medro, hipotonía y retraso de desarrollo. Los signos físicos más frecuentes al ingreso fueron: hepatomegalia (13/18; 72%); ictericia (12/18; 67%); encefalopatía (8/18; 44%); ascitis (7/18; 39%) y esplenomegalia (3/18; 17%). Cinco de 8 pacientes presentaron con encefalopatía grave (grados 3 a 4). La tabla 2 presenta los signos físicos al ingreso según la EMH de base.
Signos físicos de FHA al ingreso según la etiología
| EMH | n | Ictericia | Hepatomegalia | Esplenomegalia | Encefalopatía | Ascitis |
|---|---|---|---|---|---|---|
| Galactosemia | 4 | 4 | 4 | 0 | 2 | 0 |
| SDM | 3 | 3 | 3 | 0 | 0 | 2 |
| Deficiencia OTC | 3 | 1 | 1 | 0 | 2 | 0 |
| CDG | 2 | 1 | 2 | 2 | 1 | 2 |
| Tirosinemia tipo 1 | 2 | 1 | 0 | 0 | 0 | 2 |
| Deficiencia LCHAD | 1 | 0 | 1 | 1 | 1 | 0 |
| Intolerancia hered. fructosa | 1 | 0 | 1 | 0 | 0 | 1 |
| AMMc | 1 | 1 | 1 | 0 | 1 | 0 |
| Citrulinemia tipo 1 | 1 | 1 | 0 | 0 | 1 | 0 |
AMMc: aciduria metilmalónica clásica; CDG: defecto congénito de la glucosilación; EMH: enfermedad metabólica hereditaria; FHA: fallo hepático agudo; LCHAD: deficiencia de 3-hidroxi-acil-CoA deshidrogenasa de cadena larga; OTC: ornitina transcarbamilasa; SDM: síndrome de depleción de ADN mitocondrial.
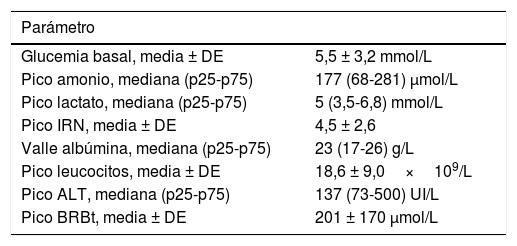
Los datos correspondientes a los valores medios o medianos del recuento leucocitario, INR, lactato, amonio, bilirrubina total, ALT, glucosa basal y niveles valle de albúmina se presentan en la tabla 3. El diagnóstico etiológico se confirmó por medio de estudios adicionales solicitados de manera individualizada, excepto en 5 casos en los que el diagnóstico se realizó durante el estudio post mortem (2 SDM, una deficiencia de 3-hidroxi-acil-CoA deshidrogenasa de cadena larga [LCHAD], un defecto congénito de la glucosilación [CDG] y una galactosemia por deficiencia de GALE).
Parámetros analíticos según la etiología
| Parámetro | |
|---|---|
| Glucemia basal, media ± DE | 5,5 ± 3,2 mmol/L |
| Pico amonio, mediana (p25-p75) | 177 (68-281) μmol/L |
| Pico lactato, mediana (p25-p75) | 5 (3,5-6,8) mmol/L |
| Pico IRN, media ± DE | 4,5 ± 2,6 |
| Valle albúmina, mediana (p25-p75) | 23 (17-26) g/L |
| Pico leucocitos, media ± DE | 18,6 ± 9,0×109/L |
| Pico ALT, mediana (p25-p75) | 137 (73-500) UI/L |
| Pico BRBt, media ± DE | 201 ± 170 μmol/L |
ALT: alanina aminotransferasa; BRBt: bilirrubina total; DE: desviación estándar; IRN: international normalized ratio (razón internacional normalizada).
El análisis retrospectivo de los hallazgos del examen físico y de laboratorio permitió la identificación de características particulares en pacientes con la misma EMH. Los 2 pacientes con tirosinemia tipo 1 presentaron tubulopatía renal. Los 4 casos de trastornos del ciclo de la urea (deficiencia de OTC y citrulinemia tipo 1) presentaron niveles elevados de amonio en comparación con el resto de las etiologías: mediana de 333μmol/L (p25: 118; p75: 1703) vs. 172,5μmol/L (p25: 61,8; p75: 236,5), respectivamente. En la galactosemia por deficiencia de GALE, la hipotonía, la displasia de cadera bilateral y la estatura baja fueron rasgos comunes en ambos casos. Los 2 casos de CDG presentaron estatura baja, algunos dismorfismos faciales y hepatoesplenomegalia. En los 3 pacientes con SDM por deficiencia de DGUOK se encontraron antecedentes positivos de consanguinidad, hipotonía, nistagmo, hipoglucemia y elevación de ferritina (789-1.931 ng/mL [rango normal: 9-120]) y de tirosina (68-543 mol/L [rango normal: 28-96]). El caso de LCHAD presentó con shock cardiogénico.
Se realizó TH por FHA en un paciente con SDM. La decisión fue tomada por un equipo interdisciplinario a la edad de 4 meses debido a insuficiencia hepática recurrente y previamente a la confirmación del diagnóstico de deficiencia de DGUOK, a pesar del deterioro neurológico y el nistagmo rotatorio. No obstante, murió a la edad de 4 años por acidosis metabólica, hiperlactacidemia y parada cardiorrespiratoria intercurrentes. Otro paciente con tirosinemia fue referido posteriormente para TH a los 7 años de edad por mal control metabólico, osteopenia, tubulopatía renal y nódulos hepáticos.
En el análisis de la evolución, el tiempo mediano transcurrido hasta el alta de la UCI fue de 7 días (p25: 3; p75: 21). La mortalidad al alta y la mortalidad global fueron del 39% (7/18) y el 44% (8/18), respectivamente. Aparte del paciente con SDM ya mencionado, el diagnóstico de los otros 7 pacientes fallecidos fue: SDM (2), LCHAD (1), CDG (1), galactosemia por deficiencia de GALE (1), deficiencia de OTC (1) y AMMc (1). Las principales causas de fallecimiento fueron progresión a fallo multiorgánico y shock cardiogénico. En 3 de estos casos (CDG, deficiencia de OTC y AMMc), la limitación del esfuerzo terapéutico se decidió por consenso multidisciplinario y sobre la base de las siguientes condiciones: 1) en el caso de CDG, a pesar de la optimización del tratamiento, incluyendo consideración de TH, que se desestimó debido a la afectación multisistémica con evolución neurológica desfavorable a causa de lesiones hipóxico-isquémicas graves objetivadas mediante estudios de imagen; 2) en el caso de deficiencia de OTC, la elevación de los niveles de amonio (> 800μmol/L) fueron difíciles de controlar en las primeras 48 h, a pesar de aplicarse las medidas terapéuticas de mayor intensidad, incluyendo la hemofiltración; aunque los niveles de amonio descendieron posteriormente, no se consiguió mejoría neurológica; el paciente progresó a fallo multiorgánico exacerbado por sepsis por Klebsiella oxytoca, y 3) en el caso con AMMc, no obstante la implementación de tratamientos específicos y la reducción de los niveles de amonio tempranas, la evolución clínica fue desfavorable desde el ingreso, con progresión rápida a fallo multiorgánico.
DiscusiónLas patologías susceptibles de producir insuficiencia hepática en el grupo de niños menores de 24 meses son muy específicas y las EMH constituyen una de las etiologías más frecuentes, con una incidencia descrita del 13-43%2. En el estudio presente, del total de 34 niños con FHA de estas edades, 18 (53%) se diagnosticaron de EMH. Este elevado número de EMH detectadas es fruto de un esfuerzo progresivo en realizar investigaciones diagnósticas más exhaustivas, incluso tras la muerte. También se puede explicar por el reconocimiento de este hospital terciario como el único centro de referencia en Portugal para el TH y el FHA desde 1994 y 2008, respectivamente. En pacientes sin una causa fácilmente identificable de FHA han de considerarse pruebas de cribado para causas metabólicas de FHA, sobre todo en este grupo etario. La etiología del FHA pediátrico queda sin determinar en el 18-47% de los pacientes1, dependiendo de la exhaustividad de la investigación diagnóstica, porcentajes que pueden ser más elevados en niños pequeños y que se acercan al 54% en los menores de 3 años, como comunicaron Squires et al6.
La identificación temprana de las EMH es crucial toda vez que algunas requieren la iniciación inmediata de terapias dirigidas o dietas específicas que pueden salvar vidas, principalmente en la galactosemia, la intolerancia hereditaria a la fructosa y la tirosinemia hereditaria tipo 11, que pueden manifestarse en el período neonatal con ictericia, hipoglucemia y, en algunos casos, FHA8. En nuestra serie, el menor número de pacientes con galactosemia y tirosinemia hereditaria tipo 1 que presentaron con FHA en comparación con la experiencia del King's College9 podría atribuirse al cribado neonatal expandido, introducido en Portugal desde el año 2005, que permite el diagnóstico de estas enfermedades en el período subclínico.
La escasa información disponible sobre consanguinidad (en solo 13/18 pacientes) es una limitación asociada a la naturaleza retrospectiva del estudio. Según Hegarty et al.9, puede haber consanguinidad en los padres en hasta el 44% de los casos de FHA asociado a EMH. Ciertos detalles de los antecedentes familiares, como los abortos recurrentes, el fallecimiento de hermanos y la enfermedad en hijos previos, son también factores que han de explorarse sin excepción, ya que pueden llevar a sospechar una EMH, como ocurrió en los casos de SDM y galactosemia por deficiencia de GALE.
Los antecedentes de vómitos, fallo de medro y/o retraso de desarrollo presentes en el 67% de los casos también son manifestaciones que hacen sospechar EMH, como han indicado Alam y Lal.2 y Brett et al.4. La encefalopatía fue más frecuente (44%) en comparación con la serie con EMH de Hegarty et al.9 (19%), un hecho atribuible a las distintas proporciones de EMH específicas en cada una de las series. Otro factor que podría contribuir a esta diferencia es el posible retraso en la remisión a nuestro hospital en los años previos al 2008, fecha en la que se estableció como centro nacional de referencia para el FHA10. Algunos rasgos clínicos pueden indicar diagnósticos específicos, guiando la investigación etiológica11. La afectación cardíaca puede llevar a sospechar LCHAD. Se sabe que ciertos dismorfismos ocurren con mayor frecuencia en contexto de CDG y de galactosemia por deficiencia de GALE11,12. La tríada de hipotonía, nistagmo e hipoglucemia suele manifestarse en casos de SDM por deficiencia de DGUOK. Estos pacientes también pueden presentar con niveles elevados de ferritina y tirosina, llevando al diagnóstico diferencial de hemocromatosis neonatal y tirosinemia tipo 113. Otros indicios obtenidos mediante pruebas de laboratorio son los signos de tubulopatía, que pueden ser indicativos de tirosinemia tipo 1, galactosemia clásica o intolerancia hereditaria a la fructosa, y la hiperamoniemia grave, que es un hallazgo típico en los trastornos del ciclo de la urea11.
En cuanto a los parámetros analíticos encontrados en el estudio, la elevación de leve a moderada de ALT fue consistente con los niveles descritos en otros estudios de FHA asociado a EMH9,14 e inferior a los descritos en FHA de otras etiologías, especialmente la viral14. La elevación moderada de la bilirrubina también fue similar a la encontrada en otros estudios9,14. En 2 series pequeñas4,15 que compararon el perfil analítico en casos de FHA asociado a EMH con los de otras etiologías, los picos de lactato y de amonio fueron similares en ambos grupos y también los mismos que los encontrados en este estudio, lo que indica que son de escasa utilidad como marcadores aislados de EMH en el FHA.
La detección de la EMH es crucial para optimizar el manejo con intervenciones específicas, evaluar la necesidad de TH y proporcionar un asesoramiento adecuado a los padres en gestaciones futuras16. Las decisiones concernientes al trasplante son muy difíciles, hecho corroborado por la falta de modelos pronósticos de FHA validados en la población pediátrica17, y en particular de FHA secundario a EMH. El TH puede estar contraindicado en casos en que el FHA se puede resolver con terapias farmacológicas o nutricionales específicas, en que la enfermedad de base afecta a otros órganos o sistemas, o con progresión rápida del FHA con fallo multiorgánico o sepsis5,18. De los 18 pacientes incluidos en este estudio, solo uno se remitió para TH en contexto de FHA: un paciente con deficiencia de DGUOK que recibió un TH a pesar de presentar con disfunción neurológica y nistagmo rotatorio. La medida no consiguió prevenir la progresión de la enfermedad, con deterioro neurológico y visual, retraso psicomotor grave y fallo de medro. La contraindicación del TH no estaba clara en este paciente con SDM hepatocerebral, ya que la literatura también describe a una proporción considerable de pacientes con resultados favorables del TH, con buena supervivencia a largo plazo y estabilización neurológica en pacientes con afectación neurológica al inicio19,20.
La tasa de mortalidad global fue del 44%, inferior al 70% comunicado por Alam et al.15, pero superior al 19% comunicado por Hegarty et al.9. Sundaram et al.14 comunicaron una mortalidad del 24% sin TH. No obstante, la heterogeneidad entre muestras en lo concerniente a la edad, las diversas etiologías del FHA y el pequeño tamaño de las series dificultan la comparación de las tasas de mortalidad. Así, mientras que Sundaram et al.14 estudiaron FHA en lactantes de hasta 3 meses de edad, en la serie de Hegarty et al.9 los pacientes tenían hasta 5 años, y la mayoría de ellos estaban diagnosticados de galactosemia o tirosinemia, a las que se atribuye un pronóstico generalmente bueno. En nuestro estudio, el alto porcentaje de pacientes con afectación multisistémica o con progresión rápida a fallo multiorgánico podría explicar la elevada mortalidad. Además, el 44% de los pacientes presentaron con encefalopatía, que generalmente se considera un marcador de pronóstico desfavorable en el FHA21.
En los últimos años, la implementación de programas expandidos de cribado neonatal en varios países y la disponibilidad de nuevas terapias dirigidas para algunas enfermedades, así como de dispositivos de soporte avanzados, han contribuido a modificar en cierto grado el curso natural y la progresión de las EMH1,4. Como muestra la serie de Couce et al.22 sobre la incidencia de EMH (no necesariamente asociada a FHA) en una unidad neonatal, la inversión en el cribado neonatal permitió el diagnóstico y el tratamiento tempranos en un grupo de pacientes antes de la aparición de síntomas, evitándose en varios casos complicaciones clínicas graves y el ingreso hospitalario.
Las limitaciones principales de este estudio están relacionadas con el pequeño número de casos, el largo período de estudio y la recogida retrospectiva de datos, que podría haber limitado la información obtenida. Además, solo se incluyeron en la serie casos de FHA en pacientes ingresados en la UCI, que corresponden a los más graves. Este criterio puede haber dado lugar a un sesgo de selección.
Nuestro estudio evidencia la alta frecuencia del FHA asociado a EMH en este grupo etario en particular y la importancia de realizar una investigación diagnóstica exhaustiva para llegar a un diagnóstico correcto. La realización del diagnóstico post mortem permite el asesoramiento adecuado a las familias. Algunos rasgos clínicos y de laboratorio, como la menor edad y la elevación moderada de ALT y los niveles pico de bilirrubina, pueden llevar a sospechar FHA asociado a EMH. El diagnóstico temprano de esta etiología puede permitir una pronta toma de decisiones en lo concerniente al tratamiento, lo que contribuye a mejorar el pronóstico. En el futuro habría que esforzarse en la optimización de la toma de decisiones clínica respecto al TH, lo que podría conseguirse mediante estudios multicéntricos amplios y homogéneos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

