


La citogenética convencional detecta un 3-5% de los pacientes con retraso global del desarrollo/discapacidad intelectual y/o malformaciones congénitas. La amplificación de sondas múltiples dependientes de ligación permite incrementar la tasa diagnóstica entre 2,4-5,8%. Actualmente, los arrays de hibridación genómica comparada o aCGH son la herramienta diagnóstica con mayor rendimiento en estos pacientes, en malformaciones congénitas y trastornos del espectro autista. El objetivo del presente trabajo ha sido evaluar la eficiencia del uso del aCGH como técnica de primera línea diagnóstica en estas y otras indicaciones (epilepsia, talla baja).
Pacientes y métodoSe ha estudiado a 1.000 pacientes afectados por las patologías mencionadas mediante la técnica de aCGH.
ResultadosSe detectaron desequilibrios de efecto patogénico en un 14% de los pacientes (140/1.000). Según el fenotipo, se diagnosticaron un 18,9% de los pacientes afectados de retraso global del desarrollo/discapacidad intelectual; un 13,7% de las malformaciones congénitas; un 9,76% de las patologías psiquiátricas, un 7,02% de los casos con epilepsia y un 13,3% de los pacientes con talla baja. Dentro de las malformaciones congénitas destacan las del sistema nervioso central con un 14,9% y las cardiopatías congénitas con un 10,6% de diagnósticos. En las patologías psiquiátricas destacan los pacientes con trastornos del espectro autista, con un 8,9% de diagnósticos.
ConclusionesNuestros resultados demuestran la efectividad y la eficiencia de la utilización del aCGH como test de primera línea en el diagnóstico genético de los pacientes con sospecha de desequilibrios genómicos. Todo ello avala su inclusión dentro del Sistema Nacional de Salud.
Conventional cytogenetics diagnoses 3-5% of patients with unexplained developmental delay/intellectual disability and/or multiple congenital anomalies. The Multiplex Ligation-dependent Probe Amplification increases diagnostic rates from between 2.4 to 5.8%. Currently the comparative genomic hybridisation array or aCGH is the highest performing diagnostic tool in patients with developmental delay/intellectual disability, congenital anomalies and autism spectrum disorders. Our aim is to evaluate the efficiency of the use of aCGH as first-line test in these and other indications (epilepsy, short stature).
Patients and methodA total of 1000 patients referred due to one or more of the abovementioned disorders were analysed by aCGH.
ResultsPathogenic genomic imbalances were detected in 14% of the cases, with a variable distribution of diagnosis according to the phenotypes: 18.9% of patients with developmental delay/intellectual disability; 13.7% of multiple congenital anomalies, 9.76% of psychiatric pathologies, 7.02% of patients with epilepsy, and 13.3% of patients with short stature. Within the multiple congenital anomalies, central nervous system abnormalities and congenital heart diseases accounted for 14.9% and 10.6% of diagnoses, respectively. Among the psychiatric disorders, patients with autism spectrum disorders accounted for 8.9% of the diagnoses.
ConclusionsOur results demonstrate the effectiveness and efficiency of the use of aCGH as the first line test in genetic diagnosis of patients suspected of genomic imbalances, supporting its inclusion within the National Health System.
Se estima que la prevalencia en la población general del retraso global del desarrollo/discapacidad intelectual (RGD/DI) es de un 1-3%1, de los trastornos del espectro autista (TEA) del 0,7%2 y de las malformaciones congénitas (MC) de un 2-3%1-3, suponiendo un grave problema sanitario y social. El diagnóstico genético de estos pacientes es de gran utilidad en su manejo clínico, permite un pronóstico preciso y, sobre todo, es fundamental para la prevención de la aparición de nuevos casos (asesoramiento genético familiar). Hasta recientemente, el estudio de estos pacientes se ha venido realizando mediante un conjunto de técnicas de laboratorio de bajo rendimiento individual. La técnica más utilizada, el análisis cromosómico convencional o cariotipo, detecta grandes pérdidas o ganancias de material genético y reorganizaciones estructurales en un 3-5% de los pacientes con RGD/DI y/o MC1,4-6. Para mejorar este rendimiento diagnóstico, se complementó la citogenética convencional con las técnicas de hibridación «in situ» fluorescente (FISH) y amplificación de sondas múltiples dependientes de ligación (MLPA). Gracias a estas técnicas, en pacientes con RGD/DI y MC con un resultado previo de cariotipo aparentemente normal, se han comunicado un 2,4-3,7% de diagnósticos adicionales con el estudio de las regiones subteloméricas7,8 y un 5,8% de diagnósticos adicionales gracias al estudio de los principales síndromes de microdeleción o microduplicación9.
El reciente desarrollo de los arrays de dosis para la detección de anomalías de copia (de polimorfismos de un solo nucleótido o SNP arrays y de hibridación genómica comparada o aCGH) ha incrementado muy significativamente el número de diagnósticos, permitiendo el diagnóstico de un 15-20% de los pacientes con RGD/DI, MC o TEA5. Los arrays de SNP, inicialmente diseñados para interrogar miles de polimorfismos de un nucleótido a lo largo del genoma, también permiten evaluar el número de copias de ADN comparando la intensidad de la hibridación del ADN del paciente con las sondas inmovilizadas en el array o matriz de puntos.
Los aCGH permiten explorar de manera simultánea la dosis de ADN en múltiples loci del genoma al comparar las cantidades relativas de ADN de 2 genomas (control y paciente), marcados con fluorocromos distintos, que se unen con fragmentos de ADN de secuencia conocida o «sondas» que están fijados a un portaobjetos o soporte de vidrio. El color de la fluorescencia en cada punto del aCGH informa sobre la cantidad relativa de cada ADN y permite inferir la presencia de ganancias o de pérdidas en regiones concretas del genoma. Existe un gran número de plataformas de aCGH, con diferencias notables en su rendimiento diagnóstico. Las hay basadas en sondas de bacterial artificial chromosomes (BAC) y en sondas de oligonucleótidos. Las sondas pueden investigar zonas localizadas a intervalos regulares del genoma humano (esqueleto o backbone) o regiones de las que se sabe están relacionadas con patología, o ser una mezcla de las 210. El consorcio International Standard Cytogenetic Array (ISCA) proporciona recomendaciones que han sido adoptadas por la mayoría de los fabricantes de arrays. La comparación de las principales plataformas comerciales llevada a cabo en un estudio multicéntrico11 concluyó que los arrays de oligonucleótidos CGH con diseño ISCA y formato 8×60K son los que proporcionan mayor rendimiento coste/beneficio. Debido a consideraciones clínicas y económicas, las guías científicas recomiendan el uso de los arrays como técnica de primera línea diagnóstica en pacientes con DI/RGD, MC y/o TEA, en sustitución de las técnicas de cariotipo, MLPA y FISH5,12-16.
Presentamos nuestra experiencia en el diagnóstico mediante aCGH de 1.000 pacientes con RGD/DI, MC, trastornos psiquiátricos (TEA, TDAH, esquizofrenia, trastorno bipolar), epilepsia, talla baja, y otros (dismorfias, fenotipo peculiar, miopatías, hipercrecimiento) como técnica de primera línea, en sustitución de las técnicas de cariotipo, MLPA y FISH. Se trata de la mayor serie con estas características descrita en nuestro país, ya que hasta ahora se han publicado series de menor tamaño o utilizando los aCGH como técnica de tercera línea, en pacientes con resultado normal en otras técnicas de diagnóstico genético tales como el cariotipo y la MLPA17.
Pacientes y métodoPacientesEn el periodo de enero del 2012 a enero del 2015 se realizaron 1.222 estudios con la técnica de aCGH en la Unidad de Arrays del Área de Genética Clínica y Molecular del Hospital Universitario Vall d’Hebron de Barcelona. En todos los casos se realizó una visita clínica antes y después del estudio. De ellos, 1.000 corresponden a pacientes con una o más de las siguientes patologías: RGD/DI, MC, trastornos psiquiátricos (TEA, TDHA, esquizofrenia, trastorno bipolar), epilepsia, talla baja y otros como dismorfias, fenotipo peculiar, miopatías e hipercrecimiento, todos ellos sin estudios genéticos previos, derivados por Genética Clínica, Neurología y Endocrinología Pediátrica del hospital. El resto de los estudios corresponden a 4 grupos: 1) estudios familiares (n = 167); 2) fetos con anomalías ecográficas (n = 41); 3) caracterización de anomalías previamente diagnosticadas con otras técnicas, principalmente el cariotipo (n = 4), y 4) pacientes (n = 10) con antecedentes de otros estudios genéticos previos.
MétodoPara conocer la tasa de diagnósticos en relación con las alteraciones fenotípicas, se procedió a la exhaustiva revisión de las historias clínicas de todos los pacientes para permitir la correcta interpretación de los estudios de aCGH. Gracias a ello se estableció una clasificación de las patologías y se contabilizó el número de pacientes cuyo fenotipo se ajustaba a cada una de ellas. Los pacientes podían presentar una o más de las patologías de la clasificación (tabla 1).
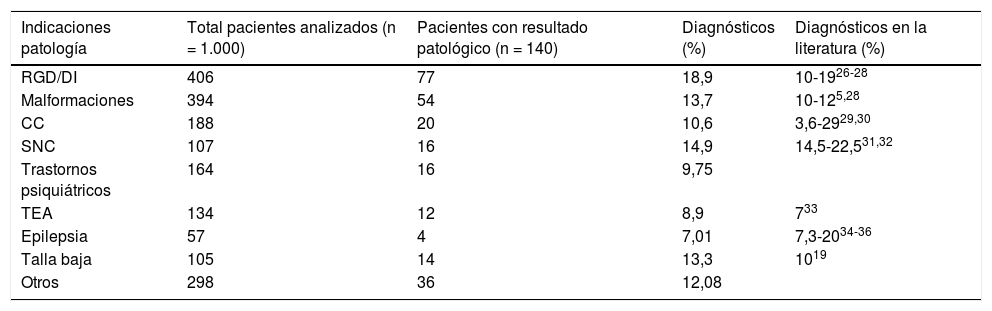
Tasa diagnóstica alcanzada con la técnica de aCGH en relación con los distintos fenotipos estudiados. Comparación con datos procedentes de la literatura
| Indicaciones patología | Total pacientes analizados (n = 1.000) | Pacientes con resultado patológico (n = 140) | Diagnósticos (%) | Diagnósticos en la literatura (%) |
|---|---|---|---|---|
| RGD/DI | 406 | 77 | 18,9 | 10-1926-28 |
| Malformaciones | 394 | 54 | 13,7 | 10-125,28 |
| CC | 188 | 20 | 10,6 | 3,6-2929,30 |
| SNC | 107 | 16 | 14,9 | 14,5-22,531,32 |
| Trastornos psiquiátricos | 164 | 16 | 9,75 | |
| TEA | 134 | 12 | 8,9 | 733 |
| Epilepsia | 57 | 4 | 7,01 | 7,3-2034-36 |
| Talla baja | 105 | 14 | 13,3 | 1019 |
| Otros | 298 | 36 | 12,08 |
Trastornos psiquiátricos incluyen: TEA, TDAH, psicosis esquizofrénica, trastorno bipolar. Otros: incluye dismorfias, fenotipos peculiares, miopatías, paresias, hipercrecimiento. Los pacientes presentaban uno o más de los fenotipos descritos.
CC: cardiopatías congénitas; RGD/DI: retraso global del desarrollo/discapacidad intelectual; CC: cardiopatías congénitas; SNC: sistema nervioso central; TDAH: trastorno por déficit de atención e hiperactividad; TEA: trastornos del espectro autista.
El ADN de los pacientes se obtuvo a partir de 3ml de sangre periférica con el Kit Gentra Puregene Blood (Quiagen, EE. UU. n. cat 158422), siguiendo el protocolo suministrado por el fabricante. En unos pocos casos, el ADN había sido extraído por diversos métodos en otros centros o laboratorios hospitalarios.
Los procedimientos utilizados fueron realizados tras obtención de un consentimiento informado de los padres.
La validación de la técnica de aCGH se efectuó con 2 plataformas distintas de arrays de oligonucleótidos: CGH Microarray Kit 4×180K (Agilent Technologies, EE. UU., n. cat G4426B) con ∼180.000 sondas distribuidas de forma aproximadamente equidistante o backbone y CGH Microarray Kit 8×60K diseño ISCA (Agilent Technologies, EE. UU., n. cat G4827A) con menor número de sondas (∼60.000). de las cuales 40.208 están distribuidas de forma uniforme (backbone) y 18.851 distribuidas a una mayor densidad y dirigidas a regiones de interés recomendadas por el consorcio ISCA. Se procedió de acuerdo con las recomendaciones del fabricante. Debido a su mayor rendimiento, se decidió realizar todos los estudios de aCGH con la plataforma de 8×60K diseño ISCA. Las imágenes obtenidas con una resolución de 3 micras (DNA microarray scanner, Agilent Technologies, EE. UU., G2505C) se analizaron con el programa Cytogenomics 2.1. (Agilent Technologies, EE. UU.). Para facilitar la evaluación del significado patológico de las anomalías de número de copia (CNV) detectadas y su contenido génico, se utilizó el software de desarrollo propio «EasyArray», escrito en los lenguajes de programación Phyton y VisualBasic. EasyArray compara cada una de las CNV detectadas con los estudios de controles normales incluidos en Database of genome variants (http://dgv.tcag.ca/dgv/app/home) y una base de datos de elaboración propia con los principales síndromes de microdeleción/microduplicación, así como si su contenido génico está incluido en la base de datos de enfermedades génicas de UNIPROT (http://www.uniprot.org/). Finalmente. EasyArray facilita la consulta de las bases de datos del consorcio ISCA (https://www.iscaconsortium.org), GeneCards (http://www.genecards.org), OMIM (http://www.omim.org) y Decipher (https://decipher.sanger.ac.uk). El programa orienta hacia una clasificación de las CNV como patogénicas, inciertas, o benignas y facilita la redacción de los informes de acuerdo con las recomendaciones del American College of Medical Genetics13. A fin de reducir el número de estudios familiares y lograr un equilibrio entre sensibilidad analítica y clínica, no se informan las variaciones de copia de significado incierto de tamaño menor de 400 Kb5. Todas las CNV inciertas y patogénicas detectadas se confirmaron mediante FISH, MLPA, cariotipo o un segundo estudio de aCGH.
Se realizó análisis estadístico con el programa SPSS versión 15.0 aplicando el test de la chi al cuadrado para valorar si existía una asociación entre el potencial diagnóstico y el número de alteraciones fenotípicas en un mismo paciente.
Finalmente, a fin de poder comparar la tasa diagnóstica y el coste de las diversas técnicas en nuestra población, todas las anomalías detectadas por aCGH como técnica de primera línea fueron revisadas a fin de determinar si habrían sido detectadas mediante las técnicas de MLPA subtelomérica (P070, MRC Holland, Holanda), MLPA de síndromes recurrentes (P245 y P297, MRC Holland, Holanda) y cariotipo convencional (anomalías de más de 6Mb)18 con un cambio de patrón de bandas juzgado como suficiente por un citogenetista experto. Para el análisis del coste por ensayo, se siguieron las directrices del Consenso para la implementación de los arrays en la genética clínica16.
ResultadosLos resultados de la comparativa entre las 2 plataformas de aCGH (4×180K y 8×60K ISCA v2, ambas de Agilent Technologies) mostraron claramente una mayor especificidad de la plataforma 8×60K ISCA v2 en la detección de CNV patogénicas. Así, por ejemplo, una deleción patogénica de 0,043 Mb fue detectada por solo 4 sondas con la plataforma de 4×180K y con 11 sondas utilizando la plataforma 8×60K ISCA v2. En otro caso, una duplicación patogénica afectando a la totalidad del gen RAF1 se detectó con 10 sondas con la plataforma 4×180K y con 18 sondas con la plataforma 8×60K ISCA v2. Como consecuencia de estos resultados, se decidió utilizar la plataforma 8×60K ISCA v2 detectándose desequilibrios de efecto patogénico en un 14% de los pacientes (140/1.000), en 17 casos se detectó más de una CNV patogénica. Se detectaron desequilibrios de significado incierto de más de 400 Kb (variants of uncertain significance [VOUS]) en un 6,1% de los pacientes (61/1.000).
Se detectaron anomalías de copia patogénicas en un 18,9% de los pacientes afectados de RGD/DI (77/406), un 13,7% de las MC (54/394), un 9,76% de las patologías psiquiátricas (16/164), un 7,02% de los casos con epilepsia (4/57) y un 13,3% de los casos remitidos por talla baja (14/105). El rendimiento diagnóstico fue del 12,08% (36/298) para las diversas patologías clasificadas en la categoría «otros» (incluyendo dismorfias, fenotipos peculiares, miopatías, paresias, hipercrecimiento). Dentro de las MC destacan las anomalías del sistema nervioso central, con un 14,9% de diagnósticos (16/107), y las cardiopatías congénitas, con un 10,6% de diagnósticos (20/188). En el grupo de las patologías psiquiátricas, destacan los pacientes con TEA, con un 8,9% (12/134) de diagnósticos (tabla 1).
Al agrupar los pacientes en función del número de anomalías fenotípicas que presentaban, se observó que la tasa diagnóstica se incrementaba de forma estadísticamente significativa (p = 0,0223) en los pacientes con un mayor número de anomalías fenotípicas (tabla 2).
Porcentaje de diagnósticos mediante la técnica de aCGH en función del número de anomalías fenotípicas presentes
| N.° de anomalías fenotípicas registradas en la historia clínica | Total pacientes analizados (n = 1.000) | Pacientes con resultado de aCGH patológico (n = 140) | Diagnósticos (%) |
|---|---|---|---|
| > 2 | 68 | 15 | 20 |
| 2 | 288 | 48 | 16,6 |
| 1 | 644 | 77 | 11,9 |
Las diferencias entre los grupos son estadísticamente significativas (p = 0,0223).
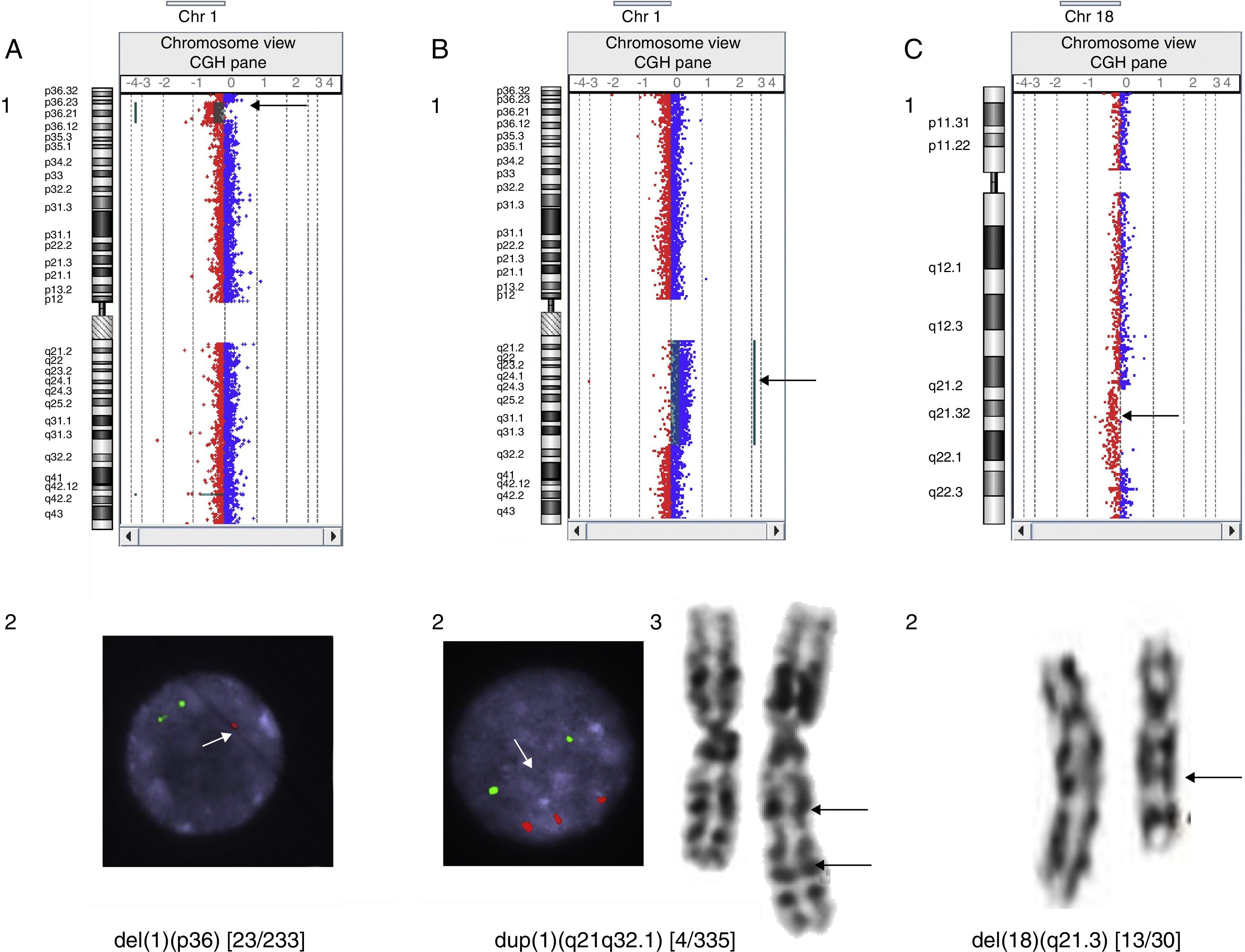
En nuestra serie se han detectado microdeleciones y microduplicaciones en mosaico, como, por ejemplo, una deleción 1p36 de 12.6 Mb en un paciente con cardiopatía, confirmada mediante técnicas de FISH en un 9,8% de las células de un cultivo de sangre; una duplicación 1q21.1-q32.1 de 61,38 Mb en un paciente con epilepsia confirmada por cariotipo y FISH en un 1,2% de las metafases y una deleción intersticial 18q21.31-q22.2 de 14,6 Mb en un paciente con RGD, no detectada por el programa, pero sí presente en el gráfico que proporciona el mismo y que se contabilizó en un 43,3% de las metafases analizadas (fig. 1).
 Deleción 1p36 en mosaico en un paciente con cardiopatía: detectada mediante aCGH (1) y confirmada en un 9,8% de las células analizadas por FISH. Solo una única señal en el núcleo de la fotografía, indicada por la flecha, corresponde a la región 1p36 (2). B) Duplicación 1q21.1-q32.1 en un paciente con epilepsia: detectada mediante aCGH (1), confirmada en un 1,2% de las células analizadas por FISH. Las tres señales inferiores del núcleo de la fotografía, a las que se dirige la flecha, corresponden a la región 1q21q32.1 (2) y cariotipo (3). C) Deleción 18q21.31-q22.2 en un paciente con RGD: detectada mediante aCGH(1) y confirmada en un 43,3% de las células analizadas por cariotipo (2).")
Casos de mosaicismos detectados por hibridación genómica comparada. A) Deleción 1p36 en mosaico en un paciente con cardiopatía: detectada mediante aCGH (1) y confirmada en un 9,8% de las células analizadas por FISH. Solo una única señal en el núcleo de la fotografía, indicada por la flecha, corresponde a la región 1p36 (2). B) Duplicación 1q21.1-q32.1 en un paciente con epilepsia: detectada mediante aCGH (1), confirmada en un 1,2% de las células analizadas por FISH. Las tres señales inferiores del núcleo de la fotografía, a las que se dirige la flecha, corresponden a la región 1q21q32.1 (2) y cariotipo (3). C) Deleción 18q21.31-q22.2 en un paciente con RGD: detectada mediante aCGH(1) y confirmada en un 43,3% de las células analizadas por cariotipo (2).
Finalmente, tras revisar a los 140 pacientes cuyas anomalías fueron detectadas con aCGH, se concluyó que 43 (30,7%) habrían sido detectadas también por otras técnicas: 6 (4,2%) mediante cariotipo convencional, 6 (4,3%) mediante cariotipo y MLPA, y, finalmente, 31 (22,1%) mediante MLPA.
DiscusiónEste trabajo presenta la mayor serie de estudios de aCGH como primera elección en pacientes con diversas patologías descrita en nuestro país, ya que hasta ahora se habían publicado series de menor tamaño o utilizando los aCGH como técnica de tercera línea, en pacientes con resultado normal en otras técnicas de diagnóstico genético, tales como el cariotipo y la MLPA17.
Con un 14% de pacientes con anomalías detectadas (tabla 1), nuestros resultados confirman que actualmente el aCGH es la técnica individual con un mayor rendimiento diagnóstico en los pacientes con RGD/DI, MC y TEA5. El rendimiento diagnóstico no es uniforme y varía mucho en función del tipo y el número de anomalías fenotípicas que se observan en los pacientes (tablas 1 y 2), coincidiendo nuestros resultados con los estudios publicados por otros autores (referencias en la tabla 1).
Las indicaciones clásicas del estudio de aCGH incluyen RGD/DI, TEA y MC5, con unas tasas diagnósticas del 18,9, el 14,8 y el 13,7%, respectivamente. Nuestros resultados y los de otros autores19 indican la conveniencia de incluir la talla baja como nueva indicación del estudio del aCGH, ya que se ha detectado aproximadamente un 13,3% (14/105) de anomalías, de las cuales solo un 21,42% (3/14) afectan al gen SHOX. Nuestros resultados también señalan la conveniencia de incluir como indicación del estudio de aCGH las epilepsias aisladas, sin malformaciones del sistema nervioso central, con un 7% de diagnósticos.
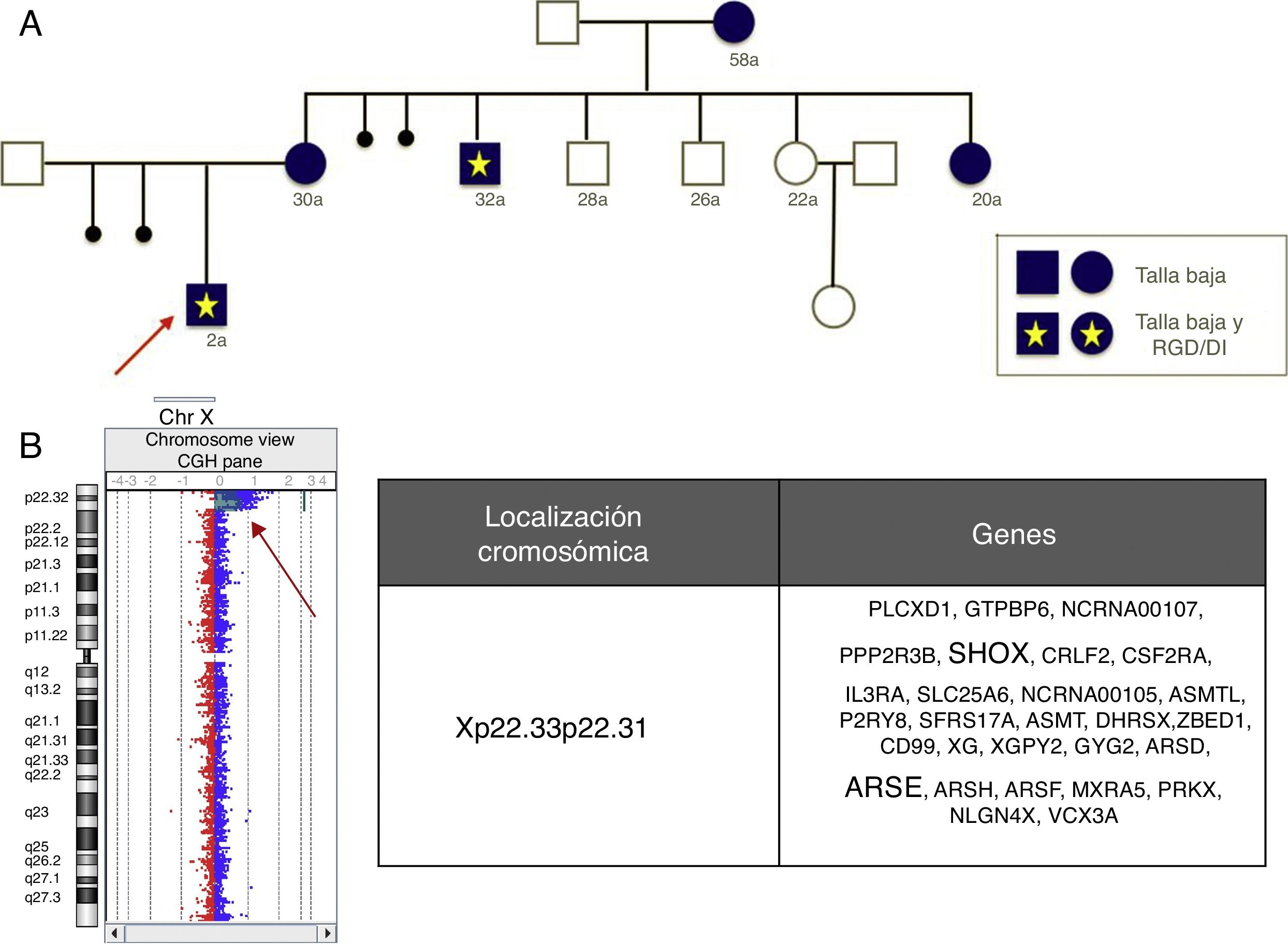
Como beneficio adicional, los estudios de aCGH permiten una caracterización de las anomalías más precisa que la FISH o la MLPA comercial, a veces con implicaciones de asesoramiento reproductivo importantes, por ejemplo, la figura 2 muestra una deleción que incluye el gen SHOX y otros genes no seudoautosómicos, como el ARSE, en portadoras con talla baja y varones afectados de un fenotipo mucho más grave incluyendo talla baja, condrodisplasia punctata y RGD/DI.
 Pedigrí de una familia portadora de una deleción Xp22.33-p22.31. B) Resultado del análisis mediante aCGH, que implica la pérdida de los genes SHOX y ARSE, principales responsables de las alteraciones fenotípicas.")
Los aCGH permiten el diagnóstico de síndromes de microdeleción con puntos de rotura atípicos que pueden ser invisibles por FISH, el método diagnóstico más extendido. En particular, nuestra serie ha permitido detectar un nuevo punto de rotura recurrente en la región crítica del síndrome de Williams que en los casos de deleción permite la hibridación parcial de las sondas de FISH comerciales más utilizadas, produciendo falsos negativos20.
Los aCGH detectan las ganancias y las pérdidas de material genómico con mucha más precisión que el cariotipo, pero son incapaces de detectar las anomalías equilibradas (principalmente translocaciones e inversiones) que en algún caso pueden ser causa de patología. Sin embargo, hay que tener en cuenta que estas representan solamente una muy pequeña proporción de las alteraciones clínicamente relevantes5,21.
Los estudios de aCGH indican la presencia de un mosaicismo cuando se observan valores intermedios de fluorescencia, si bien es difícil deducir el grado del mismo. En este sentido, nuestros resultados han sido dispares, ya que mientras se diagnosticaron fácilmente anomalías que solo se observaron en un 9,8 y un 1,2% de metafases analizadas (fig. 1 A y B), mosaicos observados en más de un 40% de las metafases provocaron cambios de fluorescencia tan sutiles que no fueron detectados por el programa de análisis Cytogenomics, aunque sí se visualizaban en el gráfico proporcionado por el mismo (fig. 1 C). Una posible explicación es que las diversas técnicas analizan poblaciones celulares diferentes: linfocitos T procedentes de cultivos celulares estimulados con fitohemaglutinina en los estudios de cariotipo/FISH y todas las células sanguíneas nucleadas en los aCGH.
La diferencia entre la tasa diagnóstica en función de las diversas indicaciones puede justificar las disparidad de los resultados publicados, con tasas diagnósticas tan extremas como el 7,6% de Shen et al.22 y el 35% de Aradhya et al.23. Ambos estudios analizan el genoma con una resolución similar (35 Kb) pero difieren en la población analizada: poco definida fenotípicamente en el primer estudio y pacientes seleccionados por presentar un fenotipo considerado muy indicativo de tener una anomalía de copia (CNV) en el segundo. Las grandes series publicadas utilizando la técnica de aCGH como primera opción diagnóstica, como por ejemplo las de Ahn et al.24 con 8.300 pacientes y Shaffer et al.6 con 8.789 pacientes, aportan la detección de un gran número de CNV patogénicas pero detallan poco el fenotipo de las mismos. Por otro lado, las series de pacientes bien caracterizadas fenotípicamente frecuentemente son de pequeño tamaño y focalizadas en un tipo concreto de patología, como por ejemplo la de Roselló et al.25, con 246 pacientes con DI. Nuestra serie aporta los resultados del estudio de aCGH como primera opción diagnóstica en 1.000 pacientes con una detallada clasificación fenotípica.
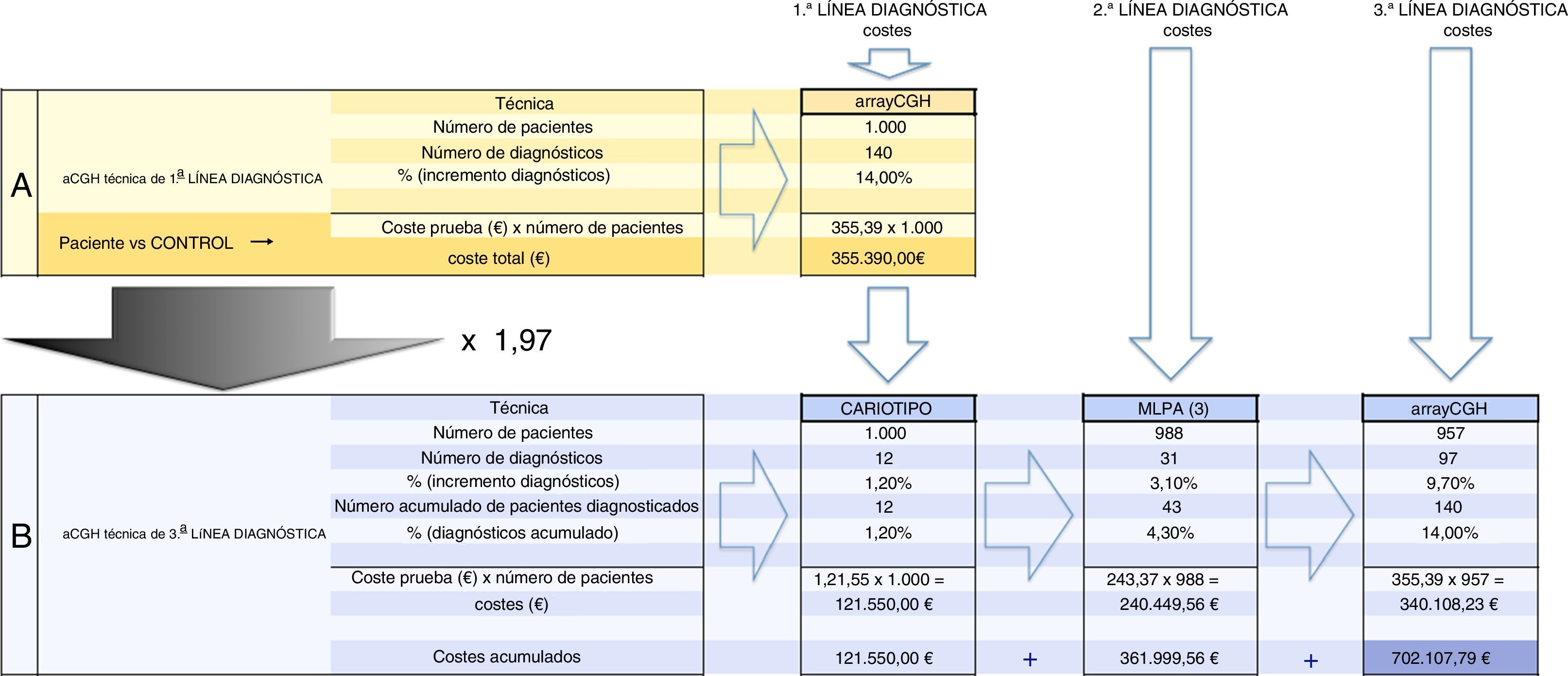
En consecuencia, el presente estudio permite contrastar con fiabilidad en términos de efectividad y eficiencia la estrategia del aCGH como primera opción diagnóstica en la detección de desequilibrios genómicos frente a la estrategia estándar de cariotipo, estudio de MLPA y la adición del estudio de aCGH como opción de tercera línea (fig. 2). La comparación de las series publicadas de pacientes estudiados con cariotipo, FISH, MLPA o aCGH es complicada ya que analizan poblaciones diferentes. El presente estudio permite una aproximación basada en una población única, asumiendo que el aCGH detecta la casi totalidad de las anomalías clínicamente relevantes. La estrategia tradicional de estudio de cariotipo y MLPA disponibles para estas patologías (2 kits de trastornos genómicos recurrentes y uno de anomalías subteloméricas) habría diagnosticado solo 43 (30,7%) de los 140 pacientes diagnosticados por aCGH a un coste aproximado de 362.000 € (1.000 cariotipos + 988 MLPA), al que habría que añadir el coste de 957 aCGH para llegar a diagnosticar los 140 pacientes, con un coste global de 702.107 € (fig. 3). El estudio de los mismos pacientes únicamente por aCGH sería de unos 355.390 €. En resumen, el enfoque tradicional de MLPA y cariotipo hubiera logrado menos de un tercio de los diagnósticos en nuestra población, y se alcanzaría la detección del 100% de los desequilibrios detectables por aCGH multiplicando el gasto por 1,97.
; 31 se habrían diagnosticado mediante MLPA con ensayos de anomalías subteloméricas y de síndromes genómicos recurrentes, y los 97 casos restantes se habrían diagnosticado mediante aCGH, después de un resultado normal de cariotipo y MLPA. Costes16: cariotipo 121,55 €/ensayo; MLPA 243,37 €/3ensayos MLPA; aCGH 355,39 €/ensayo. El enfoque tradicional de cariotipo y MLPA (estrategia B) hubiera conseguido menos de un tercio de los diagnósticos en nuestra población (43/140). Implementar el array para lograr el 100% de los desequilibrios multiplica por 1,97 el coste que supondría aplicar el array como primera y única opción (estrategia A).")
Cálculo de los costes para cada una de las dos estrategias diagnósticas. A: cálculo del coste del uso del aCGH como primera opción diagnóstica. B: cálculo de los costes del uso del aCGH como tercera opción, tras el cariotipo y la MLPA. De los 140 casos diagnosticados mediante aCGH, 12 se hubieran diagnosticado mediante cariotipo (de los cuales 2 eran aneuploidías y 10 desequilibrios de más de 6Mb); 31 se habrían diagnosticado mediante MLPA con ensayos de anomalías subteloméricas y de síndromes genómicos recurrentes, y los 97 casos restantes se habrían diagnosticado mediante aCGH, después de un resultado normal de cariotipo y MLPA. Costes16: cariotipo 121,55 €/ensayo; MLPA 243,37 €/3ensayos MLPA; aCGH 355,39 €/ensayo.
El enfoque tradicional de cariotipo y MLPA (estrategia B) hubiera conseguido menos de un tercio de los diagnósticos en nuestra población (43/140). Implementar el array para lograr el 100% de los desequilibrios multiplica por 1,97 el coste que supondría aplicar el array como primera y única opción (estrategia A).
Nuestros resultados ilustran la efectividad y la eficiencia de la utilización de los aCGH como primera opción en el diagnóstico genético de los pacientes con RGD/DI, MC, TEA, talla baja y epilepsia. Estos resultados y el análisis coste-beneficio avalan su inclusión dentro del Sistema Nacional de Salud en complementación y sustitución de las técnicas de cariotipo, MLPA y FISH.
La evolución en aCGH es continua y recientemente han aparecido nuevos diseños de array que incluyen sondas backbone y sondas dirigidas según consorcio ISCA, y además garantizan una cobertura a nivel de exón de un número significativo de genes. En este nuevo contexto se necesitan estudios adicionales para reevaluar el coste-beneficio entre las plataformas de aCGH 8×60K y 4×180K.
FinanciaciónEste trabajo ha sido subvencionado parcialmente con un proyecto del Fondo de Investigaciones Sanitarias del Ministerio de Sanidad y Consumo de España (PS09/00632).
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A los familiares de nuestros pacientes y a nuestros pacientes.
Parte de este trabajo ha sido objeto de parte de la tesis doctoral de Neus Castells Sarret, defendida el 1 de diciembre del 2015 en el Hospital Universitario Vall d’Hebron, Universitat Autònoma de Barcelona. Título: Array CGH como primera opción en el diagnóstico genético postnatal.
Este trabajo ha sido objeto de una presentación tipo póster en el XXVIII Congreso Nacional de la Asociación Española de Genética Humana (AEGH), 13-15 mayo del 2015. Título: 1.000 estudios de aCGH como técnica de primera línea: la experiencia del Hospital Vall d’Hebron.
Parte de este trabajo ha sido objeto de la publicación del artículo: A novel recurrent breakpoint responsible for rearrangements in the Williams-Beuren region. Cytogenet Genome Res. 2015;146(3):181-186.
- Documento de manejo clínico del paciente pediátrico con infección por SARS-CoV-2.
(Actualizado el 26 de Noviembre de 2020) - Test de diagnóstico rápido en las consultas de Pediatría de Atención Primaria y Urgencias Pediátricas en la era COVID-19: más que una recomendación
(Actualizado el 21 de septiembre de 2020) - Consenso nacional sobre diagnóstico, estabilización y tratamiento del Síndrome Inflamatorio Multisistémico Pediátrico vinculado a SARS-CoV-2 (SIM-PedS).
(Actualizado el 27 de Julio de 2020) - Recomendaciones para el manejo del recién nacido en relación con la infección por SARS-CoV-2.
(Actualizado el 27 de Mayo de 2020) - Manejo del paciente pediátrico ante sospecha de infección por el nuevo coronavirus SARS-CoV-2 en atención primaria (COVID-19). AEPap-SEIP/AEP-SEPEAP.
(Actualizado el 27 de Abril de 2020) - Propuesta de adaptación de las recomendaciones de reanimación cardiopulmonar pediátrica avanzada a la infección por coronavirus.
(Actualizado el 11 de Abril de 2020)

- Laringitis aguda en neonato por VHS tipo 2
- Ingresos COVID-19: intentando comprender el impacto real de la infección en pacientes hospitalizados
- Úlcera de Lipschütz como posible manifestación del SARS-CoV-2
- Neurodesarrollo a los 2 años en cardiopatía congénita: utilidad pronóstica de los marcadores de daño cerebral

Anales de Pediatría sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas