

El síndrome de Loeys-Dietz es una enfermedad autosómica dominante del tejido conectivo, causado por mutaciones en los genes TGFBR1, TGFBR2, SMAD3 y TGFB21. Se caracteriza típicamente por aneurismas y tortuosidad arterial, hipertelorismo y úvula bífida o paladar hendido. Lo debemos sospechar ante un paciente con fenotipo marfanoide con las características anteriores2.
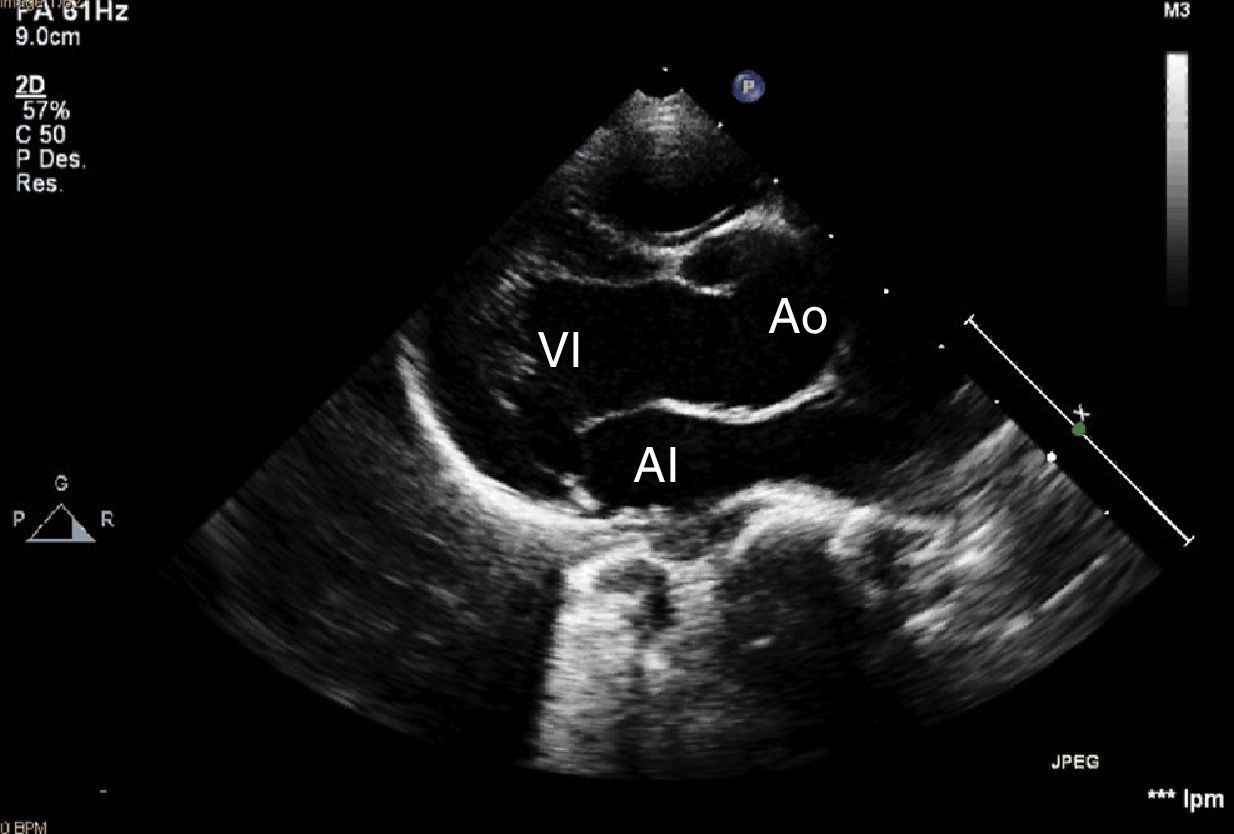
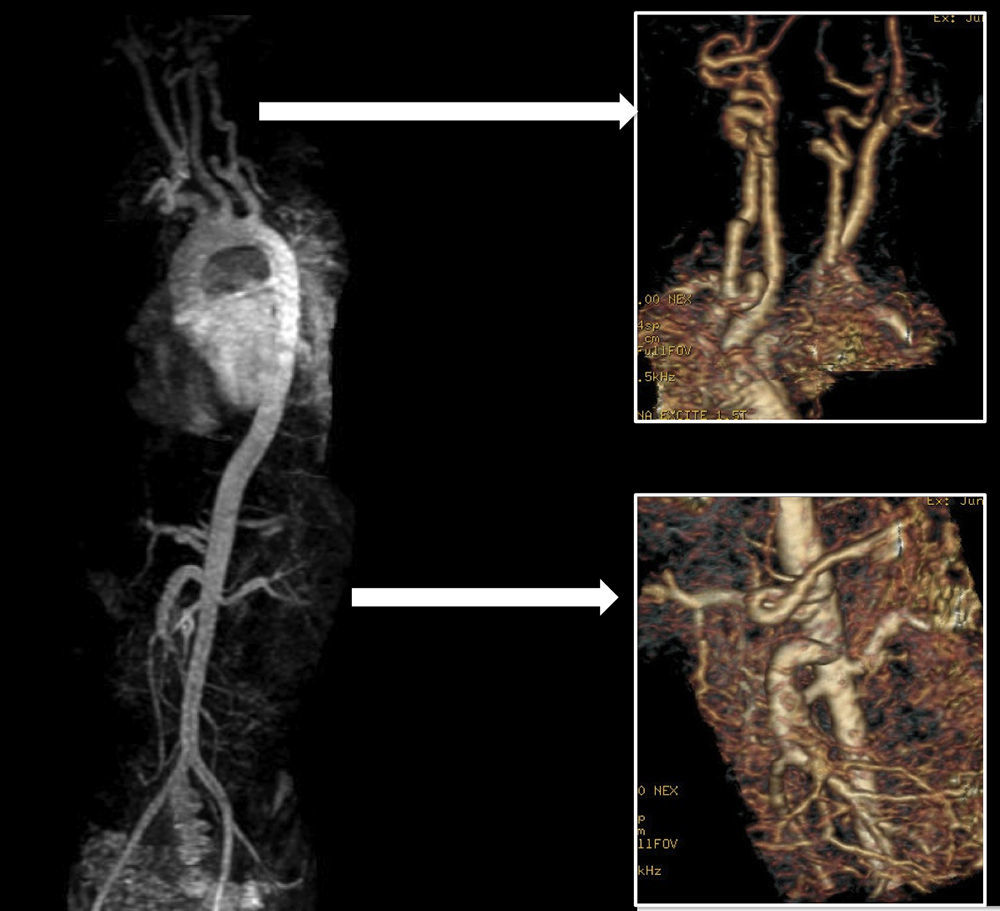
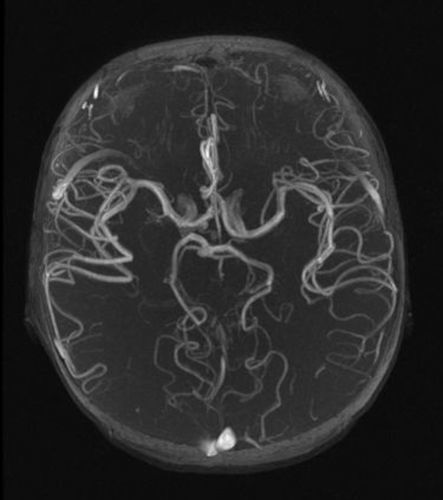
Presentamos el caso de un paciente de 2 años de edad, sin antecedentes familiares de interés, derivado para estudio por fenotipo dismórfico con hipertelorismo, craneosinostosis, hendidura palatina e hiperlaxitud articular en el que, al realizar el estudio cardiológico se evidenció dilatación de la raíz aórtica (25mm, Z-score +4) (fig. 1). En estudio genético se confirma mutación en TGFBR2, por lo que se decide ampliar estudio cardiovascular mediante angio-resonancia magnética donde se evidencia elongación y aumento de calibre generalizado en los trayectos vasculares, típico de esta entidad (figs. 2 y 3).
 en ecocardiografía realizada en paciente con síndrome de Loeys-Dietz. AI: aurícula izquierda; Ao: raíz aórtica;VI: ventrículo izquierdo.")


La afectación cardiovascular en el síndrome de Loeys-Dietz es muy frecuente. Los pacientes afectados tienen alto riesgo de disección o rotura aórtica a edades muy tempranas, incluso con diámetros aórticos no muy dilatados1,3. Se recomienda iniciar tratamiento con betabloqueante o con antagonistas de los receptores de la angiotensina, además de realizar estudios periódicos de imagen que incluyan aorta y ramas. Una de las dificultades para detectar la enfermedad es que se confunde con el síndrome de Marfan, pero con peor pronóstico, ya que los pacientes fallecen a edades más tempranas. El diagnóstico precoz es fundamental, ya que un tratamiento quirúrgico adecuado podría disminuir las complicaciones1,2.

