

Introducción
Las observaciones disponibles hoy en día sobre las anomalías caracterizadas por hipercrecimiento o velocidad de crecimiento excesiva, complementan las obtenidas en pacientes con hipocrecimiento armónico, ilustrando la complejidad del crecimiento humano y el amplio número de genes y factores reguladores que intervienen en el desarrollo de un crecimiento normal y proporcionado.
En la tabla 1 se refleja la clasificación etiológica del hipercrecimiento, de acuerdo con los principales factores que regulan el crecimiento (genéticos, nutricionales y hormonales). La mayoría de estos cuadros clínicos son primarios. No obstante, existen casos clínicos aislados de macrosomía que aún son difíciles de clasificar y, por consiguiente, no están incluidos.

En la figura 1 se refleja un algoritmo que puede ser de ayuda en el planteamiento diagnóstico de los pacientes con talla alta.

Figura 1. Algoritmo-hipercrecimiento.
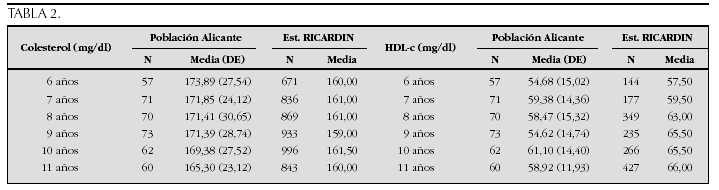
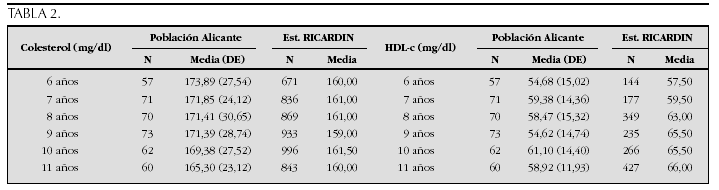
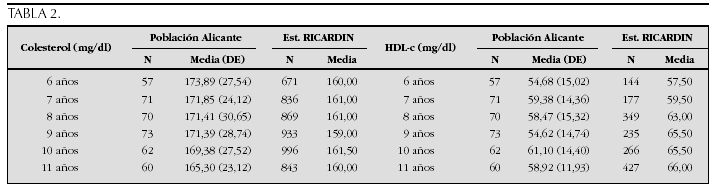
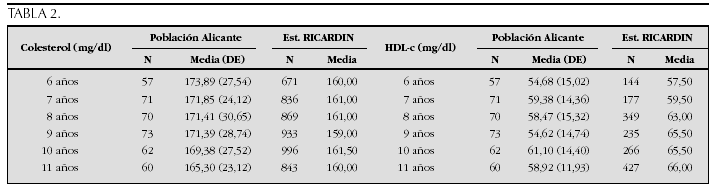
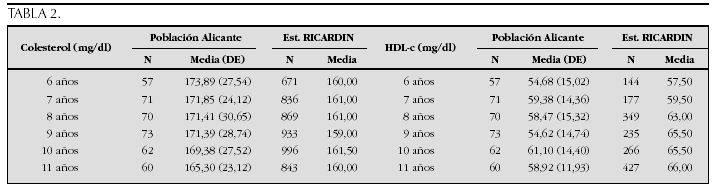
La etiopatogenia y bases moleculares de las anomalías que cursan con hipercrecimiento son complejas y parcialmente conocidas (tabla 2).

Datos recientes han incluido diferentes factores, previamente desconocidos, que resultan importantes para lograr un crecimiento lineal proporcionado. Cuando se analizan los casos disponibles, puede apreciarse un patrón parecido en diversos cuadros clínicos de hipercrecimiento.
Genes "extra" de crecimiento
Trisomía X (47,XXX), Síndrome de Klinefelter (XXY) y Varones 47,XYY
Trisomía X (mujeres 47, XXX)
Las pacientes con trisomía X tienden a ser altas. Las mujeres adolescentes y adultas 47,XXX son altas, con talla generalmente en el percentil 90 o superior (171 cm). La trisomía X es la anomalía cromosómica más frecuente, presentando una incidencia de 1:1000 recién nacidos de sexo femenino; 2-3 veces más frecuente que el síndrome de Turner. La causa que explica la existencia de talla alta es, probablemente, la presencia de un cromosoma X extra. En efecto, la existencia de cromosomas X adicionales parece influenciar la talla. Las correlaciones cariotipo-fenotipo han demostrado que los pacientes con fórmula cromosómica 45,X, síndrome de Turner, o con una deleción de uno de sus brazos cortos, 46,XisoXq, son uniformemente bajos. No obstante, se estima que el 20 % de los pacientes 45,X/46,XX y el 50 % de los sujetos con 45,X/47,XXX presentan una talla normal. Parece, por consiguiente, que la presencia de 2 o 3 cromosomas X en algunas células, conduciría a que el cromosoma X adicional compensase la pérdida de un X o el brazo corto de un X (Xp) en otras células. Además, cuando todas las células presentan 3 cromosomas X, como ocurre en la trisomía X, el 60 % de los pacientes muestran una talla por encima del percentil 75. Cada célula posee únicamente un cromosoma X activo. Los cromosomas X adicionales son inactivados y condensados en el cuerpo de Barr. Si todos los genes en el crtomosoma X inactivo permanecen inactivos, la ausencia de un X no tendrá expresión fenotípica.
Existen evidencias genéticas y citogenéticas de que la región pseudoautosómica (PAR1), así como otros genes en el brazo corto y en el brazo largo permanecen activos en un cromosoma X inactivo normal. Asimismo, existen algunas evidencias que sugieren que la deleción en el extremo de Xp con ausencia de SHOX, podría generar talla baja y que un cromosoma X inactivo adicional, como acontece en el síndrome de Klinefelter y en la trisomía X, con genes de crecimiento activos (SHOX), podría causar talla alta.
Síndrome de Klinefelter XXY
En 1959, se publicó por primera vez la fórmula cromosómica 47,XXY. Todos estos pacientes tienen en común la presencia de al menos 2 cromosomas X y un cromosoma Y, excepto algunas excepciones con 46,XX.
Las anomalías cromosómicas al nacimiento muestran una incidencia en torno a 1:500 a 1:1000. Como se indicó a propósito del síndrome de trisomía X, los genes activos en el cromosoma X inactivo pueden ser responsables de las anomalías encontradas en el síndrome de Klinefelter y, particularmente, los genes extra relacionados con el crecimiento, como el caso del gen SHOX, probablemente responsable de la talla alta.
Los pacientes con síndrome de Klinefelter con un isocromosoma Xq (47,XiXqY), en los que existe ausencia de un brazo corto extra del segundo cromosoma X y, consiguientemente del gen SHOX, muestran las manifestaciones clínicas características del síndrome de Klinefelter, salvo la talla alta.
Menos de un 10 % del número estimado de fetos afectos se detectan prenatalmente y, aproximadamente, el 75 % de los pacientes vivirán sin ser diagnosticados.
Los varones 47,XYY y 47,XYY presentan talla alta. La primera comunicación de un varón XYY se efectuó en 1961, cuando se estudió un varón, esencialmente normal, fértil, de inteligencia media, investigado por tener una hija con síndrome de Down. Posteriormente, se detectó un incremento en la prevalencia de este cariotipo XYY (20/1000 vs 1/1000 en la población general) entre los varones altos, con retraso mental y encarcelados, creándose un estereotipo para estos individuos afectos de padecer alteraciones de conducta, con agresividad física y violencia (1 % de riesgo vs 0,1 % de riesgo en varones 46,XY). No obstante, esta impresión no puede extenderse a todos los pacientes. El único factor físico consistente de este síndrome es la talla alta, situándose un 50 % de los pacientes por encima del percentil 90 en relación a la población normal.
Los varones XYY son más altos que los varones XY. Dicha afirmación está probablemente determinada por la existencia de un cromosoma Y extra.
Una región de gran importancia en el cxrecimiento y del tamaño de los dientes quedó cartografiada por diferentes investigadores en la porción más proximal del brazo largo del cromosoma Y, cercana al centrómero, denominándose "Control del crecimiento en el cromosoma Y" Growth control in the Y (GCY) o gen específico del crecimiento en el cromosoma Y. Este gen GCY podría ser el responsable de la diferencia de la longitud y tamaño de los dientes observada entre varones y mujeres.
En un estudio realizado en ocho varones XYY no seleccionados que fueron seguidos longitudinalmente, se demostró que eran más altos que los controles durante la infancia y al comienzo de la pubertad. La talla adulta fue de 188,1 cm (lo que supone el doble de la diferencia existente entre varones y mujeres (163,8 cm para mujeres y 176,5 cm para los varones). Las observaciones sugieren que la talla alta en los varones 47,XYY está relacionada con la existencia de genes extra de crecimiento en el cromosoma Y adicional: SHOX, GCY o gen del crecimiento específico del cromosoma Y.
Secreción excesiva de hormona de crecimiento
Gigantismo hipofisario y acromegalia
Ambos, gigantismo y acromegalia, son la resultante de una secreción excesiva de GH, iniciándose a partir de un adenoma eosinófilo o cromófobo, o a partir de hiperplasia de la hipófisis anterior.
El gigantismo es una entidad nosológica muy rara. La acromegalia es más frecuente, con una incidencia estimada de 3/millón/año y una prevalencia de 40-70 por millón de habitantes. Como quiera que el 10 % aproximadamente de los acromegálicos son altos, es obvio que el comienzo de la enfermedad precede a la fusión de las epífisis en la segunda década de la vida. Cabe estimar que la prevalencia de adenomas secretores de hormona de crecimiento en la adolescencia se sitúe en torno a 3 a 7 por millón. No obstante, el diagnóstico de la enfermedad suele ser más tardío. Este diagnóstico se basa en la demostración de la secreción aumentada de GH, así como por la presencia de un adenoma hipofisario o hiperplasia hipofisaria con alargamiento de la silla turca. El test definitivo para el diagnóstico de la secreción excesiva de GH consiste en la imposibilidad de reducir los niveles séricos de GH a menos de 1 ng/ml tras la realización de una prueba de sobrecarga oral de glucose (1,75 g/-g [máximo 75-100 g]). Hay que tener en consideración que los sujetos normales disminuyen la concentración sérica de GH a menos de 1 ng/ml. Los niveles séricos de IGF-1 es una prueba de "screening", encontrándose elevados entre 4-10 veces por encima de lo normal.
La cirugía transesfenoidal, en manos de un neurocirujano experto, es el tratamiento de elección. Si la secreción de GH no se normaliza tras la práctica de la cirugía, las opciones incluyen: radiación hipofisaria y tratamiento médico con octreótido (un análogo de amplia actividad de somatostatina [SMS201-995]) o bromocriptina, o ambos.
Un nuevo tratamiento, consiste en el empleo de pegvisomanto, un antagonista del receptor de GH, que compite con la GH nativa por el receptor de GH, previniendo su dimerización functional (un proceso que es prioritario para la traducción de la señal de GH y la síntesis y secreción de IGF-1).
Esta droga normaliza las concentraciones séricas de IGF-1 en más del 90 % de los pacientes, mejora la resistencia a la insulina, disminuye los niveles de glucemia en ayunas, mejorando el control diabético y, con la normalización de los niveles de IGF-1, induce una reducción rápida de los marcadores de formación y resorción ósea. En los pacientes con acromegalia, la dosis de adulto es de 10 a 30 mg, administrada de forma subcutánea una vez al día. Este tratamiento puede ser de utilidad como coadyuvante en los pacientes quienes continúan produciendo cantidades excesivas de GH y presentan niveles elevados de IGF-1 tras la cirugía o la radiación hipofisaria. La experiencia es limitada, siendo necesarios estudios a largo plazo para determinar su seguridad en relación al crecimiento del tumor hipofisario y la toxicidad hepática.
El pegvisomanto es un análogo de GH, que ha sido estructuralmente alterado para actuar como un antagonista del receptor de GH. El pegvisomanto es una proteína de ADN recombinante que contiene 191 aminoácidos a los que varios polímeros de polietinilglicol (PEG) pueden unirse covalentemente (predominantemente de 4-6 PEG/molécula de proteína).
Factores de crecimiento extra Insulina IGF-1, IGF-2
Hiperinsulinismo Lipodistrofia
La lipodistrofia incluye un grupo infrecuente de anomalías caracterizadas por la ausencia generalizada o parcial de tejido adiposo, resistencia a la insulina, hiperinsulinemia, hiperlipidemia y diabetes mellitus no cetósica. Se han identificado algunas mutaciones en genes específicos en formas de lipodistrofia generalizada transmitidas según un patron mendeliano autosómico recesivo BSCL-1 (9q34.3) y BSCL-2 (11q13); en formas parciales de lipodistrofia (tipo Dunnigan) LMNA (1q21.2) y una variante de lipodistrofia parcial familiar (brazos, cara y cuello) en el gen PPAR (tabla 3). Algunas observaciones recientes efectuadas en ratones transgénicos y en humanos, han demostrado que la resistencia a la insulina, hiperinsulinemia y diabetes mellitus son el resultado de la ausencia de grasa y la consiguiente deficiencia de leptina y adiponectina, péptidos que pueden influenciar la sensibilidad a la insulina y el balance energético. En efecto, existen evidencias que demuestran que la leptina desempeña una función relevante en la regulación de la ingesta, el gasto de energía y la función neuroendocrinológica.

Lipodistrofia congénita de Berardinelli-Seip (BSCL)
Su prevalencia se ha estimado en menos de 1 caso por millón de habitantes.
Los pacientes con lipodistrofia generalizada congenita autosómica recesiva presentan gigantismo, velocidad de crecimiento acelerada, elementos acromegaloides, grandes manos, pies y pabellones auriculares, mandíbula prominente y edad ósea acelerada. Estos pacientes pueden mostrar macrosomía en el momento del nacimiento o gigantismo postnatal.
La pérdida de la grasa subcutánea es la primera manifestación al nacimiento o en la lactancia y, gradualmente, con el paso de los años, el desarrollo completo de las anomalías descritas. La ausencia generalizada de grasa afecta al tejido subcutáneao de la cara, tronco y extremidades superiores, así como perirrenal y retroperitoneal, entre otras áreas. Aunque parece que la grasa se encuentra ausente a la inspección, los adipocitos están presentes, si bien parecen inmaduros y contienen escasa cantidad de lípidos intracelulares.
Aún se desconoce si la ausencia de grasa es primaria o secundaria a una disfunción hipotalámica. Recientemente, Agarwal et al (2002) han detectado mutaciones en el gen 9q34.3, que codifica 1-acilglicerol-3-fosfato 0-aciltransferasa 2 (AGPAT2), causantes del síndrome de lipodistrofia congénita de Berardinelli-Seip (BSCL1). La enzima AGPAT2 cataliza una reacción esencial en la vía de biosíntesis de glicerofosfolípidos y triacilglicerol, afectando a la síntesis de triacilglicerol en el tejido adiposo. Las mutaciones de este gen pueden causar lipodistrofia mediante la deplección de triglicéridos de los adipocitos. Magre et al (2001) han encontrado en varias familias mutaciones en el gen BSCL2 (11q13) que codifica una proteína denominada "seipina", una proteína de 398 aminoácidos que podría ser una proteína transmembrane. El gen BSCL2 contiene 12 exones. Magre et al han demostrado que la expression más intensa de este gen se produce en el cerebro, en la mayoría de las regiones del sistema nervioso central; por el contrario, la expresión más débil se produce en adipocitos, sugiriendo una alteración primaria del eje hipotálamo-hipofisario. Por consiguiente, es posible que la distrofia generalizada congénita sea causada por alteraciones en diferentes vías en el adipocito o en el eje hipotálamo-hipofisario.
En ausencia de depósitos de tejido adiposo, el exceso de lípidos se acumula en tejidos no adiposos, tales como los hepatocitos, el esqueleto, las células musculares cardíacas y las células b -pancreáticas. Estas células están diseñadas para almacenar grasa en situaciones extraordinarias. Los triglicéridos intramiocelulares y la esteatosis hepatica se encuentran asociados a resistencia a la insulina por razones aún no bien establecidas; si bien, posiblemente, podría estar relacionado con los ácidos grasos.
Todos los pacientes con lipodistrofia presentan niveles muy disminuidos de leptina y adiponectina, así como grados variables de resistencia a la insulina. En los últimos años, se ha demostrado que la resistencia a la insulina, la hiperinsulinemia, la diabetes mellitus y la hiperlipidemia son debidos a la ausencia de tejido adiposo y, consiguientemente, deficiencia de leptina y adiponectina. Yamauchi et al (2001) demostraron que la resistencia a la insulina, en ratones con lipodistrofia gteneralizada, podría revertirse mediante la administración sistémica de leptina y adiponectina combinadas, pero solo parcialmente si se administra leptina o adiponectina de forma aislada.
De forma similar en el ser humano, Oral et al (2002) demostraron una gran mejoría en el control de la glucemia, hipertrigliceridemia y esteatosis hepática en pacientes tratados con leptina recombinante (0,04 to 0,08 mg/kg/día) durante 4 meses. No obstante, la mejoría, aunque manifiesta, no fue completa. Es posible que la solución completa de las anomalías metabólicas pudiera producirse mediante la administración simultánea de leptina y adiponectina, como ocurre en el ratón, por lo que se requiere la realización de este experimento.
La mejoría en la acción de la insulina se ha asociado con una marcada reducción en el contenido de triglicéridos hepático y muscular. La leptina estimula la oxidación de los ácidos grasos y previene el acúmulo de lípidos en tejidos no adiposos. Estas acciones mejorarían la resistencia a la insulina. La acción de la leptina es mediada tanto periféricamente (directamente sobre el músculo), como centralmente (a través del eje del hipotálamo y el sistema nervioso simpatico).
Obesidad
Los niños obesos tienden a incrementar su velocidad de crecimiento, a ser más altos que los niños de su edad que son delgados y a presentar maduración ósea esquelética avanzada. No obstante, aunque los niños obesos prepuberales obesos son más altos que los niños de su edad no obesos, no alcanzan una talla adulta excesiva.
Recientemente, se ha comunicado que las gráficas de crecimiento para los niños obesos hasta la edad de 13 años y de niñas hasta la edad de 12,5 años, demuestran que éstos, son más altos que los niños y niñas de edades similares y no obesos hasta esas edades. Aunque los niños obesos presentan aceleración en su edad ósea, muestran, asimismo, un acortado estirón de crecimiento durante la pubertad, lo que les conduce a obtener una talla adulta normal.
La talla alta, asociada a velocidad de crecimiento incrementada, es mediada, probablemente, por la insulina e IGF-1. Los niños obesos muestran niveles séricos normales o elevados de IGF-1, pese a presentar concentraciones de GH prácticamente suprimidas, comparado con los individuos de peso normal. Es posible que la insulina y el IGF-1 (y la testosterona en algunas niñas) contribuyan a incrementar la velocidad de crecimiento, la masa magra y la maduración ósea, asociadas con sobrenutrición.
Deficiencia de factores necesarios para prevenir la elongación de los huesos
Síndrome de Marfan
Consiste en una alteración del tejido conectivo que afecta al esqueleto humano, con elongación de los huesos tubulares (dolicomorfismo), el sistema cardiovascular y el sistema ocular (subluxación del cristalino).
Se transmite siguiendo un patrón mendeliano autosómico dominante. Aproximadamente el 85 % de los pacientes presentan antecedentes familiares y, en el 15 % de ellos, la presentación es esporádica. Hoy sabemos que este síndrome es debido a una anomalía en el gen de la fibrilina 1 (FBN1) cartografiado en el cromosoma 15 (15q21.1).
Alteraciones de los genes relacionados con la regulación del ciclo celular, crecimiento y supresión tumoral
Síndrome de Sotos
Descrito en 1964, está conformado por talla alta, gran dolicocefalia, configuración facial característica (100 %), edad ósea avanzada (84 %) y anomalías neurológicas no progresivas con retraso mental. La prevalencia es desconocida, aunque se trata, probablemente, de uno de los síndromes más frecuentes de hipercrecimiento, después del síndrome de Marfan y el síndrome de Williams-Beuren.
El hallazgo clínico fundamental es el hipercrecimiento prenatal y postnatal. La talla adulta en varones se sitúan en torno a 184,3 ± 6,0 cm y en mujeres en torno a 172,9 ± 5,7 cm, lo que supone un incremento respecto de la talla genética de 11 y 6 cm, respectivamente. No obstante, se han descrito tallas adultas más elevadas: de 193 cm a 203 cm en varones y hasta 188 cm en mujeres.
La configuración cráneofacial es muy característica: frente prominente, disminución de la línea del cabello fronto-pariental, gran dolicocefalia, hipertelorismo, inclinación antimongoloide de los párpados, paladar ojival y estrecho, palatino prominente serrado y mentón prominente. Aproximadamente, el 60-80 % de los pacientes presentan una erupción prematura de sus dientes.
Las manifestaciones del sistema nervioso central son frecuentes. El retraso en las adquisiciones del desarrollo, para caminar, hablar y, en particular el lenguaje, está casi siempre presente y la torpeza es frecuente (60-80 % de los casos), como lo es la hipotonía y la laxitud articular. La deficiencia mental está presente en el 80-85 % de los casos, exhibiendo un coeficiente intelectual de 72 y un rango entre 40 y límite de la normalidad. Las convulsiones pueden estar presentes en un 30 % de los casos. Existe una incidencia incrementada de tumores cerebrales (2,2-3,9 %). La presencia de ventrículos cerebrales ligeramente alargados y la presencia de espacios subaracnoideos aumentados, son hallazgos frecuentes que, habitualmente, no requieren ningún tipo de tratamiento.
Otras anomalías que pueden presentarse, son las siguientes: defectos septales cardíacos, cifoescoliosis, alteraciones urogenitales (hidronefrosis, riñones hipoplásicos, agenesia renal, dilatación de la pelvis renal), hernias inguinales, alteraciones oftalmológicas (estrabismo, anomalías del nervio óptico o de la retina), alteraciones endocrinológicas (hipotiroidismo primario, entre otras). No obstante, no se han descrito alteraciones endocrinológicas capaces de explicar el rápido crecimiento de estos pacientes.
La existencia de una causa genética se ha sospechado desde hace mucho tiempo, habiéndose descrito varias familias con miembros afectos en dos on tres generaciones, sugiriendo un patrón de herencia autosómico dominante. No obstante, la mayoría de los casos son esporádicos, aunque podrían ser debidos a nuevas mutaciones (afectando preferentemente al cromosoma paterno).
Recientemente, se han descrito mutaciones en el gen NSD1 (5q35) en el 60-75 % de casos esporádicos, indicando que la haploinsuficiencia del gen NSD1 es la causa más importante de síndrome de Sotos (Kurotaki, 2002). No obstante, es posible que existan otras anomalías genéticas. El hallazgo de que las mutaciones en el gen NSD1, tanto heterocigotas como hemicigotas, es consistente con un patrón autosómico dominante en la mayoría de los casos de síndrome de Sotos. Hay que indicar que existen diferencias en el expectro de las mutaciones del gen NSD1 demostradas hasta la fecha entre los pacientes japoneses y los no japoneses. En efecto, la incidencia de deleciones en este gen es más elevada en los pacientes japoneses, mientras que las mutaciones intragénicas son más communes en pacientes no japoneses.
El gen humano NSD1 consta de 23 exones, codifica una proteína de 2696 aminoácidos y se expresa en el cerebro fetal humano, lo que podría explicar el gran cerebro y la deficiencia mental en estos pacientes. También se expresa en el músculo esquelético, riñón, bazo, leucocitos periféricos y timo. La proteína a la que da lugar este gen interactúa con un dominio de unión a receptores hormonales nucleares, pudiendo actuar como co-represor o como co-activador. El hallazgo de que la haploinsuficiencia del gen NSD1 induce hipercrecimiento en el síndrome de Sotos, implica que el gen NSD1 actúa como co-represor de genes que promueven el crecimiento.
Todos los ratones "knock-out" homocigotos para el gen NSD1 fallecen antes del día embrionario 10,5. Por consiguiente, se considera que el gen NSD1 es crucial para el desarrollo postimplantación. La infertilidad, el aborto y los niños nacidos muertos, podrían ser una explicación para el bajo número de casos familiares.
No existe ningún marcador bioquímico para este síndrome. El diagnóstico se basa, pereferentemente, en sus aspectos clínicos. Las manifestaciones clínicas más características son la configuración cráneofacial y el crecimiento excesivo. Sin estos datos, no puede efectuarse el diagnóstico.
La correlación genotipo-fenotipo en 88 pacientes con síndrome de Sotos queda reflejada en la tabla 4.

El tratamiento de estos pacientes debe dirigirse fundamentalmente a la deficiencia mental y a la talla alta, particularmente en niñas, las dificultades sociales y de conducta durante la infancia y la inmadurez en el adulto, así como la posibilidad del desarrollo de un tumor cerebral y el riesgo de transmisión. Los individuos afectos son fértiles, no existiendo ninguna evidencia de que su esperanza de vida esté acortada.
Síndrome de Weaver
Fue descrito en 1974. Se caracteriza por hipercrecimiento prenatal y postnatal, facies inusual, maduración ósea avanzada y camptodactilia. Se han comunicado al menos 37 casos (21 varones y 16 mujeres).
Los varones alcanzan una talla adulta entorno a 194,2 cm, un peso de 102,2 kg y un perímetro craneal de 61 cm. Las mujeres alcanzan una talla adulta alrededor de 176,3 cm, un peso de 87,6 kg and y un perímetro craneal de 59,5 cm.
El hipercrecimiento está presente al nacimiento o se inicia durante la lactancia. Aproximadamente el 80 % de los pacientes están retrasados desde el punto de vista del desarrollo o tienen retraso mental. El retraso mental puede ser desde ligero hasta severo. Muchos de estos elementos se asemejan al síndrome de Sotos. Las características cráneofaciales, aunque algo similares, son algo diferentes. En efecto, los pacientes con síndrome de Weaver presentan hipertelorismo, pabellones auriculares grandes, puente nasal deprimido, inclinación antimongoloide de los párpados, pero el cráneo, en muchos casos, no es dolicocéfalo (el occipucio es, a menudo, plano) y no tienen mentón prominente y la cara es ancha.
Otro elemento característico es que, en muchos casos, existe hipertonía más que hipotonía. Es muy llamativo la existencia de una maduración ósea avanzada, con una madurez mucho más acelerada en el carpo que en la mano. Los huesos largos están ensanchados o en bisel, y la camptodactilia es frecuente.
La mayoría de los casos son esporádicos, si bien se ha comunicado transmisión de padres a hijos en cinco ocasiones, sugiriendo un patrón de herencia autosómico dominante. Recientemente, se han descrito mutaciones en el gen NSD1 en el 46 % de pacientes (6 de 13) con síndrome de Weaver, lo que sugiere que el síndrome de Sotos y el síndrome de Weaver son síndromes alélicos.

