


Los síndromes poliglandulares autoinmunes son raras endocrinopatías en las que coexisten alteraciones de las glándulas endocrinas, basadas en mecanismos autoinmunes con otras enfermedades no endocrinas. En el tipo 1, las manifestaciones características son la candidiasis mucocutánea crónica, el hipoparatiroidismo y la insuficiencia suprarrenal.
Presentamos a una paciente que presenta la secuencia clínica típica, junto con otras alteraciones, realizando estudio genético del gen autoimmune regulator (AIRE), detectándose una mutación en homocigosis, C322fsX372.
La herencia es autonómica recesiva, asociada a mutaciones en el gen AIRE, el cual codifica una protei¿na que interviene en procesos de autoinmunidad e inmunodeficiencia.
Para el diagnóstico, se requieren al menos 2 de las 3 manifestaciones clínicas principales, aunque en el estudio de familiares de pacientes afectados solo se requiere una de ellas.
Estos síndromes deben ser diagnosticados en etapas tempranas, dada su alta morbimortalidad. Es necesario tratar cada una de las alteraciones, con el objetivo de preservar la calidad de vida.
Polyglandular autoimmune syndromes are rare diseases based on autoimmune mechanisms in which endocrine and non-endocrine disorders coexist. In type 1 the characteristic manifestations are chronic mucocutaneous candidiasis, hypoparathyroidism and adrenal insufficiency.
A case is presented of a patient with typical clinical sequence, along with other changes, and in whom a mutation in homozygosis, C322fsX372, was detected after performing a molecular analysis of autoimmunity regulator gene (AIRE).
Inheritance is autosomal recessive, associated with mutations in the AIRE gene, which encodes a protein involved in autoimmunity and immunodeficiency.
For diagnosis, At least two of the three major clinical manifestations are required for a diagnosis. However, only one of them is necessary in the study of relatives of affected patients.
These syndromes must be diagnosed early, given their high morbidity and mortality. Every manifestation needs to be treated, in order to maintain the quality of life.
Los síndromes poliglandulares autoinmunes (SPA) son raras endocrinopatías caracterizadas por la coexistencia de al menos 2 alteraciones de las glándulas endocrinas, basadas en mecanismos autoinmunes, a las que suelen unirse otras enfermedades no endocrinas1.
Se distinguen 2 tipos principales, en función del momento de presentación, el patrón hereditario y la clínica. El SPA-1 suele iniciarse en la infancia y se define por la presencia de candidiasis mucocutánea crónica, hipoparatiroidismo adquirido e insuficiencia suprarrenal (ISR). El SPA-2 suele comenzar más tardíamente y lo caracterizan la enfermedad tiroidea autoinmune, la diabetes mellitus de tipo 1 (DM1) y la enfermedad suprarrenal2. Existe un tercer subtipo, SPA-3, descrito en adultos, que implica enfermedad tiroidea autoinmune, sin afectar a la glándula adrenal, hipoparatiroidismo ni candidiasis, y el SPA-4, en el que coexisten 2 o más enfermedades autoinmunes, sin cumplir criterios de cualquiera de las anteriores3.
Caso clínicoPaciente que presenta a los a los 3 años y 11 meses un episodio de movimientos tónico-clónicos con trismus, cianosis labial y midriasis fija. En los análisis efectuados destaca calcemia de 5,5mg/dl, instaurándose tratamiento con gluconato cálcico. La exploración física y la neurológica son normales a su ingreso.
Como antecedentes familiares, el abuelo materno presentaba celiaquía y la abuela materna, hipertiroidismo y diabetes insulinodependiente. Por lo que se refiere a antecedentes personales, consta un ingreso en Unidad de Cuidados Intensivos pediátrica por deshidratación hiponatrémica secundaria a gastroenteritis con 9 meses.
Tras iniciar tratamiento con gluconato cálcico, normalización del calcio sérico, fósforo 8,39mg/dl, fosfatasa alcalina 748 UI/l y parathormona intacta 5pg/ml, con 25-OH-D3 19,9ng/ml (valores normales [VN] 12-80) y 1,25(OH)2 D3 25,4pg/ml (VN 18-78), metabolismo óseo aumentado (hidroxiprolina en orina 128,88mg/24h,VN 20-50 y osteocalcina 10,3ng/ml,VN 3,7-10). En el estudio renal, presenta calciuria 1,03mg/kg/d (VN < 4) y cociente Ca/Cr en orina 0,05 (VN < 0,2). Estudios cardiológico y oftalmológico, tomografía computarizada craneal y electroencefalograma, normales. Densitometría ósea Z-score +0,36 SDS.
Con el diagnóstico de hipoparatiroidismo, al tratamiento con carbonato cálcico se añade calcitriol, presentando varios ingresos posteriores por crisis convulsivas por hipocalcemia.
A los 4 años, se aprecia moniliasis bucal (fig. 1) por lo que se pauta nistatina, recidivando en controles sucesivos y apareciendo displasia ungueal.

Con 5 años y 6 meses, presenta transaminasa glutamicoxalaxética 662 U/l, transaminasa glutámico pirúvica 1.352 U/l (VN 0-40), sin elevación de anticuerpos anti-LKM, anti-Sm, serologías de virus hepatotropos negativas; α-1-antitripsina y pruebas de coagulación normales. En la biopsia hepática se objetiva hepatitis activa autoinmune (crónica periportal), iniciando tratamiento con azatioprina y prednisona.
Con 8 años presenta alopecia areata (fig. 2) y 7 meses después se detecta glucemia basal de 120mg/dl, con HbA1c 6,7%. Sobrecarga a la glucosa con valores alterados y anticuerpos anti-GAD en límites altos de la normalidad.

Ante sospecha de SPA-1, se realiza un estudio genético del gen autoimmune regulator (AIRE), detectándose una mutación en homocigosis, C322fsX372. Madre portadora heterocigota de la mutación, no siendo posible el análisis del padre.
Con 13 años, se detecta ACTH 87,3pg/ml (VN 5-46), con cortisol basal 15,9 μg/dl (VN 5-38), confirmándose posteriormente ACTH 263pg/ml; test de ACTH normal, indicando insuficiencia suprarrenal en estadio incipiente, por lo que se añade hidrocortisona y fludrocortisona.
Pese a la gran afectación multiorgánica, la paciente continúa controles adecuadamente, con un correcto cumplimiento de los diferentes tratamientos, lo que conlleva una satisfactoria evolución tanto desde el punto de vista clínico como psicosocial. Asimismo, mantiene un buen crecimiento, con tallas entorno al percentil 75 y peso alrededor del 50.
DiscusiónEl SPA-1 o autoimmune polyendocrinopathy, candidiasis and ectodermal dystrophy (APECED) comienza a manifestarse en la infancia, entre 3-5 años, o adolescencia2. Contrariamente a lo que ocurre en nuestra paciente, en la población general aparece primero la candidiasis (antes de los 5 años), seguida del hipoparatiroidismo (antes de los 10) e insuficiencia adrenal (antes de los 15)4. Su incidencia es 1/100.000 habitantes/año2, siendo mayor la prevalencia en finlandeses (1/25.000), sardos (1/14.500) y judíos iraníes (1/9.000)5. Es relativamente comu¿n en el norte de Italia y Suiza6, y en general en poblaciones caracterizadas por alto grado de consanguinidad1. Puede existir predominio femenino, con una proporción mujer-varón de 0, 8:1 a 2,4:11.
La primera manifestación suele ser la candidiasis (70-80% de pacientes)2, generalmente limitada a la mucosa oral, como forma leve de queilitis. Normalmente, no se afecta más del 5% de la superficie corporal, pudiendo darse formas generalizadas en inmunodeprimidos4. En la paciente, durante el seguimiento aparece candidiasis oral, que recidiva durante los primeros años, con buen control posterior.
El hipoparatiroidismo está presente en 70-93% y suele ser la primera alteración endocrina en producirse. Su aparición varía en función del sexo, afectando al 98% de las mujeres frente al 71% de los varones4. Como ocurre en nuestro caso, puede originar complicaciones (crisis convulsivas, miocardiopatía dilatada, calcificaciones cerebrales, vasculares…) debidas al disbalance del metabolismo fosfocálcico5,7,8.
La ISR tiene una prevalencia del 60-100%; normalmente comienza antes de los 15 años4. La presencia de avidez por la sal, la deshidratacio¿n hiponatrémica e hiperpotasémica y la hiperpigmentacio¿n cutánea deben hacer sospecharla9. Cabe destacar en este caso el diagnóstico casual al realizar un control analítico, con clínica silente, únicamente apreciable en descompensaciones.
Puede aparecer hipogonadismo hipergonadotropo (12%-60%), con prevalencia 3 veces mayor en las mujeres4.
La enfermedad tiroidea autoinmune es mucho menos frecuente en el SPA-1 que en el SPA-2, siendo la prevalencia de hipotiroidismo del 3-10%10.
La DM1 estaría presente en el 18%. Tal y como se aprecia en el caso descrito, son frecuentes anomalías ectodérmicas (queratoconjuntivitis e hipoplasia del esmalte dental), vitíligo y alopecia. Con menor frecuencia, se encuentra déficit de somatotropina como consecuencia de fallo hipofisario, gastritis atrófica, anemia perniciosa, hepatitis autoinmune, nefritis tubulointersticial, bronquitis, hipertensión pulmonar, síndrome de Sjögren, lupus eritematoso sistémico, artritis reumatoidea, vasculitis, blefaritis, urticaria o miastenia gravis2,4,11,12. Es reseñable la afectación multiorgánica presente en nuestro caso, habiéndose realizado los distintos diagnósticos previamente a la aparición de los síntomas, que podría deberse al temprano inicio del tratamiento con inmunosupresores, unido a un adecuado seguimiento clínico y analítico de la paciente.
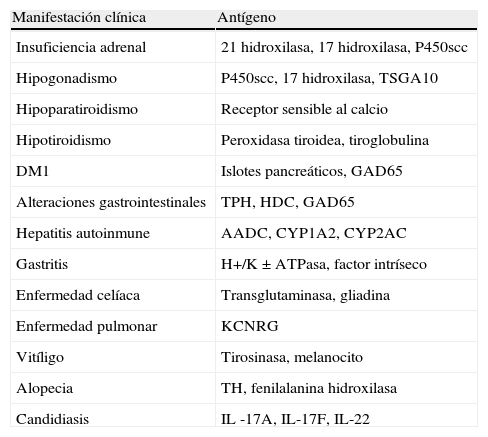
La herencia del SPA-1 tiene carácter autosómico recesivo, asociada a mutaciones en el gen AIRE. Sin embargo, se ha descrito una mutación en una familia italiana, de herencia autosómica dominante4. Pese a la herencia monogénica, no se ha encontrado relación entre el genotipo y el fenotipo, incluso dentro de miembros de la misma familia13, como se puede observar en el caso descrito, ya que la madre presenta la misma mutación que nuestra paciente, sin clínica asociada. El gen AIRE se localiza en el cromosoma 21q22.3 y se expresa mayoritariamente en el timo, los ganglios linfáticos y el hígado fetal, lo que denota el importante papel que tiene en el correcto funcionamiento del sistema inmunitario. Codifica una protei¿na que interviene en procesos de autoinmunidad e inmunodeficiencia y, por tanto, explica la variedad de manifestaciones que constituyen el síndrome6. La ausencia de esta proteína da lugar a la defectuosa selección negativa de los linfocitos T autorreactivos, causando ataque a los diversos órganos. Desempeñan, además, un papel fundamental en el SPA-1 los anticuerpos frente a antígenos de los distintos órganos, fundamentalmente enzimas intracelulares (tabla 1)4.
Antígenos en el SPA1 y su correspondiente enfermedad
| Manifestación clínica | Antígeno |
| Insuficiencia adrenal | 21 hidroxilasa, 17 hidroxilasa, P450scc |
| Hipogonadismo | P450scc, 17 hidroxilasa, TSGA10 |
| Hipoparatiroidismo | Receptor sensible al calcio |
| Hipotiroidismo | Peroxidasa tiroidea, tiroglobulina |
| DM1 | Islotes pancreáticos, GAD65 |
| Alteraciones gastrointestinales | TPH, HDC, GAD65 |
| Hepatitis autoinmune | AADC, CYP1A2, CYP2AC |
| Gastritis | H+/K±ATPasa, factor intríseco |
| Enfermedad celíaca | Transglutaminasa, gliadina |
| Enfermedad pulmonar | KCNRG |
| Vitíligo | Tirosinasa, melanocito |
| Alopecia | TH, fenilalanina hidroxilasa |
| Candidiasis | IL -17A, IL-17F, IL-22 |
Además de este gen, se ha asociado el SPA con un aumento de la frecuencia HLA-A28, HLA-A3 y HLA-DQ6 con respecto a la población general14.
Para el diagnóstico de la enfermedad, se requieren al menos 2 de las 3 manifestaciones clínicas principales3, aunque en el estudio de familiares de pacientes afectados solo se requiere una de ellas. Se ha propuesto que el análisis genético y los anticuerpos antiinterferón, presentes en la enfermedad, deben ser criterios diagnósticos4,15. El diagnóstico diferencial de la enfermedad deberá hacerse con otros síndromes autoinmunes, como el síndrome disfunción inmune-poliendocrinopatía-enteropatía ligada a X (IPEX) y el SPA-215. En el caso descrito, el diagnóstico se realizó clínicamente, al presentar las 3 manifestaciones principales y se confirmó mediante estudio genético.
Para el cribado de familiares, se recomienda estudiar los anticuerpos específicos y, si se detectan, los consiguientes test funcionales. En caso de negatividad de los tests, se debe realizar un estudio anual3. Aunque no está recomendado el estudio prenatal, debería ofrecerse a las familias consejo genético, dada su herencia autosómica recesiva15.
Los SPA deben ser diagnosticados en etapas tempranas, dada su alta morbimortalidad, tratando cada una de las alteraciones2,5. El tratamiento tiene el objetivo de preservar la calidad de vida, siendo necesario en muchas ocasiones además apoyo psicosocial5.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
