

Viernes, 20 de junio (15,30-16,30 h)
ATENCION PRIMARIA Y PEDIATRIA EXTRAHOSPITALARIA
P273 ¿COMO ACTUA NUESTRA POBLACION ANTE LA FIEBRE?
Josep-Vicent Balaguer Martínez, Francisco Codina Garacía, Inmaculada Morató Fornaguera, Sonia Sánchez Peña y Esther Caraballo Prieto
CAP de Lloret de Mar y Coorporació de Salut del Maresme i la Selva, Girona.
Introducción y objetivo:La fiebre es uno de los principales motivos de consulta en atención primaria. Nos propusimos valorar la actitud de nuestra población ante este síntoma.
Métodos:Estudio descriptivo transversal a partir de una encuesta con 10 preguntas de respuesta múltiple y cerrada. Se recogieron 100 encuestas consecutivas entre los padres de los pacientes que acuden al programa del niño sano. Estadística con SPSS 10,0 (porcentajes y X2).
Resultados:Muestra: 80 % mujeres; estudios secundarios (43 %) o primarios (37 %); con una media de 1,61 ± 0,72 hijos; inmigrantes 32 % de origen muy heterogéneo (predominio Europa del Este y Latinoamérica, 9 % y 8 % del total respectivamente), de éstos 8 personas precisaron ayuda para responder por dificultad idiomática.
Descriptivo:Tiene algún termómetro en casa el 94 %, electrónico (43 %) o de mercurio (37 %). Se coloca en axila (49 %) o recto (32 %) indistintamente de la edad y 44 % el tiempo adecuado. El 43 % de los encuestados considera fiebre a partir de 37,5 °C axilar, el 21 % 38 °C y el 19 % 37 °C. La primera medida que se adopta es dar un antitérmico (60 %) siendo paracetamol el 66 % de los casos y creyendo que es eficaz en menos de media hora el 65 %. Sólo 22 % opta por medidas físicas como primera opción y un 15 % acude directamente al médico.
Comparativo:De los cruces realizados se ha encontrado significación estadística en las siguientes situaciones. La población con estudios primarios utiliza más AAS e ibuprofeno, y cree que los antitérmicos actuan en 10 min, mientras que la población con estudios secundarios o superiores utiliza paracetamol. La población con 1 hijo opina que los antitérmicos actuan en 1h o NS/NC, los que tienen 2 o más hijos opinan que en media hora. Los inmigrantes responden significativamente más NS/NC respecto al tipo de termómetro, antitérmico a usar y tiempo en que actua éste. La población de origen español usa más paracetamol o ibuprofeno alternados.
Conclusiones:La población nativa hace un adecuado uso del termómetro para el diagnóstico de fiebre, pero observamos deficiencias en el manejo posterior de este síntoma. Entre la población inmigrante hay que reforzar la educación sanitaria tanto en el diagnóstico como en el manejo de la fiebre.
P274 DIARREA, DIETA Y LECHE
Luis Antonio García Llop, Amparo Asensi Alcoverro, Pilar Coll Más, M. del Carmen Grafiá Juan y M. Asunción Ramada Benedito
Centro de Salud de Manises, y Centro de Salud Ingeniero Joaquín Benlloch, Valencia.
Antecedentes y objetivos:Las recomendaciones actuales sobre el tratamiento de la diarrea aguda incluyen la rehidratación oral y la realimentación precoz. Sin embargo, persiste la costumbre de eliminar o diluir la leche de las denominadas "dietas astringentes". En este trabajo nos planteamos conocer el efecto de la leche en la evolución de las diarrea.
Métodos:Se ha escogido a los niños de 1 a 5 años con diarrea aguda que acuden a la consulta del centro de salud en el año 2002. Según el día de nacimiento se asigna una dieta sin lacteos (grupo A), sólo con yogur (grupo B) o con leche y yogur (grupo C).
Resultados:
Hubo presencia de fiebre en el 22,3 % de los casos, ingresó el 1,5 % de los niños, el 9,5 % tenía sangre en las heces y el 38,1 % de las diarreas se acompañó de vómitos. Se realizó Coprocultivo en el 14,4 % de los casos y se diagnosticaron 2 casos de Salmonelosis (Grupo A), 2 de diarrea por Campylobacter y 1 por Rotavirus (los tres en Grupo C).
Conclusiones:1. Mantener los lácteos (leche + yogur) en la dieta no alarga la duración de la diarrea. 2. La incidencia de intolerancia secundaria a la lactosa y/o proteinas vacunas es inferior al 2 % y no se influye por el consumo de lácteos.3. No está justificado eliminar la leche de la dieta en una diarrea.
P275 LAXITUD ARTICULAR Y ARTRALGIAS EN PEDIATRIA
Jaime de Inocencio Arocena e Inmaculada Ocaña Casas
Centro de Salud Estrecho de Corea, Madrid y Hospital Ramón y Cajal, Madrid.
Antecedentes y objetivos:Tradicionalmente se ha aceptado que existe una relación causal entre la presencia de laxitud articular y el desarrollo de artralgias.
Objetivos:1. Determinar la prevalencia de laxitud articular en niños de entre 4 y 14 años de edad sin patología orgánica del aparato locomotor. 2. Comparar la prevalencia de laxitud articular en niños con y sin artralgias.3. Analizar la contribución de las variables demográficas recogidas en el desarrollo de artralgias.
Diseño:Estudio prospectivo transversal realizado en una muestra de niños hispanohablantes de entre 4 y 14 años residentes en el Area 4 de la Comunidad de Madrid. La movilidad articular se valoró utilizando un goniómetro. La laxitud se definió como la presencia de al menos 4 de los 9 criterios propuestos por Beighton.
Resultados:Se analizaron 222 sujetos, 176 (79,5 %) procedentes de una consulta de Atención Primaria (AP) y 46 en Urgencias de Pediatría del hospital de referencia (Urg). Las características clínicas y demográficas de estas poblaciones sólo diferían en el país de origen de los niños, siendo éste distinto de España en el 36 % de la serie de AP y en el 2 % de la serie de Urg (p = 0,01).
La prevalencia de laxitud articular (≥ 4 criterios de Beighton) en el total de la serie fue del 55 % (123/222), alcanzando el 71 % (49/69) en menores de 8 años.
En la población analizada sólo 43/222 niños (19,4 %) referían artralgias. Se detectó laxitud articular en una mayor proporción de sujetos con artralgias (65 %; 28/43) que en niños sin dolor articular (53 %; 95/179), aunque las diferencias no resultaban estadísticamente significativas.
De las variables analizadas (edad, sexo, país de origen, captación en AP o Urg), sólo se detectaron diferencias significativas en el número absoluto de criterios de Beighton presentes, 3,48 ± 2,35 en niños sin dolor articular y 4,34 ± 2,47 en sujetos con artralgias (p = 0,03). Esta diferencia, sin embargo, desaparecía al comparar la proporción de niños con y sin artralgias que presentaban al menos 4 criterios y cumplían por tanto la definición de laxitud.
Conclusiones:El 55 % de la población estudiada, y hasta el 71 % de los menores de 8 años, cumple criterios de laxitud articular. En la muestra analizada no se confirma que exista relación entre la presencia de laxitud articular y el desarrollo de artralgias.
P276DIAGNOSTICO DEL SINDROME DE RETT EN ATENCION PRIMARIA
Raquel Ávila Villegas, Juan M. Ramos Navas-Parejo y Concepción Robles Vizcaino
Centro de Salud Albayzín, Granada y Hospital Clínico Universitario San Cecilio, Granada.
Introducción:El síndrome de Rett es una causa frecuente de retraso mental en las niñas. Se presenta con una incidencia de 1 por 150.000 nacidas vivas, y está provocado por una anomalía genética ligada al cromosoma X, letal en varones. El diagnóstico debemos realizarlo en base a un conjunto de signos y síntomas clínicos encontrados en las pacientes: normalidad de desarrollo psicomotor en los primeros meses, entre los 6-18 meses detención del desarrollo psíquico, 1-4 años deterioro neuropsicológico rápido, aparición de esterotipias y manifestaciones autísticas, entre 4-6 años estacionamiento de la sintomatología y por último a partir de los 7 años, deterioro motor tardío. En ocasiones el diagnóstico puede ser confirmado con técnicas de genética molecular. No se dispone en la actualidad de tratamiento específico.
Caso clínico:Recién nacida a término con peso adecuado a la edad gestacional. Apgar: 9/9. En los controles iniciales no se detectan anomalías, salvo un estrabismo por el que es remitida a la consulta de oftalmología. A la edad de 11-12 meses se observa una pérdida de las adquisiciones conseguidas, con esterotipias manuales, movimiento de "lavado de manos" y chupeteo de las mismas. Se realizan las siguientes pruebas complementarias: hemograma y bioquímica normal. Ácido láctico normal. EEG normal. RNM cerebral normal. Con la sospecha de Síndrome de Rett se deriva a la consulta de neuropediatría, confirmando el diagnóstico. El estudio genético molecular para Sdr de Rett fue positivo (delección exón 4 del gen MECP2). También se realiza a los padres siendo en ambos negativo.
Conclusiones:1. Debemos pensar en el síndrome de Rett en Atención Primaria ante toda niña con desarrollo neurológico normal que sufre un deterioro neuropsicológico.2. El diagnóstico es clínico, aunque en ocasiones puede confirmarse con técnicas de genética molecular (delección en el gen MECP2). 3. El pediatra debe coordinar la puesta en marcha de los cuidados y el trabajo del equipo multidisciplinar (neuropediatra, paidopsiquiatra, ortopeda, psicólogo, fisioterapeuta, asistenta social, etc.).
P277SINUSITIS AGUDA. TRATAMIENTO CON AMOXICILINA-CLAVULANICO Y AEROSOLTERAPIA
M. Paz González Rodríguez, José Luis Muñoz Alcañiz, Isabel Parra Martínez, M. Candela Agis Brusco, Adolfo Rodríguez Balo y Susana Ares Segura
Centro de Salud Barrio del Pilar, Madrid y Centro de Salud Fuentelarreina, Madrid.
Antecedentes y objetivos:La sinusitis es un diagnóstico frecuente en las consultas de pediatría. Entre 1-5 % de los catarros de vías altas se complican con infección de los senos. Entre los tratamientos hay diversas pautas antibióticas, medicaciones como mucolíticos, vasoconstrictores, corticoides e incluso el no administrar tratamiento alguno.
Objetivo:Comparar la efectividad amoxicilina-clavulánico por vía oral con aerosoles compuestos de corticoide, mucolítico y vasoconstrictor en el tratamiento de niños con sinusitis aguda.
Métodos:Durante un año, los niños diagnosticados de sinusitis en 6 consultas de atención primaria, se incluyeron en el estudio. Las edades fueron de 3 a 13 años. El diagnóstico de sinusitis se basó en la presencia de síntomas de infección respiratoria de vías altas, que no experimentaron mejoría a los 10 días y anomalías radiologícas en la Rx de senos (proyección de Water). Se excluyeron los niños con antecedentes de asma y/o alergia. Se distribuyeron aleatoriamente en dos grupos. Se evaluaron los síntomas y los hallazgos en la exploración tras un período de 5, 16 y 30 días.
Resultados:Se incluyeron 54 niños con edad media de 6,5 años. 25 niños y 28 niñas. 29 recibieron aerosoles y 25 antibióticos. Se excluyeron 10 pacientes de los cuales 5 eran del grupo de antibióticos y 5 del de aerosoles. Se objetivó curación clínica en 12 niños del grupo de aerosoles frente a 11 del grupo de antibióticos a los 5 días, 16 pacientes frente 15 a los 16 días, 19 frente a 15 a los 30 días. Las diferencias no fueron significativas.
Conclusiones:Considerar la posibilidad de sinusitis tras 10 días de síntomas de infección respiratoria sin mejoría es una forma simple y práctica de sospechar el diagnóstico de sinusitis. No hay diferencias significativas en cuanto a la curación, entre los que reciben antibioterapia o aerosoles.
P278HIPOFOSFATASIA ALCALINA
M. Salomé Albi Rodríguez, Cristina Ramos, Antonio Cuñarro Alonso, Marta Ortega Molina y M.ª Jesús Ceñal González-Fierro
Complejo Hospitalario de Móstoles.
Introducción:La hipofosfatasia es un trastorno infrecuente, hereditario, que consiste en la aparición de una alteración de la actividad de la fosfatasa alcalina inespecífica tisular (isoenzima L/B/K), con el consiguiente defecto en la mineralización de las estructuras óseas y dentales.
Material y métodos:Presentamos el caso de un varón de 2 años con hipofosfatasia y pérdida precoz de la dentición temporal.
Caso clínico:Varón de 2 años y 11 meses, que consulta por movilidad dentaria y caida de dientes. Antecedentes personales: no refiere la existencia de ningún traumatismo previo; es un niño sano que precisó ingreso por GEA a Yersinia enterocolítica a los 7 meses. En la historia familiar destaca la presencia de un tío materno que presentó problemas periodontales que se iniciaron a los 20 años de edad. A la exploración física presenta ausencia, por caida previa, del diente 51 y movilidad importante de los incisivos inferiores y del incisivo superior central con exposición de más de un tercio de su raiz; además de diversas caries sin signos de inflamación gingival ni otras alteraciones de la mucosa; el resto de la exploración por aparatos es rigurosamente normal, con un peso y talla dentro de percentiles normales.Exploraciones complementarias: Hitachi con iones normales: Calcio: 9,8 mg/dl (N: 8,7-11); Fosfatasa Alcalina: 168 U/l (N: 185-990); Fracción alcalina termoestable 9 % (N: 20-30); Excreción urinaria de fosfoetanolamina (PEP): 8950 u/mg Creatinina; PTH intacta RIA: 31 pg/ml (N: 9-55); Vitamina D: 18 ng/ml (N: 15-10; 0); Función renal normal; Calciuria 2,6 mg/kg/d (N: < 4); Ca/Cr: 0,10; Cultivo de flora bucal: flora saprofita. Estudio genético: pendiente.
Conclusiones:La hipofosfatasia es un error congénito del metabolismo con un amplio espectro de manifestaciones clínicas, pudiendo aparecer desde formas benignas que debútan en la edad adulta (con la aparición de pseudofracturas y dolores articulares), hasta formas letales neonatales (que cursan con hipercalcemia importante, falta de mineralización ósea...). Ante la aparición precoz de alteraciones dentarias y la caída prematura de la dentición, es importante sospechar esta enfermedad, para poder diagnosticarla, controlar los síntomas y hacer un seguimiento clínico y un consejo genético familliar adecuado.
P279DESCRIPCION DE UN BROTE DE GASTROENTERITIS POR ROTAVIRUS EN UNA COMUNIDAD
M. Pilar Lalana Josa, Juan Ignacio Martín Sánchez, M. Paz González Fernández, Joaquín Gimbao Bescós, Francisco Javier Castillo García y M. Victoria Baños Ledesma
Centro de Salud Tarazona, Zaragoza, Gerencia de Area del Servicio Aragonés de Salud, Zaragoza y Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Antecedentes y objetivo:La gastroenteritis por rotavirus es un cuadro de lactantes y niños pequeños, a menudo grave, siendo una causa frecuente de ingreso hospitalario. Se describe un brote de gastroenteritis por rotavirus en una comunidad.
Métodos:Se crea una encuesta y se entrevista por teléfono a las madres y padres de los casos recogidos y con los datos se crea una base de datos en SPSS 6.0 con el fin de realizar un descriptivo de los casos. Se solicita a la Diputación General de Aragón plano de Tarazona para ubicar los casos y los boletines de análisis de la calidad de agua de consumo desde Enero del 2002. Mediante hoja de cálculo Excel distribución de los casos según la semana de declaración y comparación con los datos de Sanidad Ambiental.
Resultados:Tarazona es una población de aproximadamente 10.000 habitantes situada en la provincia de Zaragoza, a 90 km de la capital en dirección a Soria. El 23 de Septiembre del 2002 se detectó el primer caso de gastroenteritis por rotavirus y hasta el 9 de Noviembre se recopilaron 26 casos (clínica de gastroenteritis con investigación en heces positivo a rotavirus). El 100 % eran menores de 3 años con una mediana de 1,5 años, siendo el 39,3 % varones y el 60,7 % mujeres. Sólo un caso acude al colegio y 5 a guardería. El 11,5 % (3 casos) precisaron ingreso hospitalario. El 100 % tuvieron diarrea acuosa, el 85 % vómitos y el 81 % fiebre, resolviéndose el cuadro clínico con una mediana de 7 días (rango: 2-7). En el 81 % de los domicilios hubo convivientes afectados antes y/o después de que los casos iniciasen los síntomas.
La ubicación de los casos en la población de Tarazona no sigue aparentemente ninguna pauta concreta. El 88 % de los casos consumían agua embotellada y los resultados de la calidad del agua de la red durante el brote, reunía las condiciones necesarias de potabilidad. Se construyó una tabla con calles sin número de los casos y semana de declaración por ver si existía relación entre los casos y cortes en el suministro de agua por calle, quedando sólo de manifiesto una acumulación de casos en la semana 43 y su entorno.
Conclusiones:Pese a la investigación, la fuente de infección y el mecanismo de transmisión han quedado sin determinar. Dado que los rotavirus son estables en el ambiente, la transmisión ha podido ser por fomites o persona a persona.
P280PREVALENCIA Y GRAVEDAD DEL ASMA EN NIÑOS DE 13-14 AÑOS DE DOS POBLACIONES URBANAS DE CANTABRIA
Alberto Bercedo Sanz, Luis Lastra Martínez, Carlos G. Redondo Figuero, Fco. Manuel Gómez Serrano, Elena Pérez Gil, M. Auxiliadora de Andrés Fraile, Mónica Pacheco Cumani y Elena Mora González
Hospital Universitario Marqués de Valdecilla, Santander.
Antecedentes y objetivo:El asma es la enfermedad crónica infantil más frecuente en los países desarrollados, y constituye uno de los problemas de salud pública más importantes debido a su magnitud y distribución universal, al aumento de la morbilidad y mortalidad, y a la repercusión económica y social asociada. En España, la prevalencia y gravedad del asma han sido valoradas en el estudio ISAAC (International Study of Asthma and Allergies in Childhood), demostrando una gran variabilidad geográfica entre los centros participantes, con resultados de prevalencia de asma por encuesta validada entre el 5,5 y el 14,6 %. El objetivo de este estudio es determinar la prevalencia y gravedad del asma bronquial, en los niños de 13 y 14 años de las dos comunidades urbanas de mayor población de Cantabria.
Material y métodos:Estudio transversal de prevalencia y gravedad de asma mediante cuestionario escrito y videocuestionario del estudio ISAAC, a 1813 niños de 13-14 años de edad escolarizados en 37 centros escolares de las ciudades de Santander y Torrelavega.
Resultados:El número total de encuestas validadas fue de 1813 niños que representó el 80,47 % de la población de estudio (n = 2253). La prevalencia acumulada de asma fue del 24,9 % y la prevalencia actual de asma (asma en el último año) del 16,7 % [IC-95 %: 15,0-18,5 %]. El 30,4 % de los niños que han tenido crisis en el último año afirman que han sido graves. Un 16,8 % de los niños refieren haber sido diagnosticados de asmáticos. Un 21,1 % refieren asma inducido por el ejercicio y un 28,6 % refieren tener tos seca sin estar resfriado que les despierta por la noche en el último año. La prevalencia acumulada de asma medida con el videocuestionario es de un 27,2 % y la prevalencia actual de asma es de un 14,4 %. El 9,9 % de los niños se identificaron con la escena de ataque severo de asma del videocuestionario.
Conclusiones:La alta prevalencia de asma y síntomas relacionados con asma en la población infantil de 13-14 años estudiada en Cantabria es concordante con la referida en otras zonas geográficas de nuestro país. Es necesario confirmar estos datos mediante estudios funcionales de hiperrespuesta bronquial dada la gran variabilidad geográfica del asma bronquial existente en España.
P281IMPORTANCIA RELATIVA DE LOS DISTINTOS FACTORES DE RIESGO EN LA IDENTIFICACION DE HIPERCOLESTEROLEMIAS PRIMARIAS EN LA INFANCIA
Nieves Fernández Martínez, Óscar Cristóbal Alonso, María Jesús Eiris Cambre, Ángel Lado Llerena, Augusto Nores Lorenzo, Manuel Sánchez Loureiro y Xosé Segade
Centro de Salud Municipal A Serra de Outes.
Introducción y objetivo:El proceso ateromatoso comienza en la infancia y su progresión depende de factores genéticos y ambientales. Entre los factores de riesgo susceptibles de intervención preventiva se encuentra la hipercolesterolemia. El objetivo es valorar la importancia de las distintas variables para la identificación de las hipercolesterolemias.
Métodos:Estudio descriptivo transversal (febrero 2003), cuya población objeto de estudio fueron todos los niños entre 2 y 15 con analítica de colesterol (n = 189). Se recogieron las siguientes variables: género, edad, diagnóstico o no de dislipemia, presencia de dislipemia en los progenitores y obesidad. Para el análisis univariante se emplearon proporciones y medias, estas con sus correspondientes desviaciones estándar (DE), y para el multivariante el análisis de regresión logística con sus correspondientes odds ratios.
Resultados:El 50,8 % eran del género masculino, siendo la edad media de 10,8 (DE = 3,2) años. El 28,6 % estaban diagnosticados de hipercolesterolemia. Tenían dislipemias primarias el 9,5 % (hiperlipemia familiar combinada el 4,8 %, hipercolesterolemia poligénica el 3,2 % e hipercolesterolemia familiar heterocigota el 1,6 %). Tenían antecedentes de obesidad el 11,1 %. La concetración media de colesterol total en los niños dislipémicos fue de 218 (19) mg/dl y la de LDL de 137 (24) mg/dl. La odds ratio (razón de ventajas) fue de 59,7 veces para aquellos niños con antecedentes de hipercolesterolemias familiares y de 3 veces para los que presentaban obesidad.
Conclusiones:Los antecedentes familiares de hipercolesterolemia son los que tienen mayor importancia relativa en la detencción de niños dislipémicos y en la detencción de hipercolesterolemias primarias.
La identificación de niños afectos de dislipemia de causa genética es prioritaria en los primeros años de vida.
P282PREVALENCIA DE SINTOMAS RELACIONADOS CON RINITIS ALÉRGICA EN NIÑOS DE 13-14 AÑOS DE DOS POBLACIONES URBANAS DE CANTABRIA
Luis Lastra Martínez, Fco. Manuel Gómez Serrano, Carlos G. Redondo Figuero, Alberto Bercedo Sanz, Mónica Pacheco Cumani, M. Auxiliadora de Andrés Fraile, Elena Pérez Gil y Elena Mora González
Hospital Universitario Marqués de Valdecilla, Santander.
Antecedentes y objetivo:La rinitis alérgica es una patología frecuente en la infancia con una morbilidad y prevalencia que ha aumentado en las últimas décadas como demuestran los diferentes estudios epidemiológicos. El estudio ISAAC (International Study of Asthma and Allergies in Childhood), ha establecido un método de cuestionario que permite la comparación de la prevalencia y gravedad del asma bronquial y de enfermedades alérgicas como la dermatitis atópica y la rinitis alérgica entre diferentes comunidades. El objetivo de este estudio es determinar la prevalencia de la rinitis alérgica, en los niños de 13 y 14 años de dos poblaciones urbanas Cantabria.
Material y métodos:Estudio transversal de prevalencia de rinitis alérgica mediante cuestionario escrito del estudio ISAAC, a 1813 niños de 13-14 años de edad escolarizados en 37 centros escolares de las ciudades de Santander y Torrelavega.
Resultados:El número total de encuestas validadas fue de 1813 niños que representó el 80,47 % de la población de estudio (n = 2.253). El 55,1 % de los niños manifestaron historia previa de síntomas relacionados con rinitis y el 44,3 % [IC-95 %: 42-46,6 %], refirieron síntomas en el último año (prevalencia actual de rinitis alérgica). Un 12 % de los niños de 13-14 años asociaron síntomas de rinitis y conjuntivitis en el último año, y el período del año en el que los niños presentaron con más frecuencia síntomas nasales fue en los meses primaverales de marzo, abril y mayo, con una disminución acusada en verano. Un 16,8 % de los niños refieren haber sido diagnosticados de rinitis alérgica o fiebre del heno y cuando se valoró si los problemas nasales impedían realizar las actividades diarias, el 94 % no les afectó nunca o alguna vez, y sólo al 6 % de vez en cuando o muchas veces.
Conclusiones:La elevada prevalencia de síntomas relacionados con la rinitis alérgica en la población infantil de 13-14 años estudiada en Cantabria es concordante con la referida en otras zonas geográficas de nuestro país con metodología semejante. El conocimiento de estos resultados contribuirá a no infravalorar la sintomatología nasal alérgica en la infancia y a un mejor control de los factores predisponentes de la rinitis alérgica.
P283EXAME GLOBAL DE SAUDE A CRIANÇAS DE 13 ANOS
Sandra Cristina Alves, Claudia Ferraz, Carmelina Dias y Teresa Neto
Hospital Pedro Hispano, Matosinhos (Portugal).
Objectivos:1. Avaliação das patologias com maior incidência em crianças com 13 anos.2. Orientação preventiva.
Material e métodos:Foram convocadas, por carta, 190 crianças nascidas em 1989, inscritas na Unidade de Saúde Farol, no Centro de Saúde Senhora da Hora. O estudo efectuado foi transversal e descritivo. A amostra foi obtida de forma sistemática.
Resultados:Foram avaliadas 139 crianças, 68 do sexo masculino e 71 do sexo feminino. O Índice de massa corporal oscilou entre 14,5 a 30,1 (média = 20,4), verificando-se que 32 (23,0 %) crianças se encontravam acima do Percentil (P) 85 e nenhuma abaixo do P5. Em relação à tensão arterial, constatou-se que 21 (15,2 %) crianças tinham a pressão sistólica e 7 (5,0 %) a diastólica, acima do P 95.
O Boletim Nacional de Vacinas não estava actualizado em 9 (3,0 %) casos.
Analisando os antecedentes familiares foi possível observar que estas crianças pertenciam a famílias com vários factores de risco cardiovasculares (Diabetes, Obesidade, Dislipidemia, Hipertensão e doenças cardiovasculares).
Ao exame objectivo foram diagnosticadas alterações predominantemente ortopédicas, dermatológicas, orais e visuais. Avaliando o estadio pubertário constatou-se que a maioria se encontrava no estadio 3 e 4.
Cerca de 67 (48,5 %), praticavam exercício físico extra-escolar, variando de 1x/semana a 2 h/dia. Quanto ao consumo de substâncias todos negaram o consumo de drogas, álcool e tabaco, no entanto 11 afirmaram ingestão habitual de café. O aproveitamento escolar era bom em 78 (56,1 %) crianças.
Laboratorialmente, de valorizar num hemograma o valor da hemoglobina inferior a 12 g/dl e em 4 o número de leucócitos inferior a 4.500. No perfil lipídico em 26 crianças foi detectado Colesterol Total acima do P75 e 8 hipertrigliceridemia.
Conclusão:Esta consulta contribuiu para reforçar a necessidade de um exame objectivo periódico com rastreio visual e vigilância analítica. É fundamental alertar os adolescentes para os factores de risco cardiovasculares, nomeadamente, obesidade e dislipidemia de forma a promover hábitos saudáveis.
CIRUGIA
P284TRATAMIENTO DE CIRUGIA ORTOPÉDICA EN PIES EQUINOS PARA NIÑOS CON PARALISIS CEREBRAL ESPASTICA
Igor Nazarov
Universidad Médica Estatal, Moscú (Rusia) y Academia Médica de Postgraduados, Moscú (Rusia).
Antecedentes y objetivos:Las deformidades de los pies equinos en niños con Paralisis Cerebral Infantil (PCI) son consecuencia de las contracturas de los tejidos blandos que son fibras contraídas en los músculos, fascias,aponeurosis,tendones y ligamentos, que a su vez son consecuencias de la espasticidad y del proceso degenerativo de distrofia, que se ha demostrado histológicamente. El objetivo más importante para mejorar los resultados de la rehabilitación de este colectivo de pacientes, es la eliminación de estas contracturas que permitiría aumentar la capacidad de movimiento y favorecería el desarrollo del aparato motor del niño.
Métodos:La intervención quirurgica que practico para eliminar las contracturas de los tejidos blandos,desde hace diez años, consiste en la realización de cortes en las fibras contraídas que están en los tejidos blandos, sin tocar las estructuras sanas y ensenciales. Para ello, se utiliza un escalpelo puntiagudo que permite realizar una intervención quirurgica subcutánea, bajo anestesia general. El post-operatorio es poco traumático y no requiere inmovilización.
Para resalizar esta investigación, se operaron 188 pies equinos en 120 niños y se utilizaron los métodos tradicionales de investigación médica (ortopédico, plantografía,algometría,radiografía, técnicas visuales e histiología) antes y despues de las operaciones y al cabo de 1-4 años de las operaciones realizadas.
Resultados:Los resultados de los métodos de investigación demuestran que en 181 pies operados, se consiguió disminuir el ángulo tibia-pie hasta 90 grados y menos, aumentar la superficie de apoyo plantar, eliminar el síndrome de dolor (que tambien provoca posturas incorrectas del pie), verticalizar los pacientes, mejorar el equilibrio y la capacidad de movimientos y desplazamientos.
En los 7 pies restantes, se consiguen resultados beneficiosos, pero a menor grado; el ángulo tibi-pie disminuye a 95 grados como máximo y la capacidad de apoyo plantar aumenta aunque en menor medida, debido a que estos pies ya tenían contracturas en las articulaciones y empezaban a deformarse los huesos de los pies.
Conclusiones:Los resultados de esta investigación demuestran que es preciso que la cirugía ortopédica ocupe su lugar en el tratamiento de los pies equinos para optimizar la rehabilitación de los niños con paralisis cerebral infantil espástica.
P285TUMOR GLOMICO SUBUNGUEAL
Cristina García Llopis, Francisco Javier Castejón Casado, Caridad Llopis Baño, Antonio Vicente Pintor y Ricardo Fernández Veladés
Hospital Virgen de las Nieves, Granada.
Introducción:El glomangioma o tumor glómico de la mano es una lesión bastante rara, que con frecuencia no tiene un diagnóstico temprano y tratamiento efectivo por ser inusual, como muestran las pocas series publicadas en la literatura, aunque probablemente tenga un diagnóstico erróneo inicial en mucho casos. Presentamos el caso de un paciente pediátrico, mucho más infrecuente la aparición en esta edad.
Caso clínico:Varón de 9 años de edad que acude por dolor y tumefacción en el pulpejo del tercer dedo de la mano derecha, de 4 años de evolución, con períodos de agravamiento y que le incapacita para realizar sus tareas habituales. A la exploración se aprecia en el pulpejo del dedo un nódulo subcutáneo con piel normal, de aproximadamente 1 cm de diámetro, y dolor a la palpación o compresión de la uña. La RX simple indica un ligero aumento de partes blandas, la Eco-Doppler es normal y la RMN muestra en la zona subungueal una imagen focal hiperintensa en T2 e hipointensa en T1, que mide aproximadamente 4 x2 x 3,5 mm (diámetros transverso, anteroposterior y longitudinal), de contornos netos y respetando la cortical ósea de la falange distal. Se establece diagnóstico de glomangioma o tumor glómico subungueal y se interviene, realizándose con éxito la exéresis tumoral, confirmándose el diagnóstico por la anatomía patológica. La curación se obtiene sin secuelas estéticas.
Comentario:Los tumores glómicos son realmente infrecuentes, sobre todo en la edad pediátrica, aunque no excepcionales. Deberían sospecharse en todo paciente con dolor distal de dedo sin causa aparente, mucho más si tiene una evolución larga. Presentan una serie de características: predominio en región subungueal, más frecuente en adultos, sexo femenino y diagnóstico tardío por la frecuencia de diagnósticos iniciales incorrectos. Son tumores benignos derivados de células vasculares, que forma anastomosis veno-capilares particularmente numerosas en la parte distal de los dedos. El diagnóstico es fundamentalmente clínico, siendo el dolor el síntoma más frecuente, que aumenta tras presión, trauma mínimo o frío y la existencia a menudo de una mancha azulada subungueal. Como pruebas complementarias útiles están la RX simple y la RMN. En el diagnóstico diferencial destacan el absceso subungueal y el osteoma osteoide. El abordaje quirúrgico es un tema debatido. Son complicaciones la recidiva y la deformidad ungueal entre otras.
P286FASCITIS NECROSANTE COMO COMPLICACION POSTOPERATORIA DE INVAGINACION INTESTINAL
Cristina García Llopis, Carlos Jiménez Álvarez, Antonio M. Ruiz Montes y Manuel Villegas Rubí
Hospital Virgen de las Nieves, Granada.
Introducción:La Fascitis Necrosante es una complicación postoperatoria poco frecuente en Pediatría, que puede desarrollarse tras intervenciones limpias o contaminadas en pacientes sin otros factores de riesgo.
Caso clínico:Paciente de 7 meses de edad que se interviene por invaginación intestinal tras fracasar el intento de reducción hidrostática. A las 10 horas de la intervención comienza bruscamente con signos de sepsis y shock hipovolémico, apareciendo además en la pared abdominal enrojecimiento, induración, edema y crepitación, que adquiere en pocas horas un color azulado-negruzco. Se realizan drenajes de los que se obtiene líquido serohemorrágico con cultivo positivo a Enterococo faecalis y E. coli. Se instaura tratamiento con expansores de volumen y antibióticos de amplio espectro (aminoglucósido y cefalosporinas de 3.ª generación), con lo que se consigue reponer el estado general; sin embargo la lesión abdominal progresa rápidamente con aparición de signos de necrosis que obliga a realizar un desbridamiento extenso de la piel, tejido subcutáneo y fascia (desde el pliegue inguinal hasta el reborde costal). En cultivos posteriores de la herida aparecen sucesivamente Pseudomona aeruginosa, Estafilococo epidermidis y Clostridium sp. La evolución es favorable con excelente estado general y cierre del defecto por medio de injertos de piel y plastias por deslizamiento.
Comentario:La Fascitis Necrosante es un proceso muy agresivo que puede poner en peligro la vida del paciente, por lo que saber reconocer los cambios cutáneos que aparecen rápidamente es fundamental para realizar un tratamiento precoz con desbridamiento hasta tejido sano, antibioterapia de amplio espectro y medidas de soporte, pudiendo así detener el proceso y asegurar la supervivencia del paciente.
P287DILATACION URETERAL, UN FACTOR DETERMINANTE EN EL ÉXITO DEL TRATAMIENTO ENDOSCOPICO DEL REFLUJO
Iván Somoza Argibay, Jorge Liras Muñoz, Alberto Sánchez Abuín, Roberto Méndez Gallart, Manuel Gómez Tellado, Ernesto Pais Piñeiro y Diego Vela Nieto
Hospital Juan Canalejo, A Coruña.
Introducción:El tratamiento endoscópico del reflujo vésico-ureteral (RVU) se ha convertido en una de sus principales armas terapéuticas. En 1995 se comienza a utilizar en nuestro hospital, indicándose en los RVU de grado II no curados tras 1 año de tratamiento médico, en todos los de grado III y en los de grado IV sin daño renal. Según la Clasificación Internacional del Reflujo (IRSC) los RVU de grado IV se diferencian de los de grado III básicamente por el abombamiento de los cálices. En cada uno de los grados de RVU se pueden observan distintos grados de dilatación ureteral.
Objetivo:Estudiar la relación entre el grado de dilatación ureteral y la curación endoscópica del RVU independientemente de los grados de reflujo.
Material y métodos:De una serie de 245 unidades renales refluyentes intervenidas endoscópicamente realizamos una revisión de los primeros 3,5 años (1996-1999). Hemos recogido 61 pacientes, con un total de 90 (URR) que fueron estudiadas independientemente. Se valoraron las cistografías miccionales (CUMS) y mediante un estudio de doble ciego se graduó la dilatación ureteral de todos los pacientes en tres grados: leve-normal, moderada y severa. Se compararon los porcentajes de curación tras la 1.ª inyección endoscópica en los 3 grados de dilatación ureteral. Los datos fueron analizados mediante la prueba estadística test x2 para comparación de proporciones.
Resultados:3 pacientes presentaban RVU de grado I, 10 de grado II, 54 de grado III y 23 de grado IV. Tras graduar la dilatación ureteral había 39 con grado leve-normal, 39 moderada y 12 severa. Se encontraron diferencias estadísticamente significativas entre los porcentaje de curación de cada uno de los grados de dilatación ureteral al considerar todas las URR tratadas y también al considerar únicamente los RVU de grado III y IV. Sin embargo al comparar los porcentajes de curación de los RVU de grado III con los de grado IV sin considerar el grado de dilatación ureteral no se encontró significación estadística.
Conclusiones:Los resultados muestran que la dilatación ureteral es un importante factor pronóstico del éxito del tratamiento endoscópico del RVU. La IRSC pasa por alto diferencias importantes entre reflujos del mismo grado.
P288TRATAMIENTO QUIRURGICO DEL REFLUJO GASTROESOFAGICO
Manuel Haro Gómez, M. Teresa Rojo Jurado, Pedro Terol Barrera, Orlando Farfán, Filiberto Ramírez Gurruchaga, Francisco Chaves Pecero y Federico Argüelles Martín
Hospital Universitario Virgen Macarena, Sevilla.
Introducción y objetivos:El reflujo gastroesofágio (RGE) es una entidad frecuente en pediatría y suele cursar con clínica banal, sin embargo en ocasiones se complica con desnutrición, apneas respiratorias, hemorragias, estenosis esofágica, anemia, esófago de Barret,.. En los casos resistentes al tratamiento médico puede ser necesario el tratamiento quirúrgico, siendo la vía laparoscópica una opción posible que permite acortar el postoperatorio y disminuir la mormilidad. El objetivo es valorar la eficacia del tratamiento quirúrgico en pacientes pediátricos con RGE patológico refractario al tratamiento médico.
Material y método:Se realiza un estudio descriptivo retrospectivo de 23 pacientes diagnosticados de RGE patológico refractario a 6-12 meses de tratamiento médico. Se revisan la edad de inicio, edad de cirugía, síntomas y complicaciones. Se ha realizado a todos los niños 2 phmetrías y 1 endoscopia antes de la cirugía, y 1 phmetría postoperatoria.
Resultados:La edad media de comienzo de síntomas es 15 meses (0 meses-24 meses), la edad media de la cirugía es 2 años (1 año-4 años). Los síntomas y complicaciones presentados son vómitos y regurgitaciones en 15 niños, dolor retroesternal en 11 niños, hipo persistente en 6, bronquitis en 6, neumonía en 3, apneas en 2 y anemia ferropenica en 2 niños. Se ha realizado funduplicatura tipo Nissen por laparotomía a 18 niños y por vía laparoscopica a 5 niños (> 2 años). La evolución clínica a largo plazo de los niños operados por laparotomía ha sido buena. En los 3 meses de postoperatorio 2 niños han presentado disfagia y 1 de ellos ha precisado dilatación con balón por presentar estenosis esofágica. La evolución a corto plazo de los niños operados por vía laparoscopica ha sido buena, excepto a 1 niño que ha tenido que ser reintervenido por reaparición de los síntomas tras la dehiscencia de las suturas. Los niños operados por laparoscopia están pendientes de evolucionar a largo plazo.
Comentario:Hemos tenido buenos resultados al realizar el Nissen tanto por vía laparoscópica como por laparotomia en los niños con RGE refractario al tratamiento médico. Estos resultados coinciden con los existentes en la literatura, llegando a un 80-90 % de efectividad.
P289PROTOCOLO DE ACTUACION EN EL RECIÉN NACIDO Y LACTANTE CON VALVULAS DE URETRA POSTERIOR EN LOS ULTIMOS 5 AÑOS
Jorge Liras Muñoz, Iván Somoza Argibay, Alberto Sánchez Abuín, Ernesto Pais Piñeiro, Diego Vela Nieto, Manuel Gómez Tellado y Roberto Méndez Gallart
Hospital Juan Canalejo, A Coruña.
Introducción:La ablación transuretral constituye el tratamiento de elección en los pacientes que presentan válvulas de uretra posterior (VUP), existiendo todavía gran controversia acerca de la utilidad de la derivación urinaria.
Material y métodos:De un total de 42 pacientes tratados en nuestro servicio desde 1985 por VUP, revisamos aquellos niños menores de dos años tratados desde 1997. Un total de 20 pacientes fueron divididos en dos grupos. El grupo I lo constituyeron los tratados inicialmente con ablación de VUP y el grupo II los pacientes que tuvieron que ser derivados por bajo peso al nacimiento (< 3 kg) o alteración de la función renal (> 1,2 aclaramiento de creatinina).
Resultados:Los 13 pacientes del grupo I fueron tratados inicialmente mediante ablación de las válvulas de uretra posterior, 5 mediante cuchilla fría y 8 con RTU, el resector utilizado fue el número 9.º Once pacientes no tuvieron ninguna complicación, 1 paciente tuvo un megauréter obstructivo en el posoperatorio, y a otro se le apreció una RTU incompleta. En el grupo II, de los 7 pacientes con derivación urinaria (6 ureterostomías, 1 vesicostomía) fueron desderivados mediante cierre de ureterostomía, colocación de doble J, ablación con cuchilla fría en cuatro casos y RTU en tres casos en el mismo acto operatorio. Uno de los pacientes tuvo un sangrado posoperatorio que cedió con sondaje transitorio. El resto de los niños evolucionan favorablemente.
Conclusiones:Hemos observado buena capacidad vesical y resolución del reflujo (60 %) mediante ablación exclusivamente. No hay diferencias importantes entre cuchilla fría y RTU así como en la evolución de los niños tratados por derivación o ablación exclusivamente. Es importante investigar en el posoperatorio la aparición de fallo miogénico, vejiga hiperrefléxica y vejiga de baja acomodación.
P290HERNIA DIAFRAGMATICA CONGÉNITA: TRATAMIENTO PRENATAL MEDIANTE OCLUSION TRAQUEAL, PROCEDIMIENTO EXIT Y SUPERVIVENCIA POSNATAL
Jorge Liras Muñoz, Iván Somoza Argibay, Alberto Sánchez Abuín, Roberto Méndez Gallart, Manuel Gómez Tellado, José Luis Fernández Trisac, José Manuel García-Consuegra Gómez, Humberto Aymerich Cano y Diego Vela Nieto
Hospital Juan Canalejo, A Coruña.
Introducción:La hernia diafragmática congénita (HDC) es una patología sencilla de diagnosticar mediante ecografía fetal. Aquellos casos con mal pronóstico (severa hipoplasia pulmonar) son susceptibles de terapia antenatal. La oclusión temporal mediante clip traqueal o balón endotraqueal de la vía aérea ha resultado ser efectiva tanto a nivel experimental como en humanos a la hora de favorecer el desarrollo del pulmón hipoplásico. Presentamos la primera experiencia realizada en España de supervivencia de un feto diagnosticado de HDC tratado intraútero mediante balón endotraqueal que fue sometido posteriormente a un procedimiento EXIT para su retirada y a la corrección protésica del defecto.
Caso clínico:Primigesta de 29 años con diagnóstico de HDC en la semana 20 con visualización de estómago, asas intestinales y lóbulo hepático izquierdo en tórax. Cociente cefalo-pulmonar < 1. Ante los signos de mal pronóstico para la viabilidad postnatal del feto y la decisión materna de no interrumpir el embarazo, se decide por elequipo de intervención fetal de nuestro centro la posibilidad de corrección antenatal. Tras contactar con el Hospital Universitario de Lovaina, se inserta en este Centro un balón inflable a nivel de carina mediante fetoscopia con un solo trocar de 3,3 mm en la semana 27. Los controles realizados aseguraron la viabilidad, posición adecuada del balón y mejoría del cociente cefalo-pulmonar (1,5). En la semana 33 ante el inicio del trabajo de parto prematuro se decide realización de procedimiento EXIT para extracción del balón mediante traqueobroncoscopia bajo soporte placentario con relajación uretina completa. El procedimiento se lleva a cabo de forma satisfactoria y el neonato es intubado tras la retirada. Sometido a ventilación mecánica de alta frecuencia, fue intervenido en la UCI neonatal en las 24 horas iniciales de vida reparando el defecto con un parche deGore-Tex. La evolución posterior fue favorable, en la actualidad tiene 6 meses de vida, no precisa ningún tipo de soporte ventilatorio, y su desarrollo psicomotor es normal.
Conclusiones:El hecho de que hasta el momento este caso sea el tercer paciente vivo sometido a este técnica le confiere un valor añadido. Creemos que pese a su naturaleza aún en fase de desarrollo experimental, esta opción terapéutica está indicada en aquellos casos de HDC con mal pronóstico antenatal en los cuales no se opte por una interrupción del embarazo. La intervención fetal es la "asignatura pendiente" en los servicios de Cirugía Pediátrica de nuestro país.
P291VALORACION ECOGRAFICA DEL NIÑO CON SOSPECHA DE APENDICITIS AGUDA A LO LARGO DE UN AÑO
Jorge Liras Muñoz, Alberto Sánchez Abuín, Iván Somoza Argibay, Roberto Méndez Gallart, Manuel Gómez Tellado, José Ríos Tallón, Ernesto Pais Piñeiro, Javier Bueno Recio y Diego Vela Nieto
Hospital Juan Canalejo, A Coruña.
Antecedentes y objetivo:La apendicitis aguda es la urgencia quirúrgica más frecuente en edad pediátrica. Muchos han sido los trabajos y estudios en los últimos años encaminados a valorar el papel de la ecografía abdominal en su diagnóstico, tanto en población adulta como pediátrica. Sin embargo, los resultados siguen siendo contradictorios. El objetivo de nuestro trabajo es analizar de forma descriptiva y preliminar el papel que la ecografía abdominal juega en nuestro servicio para el diagnóstico de apendicitis aguda.
Material y método:Estudio retrospectivo de las 88 apendicectomías realizadas en nuestro servicio en el año 2000, recogiendo además de datos generales (edad, sexo, técnica quirúrgica, pruebas complementarias, antibioterapia, cultivos, días de estancia, complicaciones), información sobre estudios ecográficos practicados y su relación con la clínica del niño (típica o atípica), tiempo de evolución al diagnóstico, diagnóstico anatomopatológico definitivo y peso sobre la decisión quirúrgica final.
Resultados:29 niños fueron estudiados ecográficamente (33 % del total), de los cuales 15 presentaban una clínica típica (52 % de los niños estudiados con ECO cuando la clínica típica supuso el 77,2 % del total) y 14 una clínica atípica (48 % de las ECOs frente al 22,7 % del total). La evolución media del cuadro en el momento del diagnóstico fue similar en los niños sometidos y no sometidos a estudio ecográfico (36 horas) De los 29 estudios ecográficos, 10 fueron informados como normales, comprobándose después apendicitis en 8 casos (30 % perforadas). De los 19 estudios ecográficos con datos sugestivos (apéndice tubular dilatado, líquido libre, plastrón o dilatación de asas), sólo 1 caso fue negativo, cuando la incidencia global de apendicectomías en blanco fue del 8 %. Finalmente, los hallazgos de 15 de los 29 estudios ecográficos hechos (52 %) fueron considerados concluyentes.
Conclusiones:La ecografía abdominal para el diagnóstico de apendicitis aguda fue poco utilizada en nuestro servicio (33 % de niños atendidos), siendo la valoración clínica suficiente en la mayoría de casos, manteniéndo índices de apendicectomías en blanco razonables (8 %). Su uso es mayor con significación estadística en niñas, menores de 6 años y en aquellos casos clínicos atípicos o dudosos, pero no rutinariamente. En cuanto a sus resultados, el diagnóstico ecográfico positivo nos parece fiable y certero (VPP 95 %), no siendo así el diagnóstico negativo (VPN 80 %).
P292 SIMPATECTOMIA POR VIDEOTORACOSCOPIA EN HIPERHIDROSIS PALMAR PRIMARIA
Luis Felipe Ávila Ramírez, Ana Lourdes Luis Huertas, José Luis Encinas Hernández, Susana Rivas Vila, Francisco Hernández Oliveros, Pedro Olivares Arnal y Juan Antonio Tovar Larrucea
Hospital Universitario La Paz, Madrid.
Antecedentes y objetivos:La hiperhidrosis palmar primaria (HP) forma parte de una tríada con afectación palmar, axilar y plantar, de etiología desconocida que debuta en la infancia, afectando también a adolescentes y adultos jóvenes. Constituye un problema importante en el niño, interfiriendo en sus actividades cotidianas y en el desarrollo psicológico e integración social. La simpatectomía por videotoracoscopia (SVT) es en la actualidad un tratamiento seguro y eficaz en el manejo de esta patología. Nuestro propósito es valorar los resultados a corto plazo de la SVT en niños con HP.
Métodos:Revisión retrospectiva en un período de 5 años (1998-2002) de 21 pacientes con edad media de 17 años (rango 7-34 años), 17 mujeres y 4 varones. En todos los pacientes se realizó SVT bilateral utilizando un único trocar de 10 mm de diámetro situado en el hueco axilar. Con un neumotórax de 10 mmHg se consigue una adecuada visibilidad del campo quirúrgico. Los ganglios T2, T3 y T4 se localizan sobre la cabeza de la 2.ª, 3.ª y 4.ª costilla. Si presenta HP aislada se realiza simpatectomía de T2 y T3, si existe hiperhidrosis axilar se lleva a cabo también simpatectomía de T4. Es importante la identificación y preservación de T1, que se encuentra rodeado de grasa para evitar complicaciones posteriores (signo de Horner). La duración total aproximada de la intervención es de 20 minutos.
Resultados:En todos nuestros pacientes se produjo un cese inmediato de la sudoración. En el postoperatorio precoz no se presentaron complicaciones derivadas de la técnica (neumotórax, enfisema subcutáneo, miosis y ptosis palpebral). Sudoración compensatoria en tronco y/o miembros inferiores la refirieron solo un tercio. Los 15 pacientes se encuentran totalmente satisfechos con el resultado de la SVT.
Conclusiones:La SVT es la alternativa terapéutica con un alto grado de eficacia y seguridad en la HP. Su realización precoz en los niños afectados por esta patología supone un importante beneficio e incremento de su calidad de vida.
P293HERNIA DIAFRAGMATICA DE APARICION TARDIA. DEBUT CLINICO Y ERRORES DIAGNOSTICOS
Pedro Cortés Mora, Raúl Sánchez Pérez, Manuel González-Ripoll Garzón, José Luis Gómez Llorente, Fco. Javier Aguirre Rodríguez, Moisés Leyva Carmona, M. Ángeles Llamas Guisado, José Vargas Vallejo, Eduardo López Candel y M. del Rosario Jiménez Liria
Hospital Torrecárdenas del SAS, Almería.
Introducción:La hernia diafragmática congénita (HDC) se origina por ausencia o defecto en la fusión de los esbozos diafragmáticos anterior y posterior entre la semana 8.ª y 10.ª del desarrollo embrionario. Aunque la mayoría se detectan precozmente intraútero o inmediatamente después del nacimiento, presentando pocos problemas diagnósticos, hasta el 5 % pueden no tener sintomatología en el período neonatal. Debido a la rareza de la presentación clínica tardía y a los errores diagnósticos que pueden generar, presentamos dos nuevos casos.
Casos clínicos:
Caso 1:Niño de 3 años con período neonatal normal que durante el estudio de 2.º episodio de dolor abdominal se detectan hipoventilación en tercio inferior de hemitórax izquierdo, generando diagnóstico diferencial con neumonía, derrame y malformación. La confirmación de HDC se realiza mediante tránsito gastroesofágico con bario. Evolución satisfactoria en controles posteriores.
Caso 2: Niño de 4 años con período neonatal normal. En el estudio de un catarro de vías altas se detecta masa en mitad inferior de hemitorax izquierdo en la radiografía de tórax. La confirmación de HDC se realiza mediante tránsito gastroesofágico con bario. Durante el acto quirúrgico se halló malrotación intestinal asociada. Sufre neumotórax tras la intervención que se resuelve espontáneamente. Evolucionando satisfactoriamente en controles posteriores.
Comentario:Con estos dos casos queremos recordar que las HDC pueden manifestarse clínica y radiológicamente de forma tardía y heterogénea, suponiendo por lo tanto una entidad a tener en cuenta en el diagnóstico diferencial de las neumopatías del niño.
CUIDADOS INTENSIVOS
P294ACLARAMIENTO DE CREATININA EN EL POSTOPERATORIO DE CIRUGIA CARDIACA INFANTIL
Patricia Aparicio, Andrés José Alcaraz Romero, M. del Mar Guerrero Soler, Esther Panadero Carlavilla, Carlos Romero Román, Rafael Muñoz-Pacheco Román y Amaya Bustinza Arriortua
Hospital General Universitario Gregorio Marañón, Madrid.
Objetivo:Comparar el aclaramiento de creatinina calculado con recogida de orina de 24 horas (CCr 24 h) con el aclaramiento de creatinina teórico en relación a la talla (calculado según la fórmula de Schwartz [CCr FS]), en niños que reciben tratamiento con furosemida en el postoperatorio de cirugía cardiaca.
Métodos:Estudio prospectivo observacional, realizado sobre 40 niños intervenidos de patología cardiaca, ingresados en Unidad de Cuidados Intensivos Pediátricos, que clínicamente requieren tratamiento diurético con furosemida. Se recoge orina de 24 horas y se mide la creatinina sérica durante cuatro días consecutivos, para el cálculo de CCr 24h. El cálculo teórico se realiza empleando la fórmula de Schwartz (CCr FS = K. Talla/creatinina sérica). Para determinar la concordancia entre ambos métodos empleamos el análisis de Bland y Altman.
Resultados:Los valores del CCr calculados con cada uno de los métodos diariamente, así como su diferencia (Dif CCr) y la correlación entre las dos mediciones se presentan en la siguiente tabla:

Utilizando el método de Bland y Altman apreciamos que, en los casos de CCr < 60 ml/min/1,73 m2, todos los valores se encuentran dentro de un rango de diferencia menor de 20 %.
Conclusiones: 1.La fórmula de Schwartz sobreestima el valor del CCr 24 h.2. Los valores de CCr FS se aproximan más a los obtenidos con el CCr 24 h cuando la cifra es inferior a 60 ml/min/1,73 m2.3. Ambos métodos se pueden utilizar en el cálculo del CCr en niños que reciben tratamiento con furosemida durante el postoperatorio de cirugía cardiaca, porque las diferencias entre ellos no tienen repercusión sobre la actitud terapéutica.
P295
BALANCE DE SODIO CON PERFUSION CONTINUA DE FUROSEMIDA DURANTE EL POSTOPERATORIO DE CIRUGIA CARDIACA INFANTIL
Patricia Aparicio, Andrés José Alcaraz Romero, Carlos Romero Román, Susana Elena Zeballos Sarrato, Rafael Muñoz-Pacheco Román, Esther Panadero Carlavilla y Carlota Seriñá Ramírez
Hospital General Universitario Gregorio Marañón, Madrid.
Objetivo:Determinar el balance de sodio (Na) en niños que reciben perfusión continua de furosemida durante el postoperatorio de cirugía cardiaca con circulación extracorpórea (CEC).
Métodos:Estudio prospectivo observacional, realizado sobre 20 niños intervenidos con CEC, ingresados en Cuidados Intensivos Pediátricos, que clínicamente requieren tratamiento diurético con furosemida. Se contabilizan los aportes diarios de Na, así como las pérdidas de Na renales y extrarrenales (digestivas y por drenajes), durante siete días consecutivos (días 2.º a 8.º de postoperatorio). Se recogen además los valores diarios de Na sérico y la dosis de furosemida administrada. Análisis de datos con SPSS 10,0 para Windows.
Resultados:Los valores de Na (mEq/l), aportes de Na (mEq/kg/día), balance de Na (mEq/kg/día), y dosis de furosemida (mg/kg/día) diarios se presentan en la siguiente tabla:

El Na sérico descendió significativamente durante el tratamiento con furosemida en perfusión. El balance acumulado de Na de los días 2, 3 y 4 fue 20,8 ± 12,7 mEq/kg. El balance acumulado de Na hasta el 4.º día más negativo (< P50) se asoció con la dosis de furosemida, y no se relacionó con el Na sérico, los aportes de Na ni la osmolaridad sérica. Se encontró correlación entre la dosis de furosemida y el balance de Na a partir del 8.º día de tratamiento.
Conclusiones: 1.El empleo de furosemida en perfusión continua durante el postoperatorio de cirugía cardiaca produce un balance negativo de Na e hiponatremia.2. El balance de Na negativo está en relación con la dosis de furosemida administrada.
P296
A PROPOSITO DE UNA OBSERVACION DE INSUFICIENCIA RESPIRATORIA AGUDA TRAS EXTUBACION POSTANESTÉSICA
Gemma Novoa Gómez, Susana Rey García, Cristina Lorenzo Legeren, Clara García Cendón, Celia M. Rodríguez Rodríguez, Federico Martinón Sánchez, Pilar Fenández Eire y M. de los Ángeles Montes Deza
Complejo Hospitalario de Ourense.
La insuficiencia respiratoria grave de inicio brusco tras extubación postanestésica, sin resolución mediante reintubación, plantea la necesidad de establecer un diagnóstico diferencial de carácter urgente que permita el tratamiento inmediato.
Una observación singular de este proceso acompañado de ocupación alveolar justifica esta presentación.
Aportamos un caso de un varón de 2 años de edad desprovisto de antecedentes de interés, con exploración física normal, salvo hernia inguinal derecha, por la que fue sometido a cura quirúrgica simple bajo anestesia general, y que al ser extubado presentó signos clínicos de insuficiencia respiratoria aguda grave, acidosis respiratoria e hipoxemia, con una radiografía de tórax con ocupación difusa pulmonar con patrón alveolar de predominio medial compatible con edema agudo de pulmón. Bajo tratamiento médico con oxígeno, adrenalina, corticoides y diuréticos se alcanzó la normalización clínica y radiológica 24 horas más tarde.
La historia natural del proceso, los hallazgos radiológicos y la propia evolución permitieron el diagnóstico de edema pulmonar agudo secundario a laringoespasmo, proceso que ha de ser considerado como complicación postanestésica por obstrucción alta, para diferenciarlo de otras anomalías obstructivas, infecciosas o broncoaspirativas, así como, de otras etiologías de edema pulmonar, dado que la actitud terapéutica sería otra y podría conducir a acciones yatrogénicas incluso irreversibles.
P297DEPURACION HEPATICA EXTRACORPOREA (SISTEMA MARS)
Kay Boris Brandstrup Azuero, Susana Elena Zeballos Sarrato, Andrés José Alcaraz Romero, Jesús López Herce, M. Elena Cela de Julian y Raúl Roberto Borrego Domínguez
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción:La diálisis con albúmina (sistema MARS) se caracteriza por la retirada especifica de sustancias unidas a la albúmina y puede constituir un reemplazamiento eficaz de la función excretora y detoxificadora hepática. Es una técnica con poca experiencia en niños que puede ser útil, entre otras en el tratamiento de la insuficiencia hepática aguda.
Caso clínico:Varón de 14 años de edad con Linfoma Linfoblástico que a los 10 días de recibir trasplante de progenitores hematopoyéticos presenta cuadro de shock con afectación multiorgánica (hemodinámica, respiratoria, renal y hepática). Precisa tratamiento con inotrópicos, ventilación mecánica, hemodiafiltración veno-venosa continua y terapia antiinfecciosa de amplio espectro. Tiene deterioro hepático progresivo con unos parámetros máximos: GOT 2155 U/l, GPT U/l 1360 U/l, GGT 305 U/l, FA 1150 U/l, Amonio 156 μg/dl, Bilirrubina directa 15 mg/dl, Albúmina 2,5 g/l y coagulopatía (INR 1,8). Se decide comenzar tratamiento con soporte extracorpóreo hepático (sistema MARS) al 7.º día del ingreso que se mantiene de forma ininterrumpida durante 3 días con buena tolerancia hemodinámica y sin presentar complicaciones, apreciándose mejoría progresiva de la función hepática hasta GOT 64 U/l, GPT 100 U/l, GGT 114 U/l, Bilirrubina directa 5 mg/dl, Amonio 41 μg/dl, Albúmina 2,8 g/dl y mejoría de la coagulación (INR 1,25). En biopsia hepática vía transyugular se observan alteraciones compatibles con enfermedad venooclusiva hepática sin infiltración tumoral. Al ingreso presenta estado de conciencia normal, iniciándose sedoanalgesia y relajación, retirándose para valoración neurológica durante el deterioro hepático. En el EEG se aprecian alteraciones compatibles con encefalopatía hepática. Con el tratamiento MARS presenta ligera mejoría, con respiración espontánea, pero persiste coma sin apertura ocular. Se realiza TAC cerebral donde se observa masa en región frontal y TAC abdominal, existiendo múltiples lesiones tumorales intestinales. Ante la irreversibilidad de la enfermedad de base no se llevan a cabo medidas extraordinarias falleciendo al 13.º día del ingreso.
Conclusiones:La terapia con sistema MARS puede ser una técnica útil como soporte transitorio de mantenimiento de pacientes con fallo hepático agudo.
P298 SINDROME COMPARTIMENTAL ABDOMINAL. UN DIAGNOSTICO A TENER EN CUENTA
Ana Arévalo Garrido, Antonia Solas Beltrán, Irene Peláez Pleguezuelos, Rosa M. Rodríguez García, M. Dolores Gámez Gómez, F. Javier Navarro Barragán, Jesús de la Cruz Moreno y Felipe González Rivera
Hospital Universitario Ciudad de Jaén.
Introducción:El síndrome compartimental abdominal (SCA) se caracteriza por el aumento de la presión intrabdominal (PIA) y el conjunto de alteraciones fisiopatológicas secundarias a la misma.
Caso clínico:RN que ingresa a las 24h de vida por distensión abdominal. A. Perinatales: sin interés; parto eutócico a las 41s, Apgar 8/8/9. Exploración:sólo destaca el abdomen distendido, globuloso, blando, no doloroso a la palpación, ano perforado. A su ingreso presenta mala tolerancia oral, con expulsión de meconio a las 48h de vida. E. Complementarios: radigrafía de abdomen: dilatación del colon; Enema opaco: colon distendido con segmento distal estenótico compatible con enferdad de Hischprung. A las 24h de realizarse el enema presenta fiebre,signos de shock y heces mucosanguinolentas, sugiriendo ECN perforada asociada a. A los 9 días de vida se practica 1.ª intervención quirúrgica abdominal apreciando abundante líquido libre peritoneal, intestino delgado con zonas equimóticas, colon dilatado y de aspecto inflamado, practicando colostomía disociada. A los 13 días de vida se realiza la 2.ª intervención por mayor distensión abdominal y oligoanuria dejando ileostomía de descarga. En el postoperatorio presenta insuficiencia respiratoria que precisa de ventilación mecánica, insuficiencia renal aguda resistenta a expansión de volumen, diuréticos y hemofiltración y PIA entre 40-60 mmHg medida en vejiga y con cateter venoso intrabdominal. Ante el diagnóstico de SCA se procede a la tercera intervención, con descompresión quirúrgica abdominal, dejando placa de silicona. PIA postoperatoria descenció a 15, mejorando la función respiratoria y restableciendo diuresis a las 48h. Se practica cierre quirúrgico definitivo de la pared abdominal 21 días más tarde.
Conclusiones:1. La elevación de la PIA comprende alteraciones que afectan al sistema cardiovascular, pulmonar, renal, esplácnico y nervioso central. 2. La elevación de la PIA iniciaría la hipoperfusión esplácnica que, de no detectarse precozmente provocaría el SCA y finalmente fallo multiorgánico.3. Es importante, no sólo un diagnóstico precoz, sino también la realización de un tratamiento descompresivo inmediato.
P299
INTOXICACIONES: REVISION DE CASOS EN LOS ULTIMOS 15 AÑOS
Raquel Gómez Casares, José Luis Ruibal Francisco, Bruno Nievas Soriano y Olga Pérez Rodríguez
Hospital Clínico San Carlos, Madrid.
Introducción:Las intoxicaciones son una consulta común en nuestras salas de Urgencia. En algunos casos son accidentales, pero en otros son voluntarias. Los rangos de edades son muy variados, así como el compuesto que ha producido la intoxicación, lo cual nos indica el tratamiento a elegir. La clínica secundaria y la gravedad del cuadro producido dependen de todos esos factores.
Objetivos:Estudiar los casos de población pediátrica que han sufrido una intoxicación y que han precisado ingreso en la Unidad de Cuidados Intensivos en los últimos 15 años en nuestro Centro. Se desea conocer la causa de la intoxicación, el producto causante de la misma, la clínica presentada en el momento de su ingreso junto con las posibles complicaciones posteriores, y el tratamiento empleado durante su ingreso, a fin de mejorar nuestro protocolo de actuación en cada caso.
Métodos:Recogida de datos de las historias de los niños ingresados en la Unidad de Cuidados Intensivos de nuestro Centro. Se realiza un tratamiento estadístico para exponer los distintos resultados, y se compara con los datos publicados por otros estudios.
Resultados:Tal y como se propugna en muchos estudios, en general las intoxicaciones accidentales son más comunes entre los niños menores de 5 años, y las voluntarias entre los adolescentes, predominando el sexo masculino en las primeras y el femenino en las segundas. Las benzodiacepinas parecen ser el producto más frecuentemente implicado en las intoxicaciones, tanto voluntarias como accidentales, y la intoxicación etílica muestra un gran predominio en las intoxicaciones voluntarias entre los adolescentes. En general la clínica ha sido leve, afectando al SNC principalmente y no han presentado complicaciones posteriores. El tratamiento empleado ha sido el adecuado para cada agente.
Conclusión:Las intoxicaciones en los últimos 15 años en nuestro área no han variado de manera importante en cuanto a epidemiología y a clínica, aunque sí han cambiado ligeramente los tóxicos implicados, obligando a modificar las pautas de tratamiento.
P300TERLIPRESINA EN EL TRATAMIENTO DEL SHOCK SÉPTICO REFRACTARIO A CATECOLAMINAS
Manuel Fernández Sanmartín, Federico Martinón Torres, Antonio Rodríguez Núñez, Montserrat López Franco, Sonia Marcos Alonso, José M. Iglesias Meleiro y José M.ª Martinón Sánchez
Hospital Clínico Universitario. Complejo Hospitalario Universitario, Santiago de Compostela.
Introducción:En la infancia, el shock séptico puede cursar con hipotensión severa, que no responde a dosis crecientes de catecolaminas. En este contexto, se han buscado fármacos alternativos que permitan mejorar la situación hemodinámica del paciente, existiendo experiencias positivas con la utilización de vasopresina. Sin embargo, la disponibilidad actual de este fármaco en nuestro medio es limitada lo que dificulta su utilización. La terlipresina, un análogo sintético de vida media larga de la vasopresina que al igual que esta se utiliza en el tratamiento de las varices esofágicas y la diabetes insípida, se ha empleado ocasionalmente en adultos con shock séptico refractario a noradrenalina. Describimos un caso de shock séptico con hipotensión refractaria a altas dosis de aminas que respondió positivamente a la administración de terlipresina.
Caso clínico:Paciente de 2 años diagnosticado de neuroblastoma abdominal estadio IV que fue sometido a quimioterapia mieloablativa y rescate mediante autotrasplantre de progenitores hematopoyéticos, desarrollando en el período inicial postrasplante un fallo multiorgánico con insuficiencia renal aguda, síndrome de distress respiratorio agudo, mucositis severa (grado III) y shock séptico. Progresivamente el shock se hizo refractario al tratamiento aminérgico optimizado. Se inició tratamiento con Terlipresina intravenosa a dosis de 0,3 mg cada 4 horas, observándose una mejoría hemodinámica rápida y mantenida. Una hora después de la administración de la primera dosis de terlipresina la tensión arterial media ascendió de 40 mmHg a 78 mmHg, permitiendo disminuir la infusión de noradrenalina hasta su retirada en las 24 horas siguientes. El tratamiento se mantuvo durante 5 días y se retiró de forma progresiva a lo largo de los dos días siguientes, sin que se produjera un deterioro de la situación hemodinámica, ni se observaran efectos secundarios reseñables.
Conclusión:La terlipresina puede considerarse, bajo la premisa de la necesidad de estudios aleatorizados que valoren su eficacia real y posibles secundarismos, como una alternativa eficaz en el soporte hemodinámico de los pacientes pediátricos con shock séptico refractario a catecolaminas.
P301TRASLADO INTERHOSPITALARIO DE PACIENTES PEDIATRICOS CRITICOS EN UVI MOVIL. ¿EXISTEN DIFERENCIAS CON EL ADULTO?
Pedro Antonio García Ramiro, Manuel Linares Abad, M. Dolores Benítez Redondo, Valle Yeste Jiménez, Isabel López Medina, Marta Sanjuán Peláez, Antonio Guisado Calderón e Ignacio José Serrano Díaz
Universidad de Jaén, y Transporte de Pacientes Críticos, Jaén.
Antecedentes y objetivos:El Transporte de enfermos entre hospitales constituye una necesidad de primer orden en el Sistema Sanitario, que conlleva costes humanos y material de considerable magnitud. Sin embargo, la mayor parte del material que se sitúa dentro de los vehículos sanitarios es destinado al tratamiento, estabilización y traslado del paciente adulto, con la falsa idea de que los mismos materiales pueden adaptarse al paciente pediátrico, pues es tratado injustamente como "un adulto en pequeño".
El objetivo del presente estudio es conocer las características específicas de los pacientes pediátricos trasportados por nuestros vehículos asistenciales con el fin de llevar a cabo los cambios y modificaciones adecuadas en el vehículo, mejorando de este modo, la calidad asistencial prestada.
Métodos:Se ha realizado un estudio descriptivo retrospectivo del total de las historias clínicas contenidas en nuestros archivos desde la apertura del servicio en Julio de 1999 hasta Diciembre de 2002. Se analizan datos relativos a las siguientes variables: sexo, edad, hospital de origen, hospital de destino, diagnóstico (Cie-9 y Cie-10), soporte y material empleado durante el traslado,...
Resultados:Del análisis de los datos se deriva que el 34 % del total de nuestros pacientes son menores de 14 años, sin encontrar diferencias significativas entre ambos sexos. De estos pacientes el 15 % se corresponde con pacientes lactantes y neonatos que son los que requieren mayores cambios en la adaptación de las unidades, tanto en su inmovilización como en el material electromédico y fungible contenidos en el habitáculo asistencial. Las modificaciones llevadas a cabo para adecuar nuestras unidades al transporte de pacientes pediátricos críticos se muestran en imágenes.
Conclusiones:La adecuación del vehículo asistencial, mostrada en las imágenes, han mejorado las deficiencias e incluido las carencias detectadas tras el análisis de los datos. Podemos afirmar que todas las modificaciones de las unidades asistenciales, unidas a la formación específica del personal asistencial mejoran la calidad de los servicios prestados.
P302TRATAMIENTO CON CONCENTRADO DE PROTEINA C EN PURPURA SECUNDARIA A SEPSIS NEUMOCOCICA
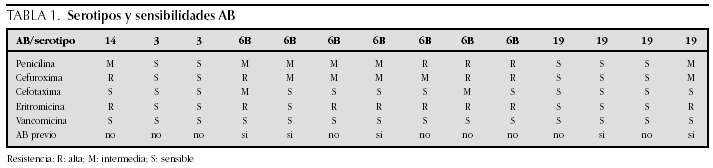
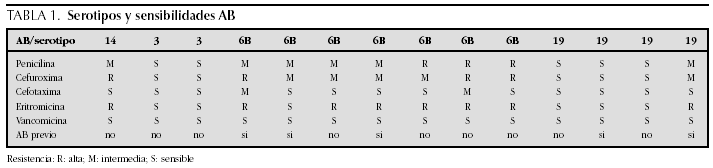
M. Ángeles Murillo Pozo, Antonio Vázquez Florido, Cristina Montero Valladares, Alberto Varona García, José Domingo López Castilla, Juan Antonio Soult Rubio, José Carlos Flores González, Miguel Muñoz Sáez, Mercedes Carranza Conde y Aníbal Tovaruelas Santos
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción y objetivo:El tratamiernto precoz con concentrado de proteina C puede limitar la activación del sistema hemostático, interrumpir la CID y finalmente normalizar la microcirculación. Describimos nuestro primer caso de uso de Concentrado de proteina C, junto a terapia convencional en una niña con Shock séptico, CID y púrpura secundarios a Sepsis neumocócica.
Caso clínico:Niña de 16 meses que ingresa en nuestra UCI-P por fiebre elevada, exantema petequial, afectación del estado general, coagulación intravascular diseminada (CID) y fallo hemodinámico, que precisa resucitación volumétrica y drogas inotrópicas. Rash purpúrico con lesiones vasculares pregangrenosas en área nasal. Iniciamos tratamiento con Proteina C 10 U.I./kg peso en 10 minutos intravenosa, después bolo de 100 U.I./kg de peso en 1 hora, seguido de 10 U.I./kg/hora en infusión intravenosa contínua. Las dosis posteriores se ajustaron para mantener unos niveles adecuados de Proteina C. En la tabla 1 podemos ver los niveles de la función de la proteina C, tiempo de protrombina (TP), INR, tiempo de tromboplastina activada (TPTA), fibrinógeno y dímeros D.

ENDOCRINOLOGIA
P303SINDROME DE MCCUNE-ALBRIGHT
M.ª José García Arias, Almudena del Pino de la Fuente, Silvia Ortega Pérez, Rafael Vera Medialdea, Lourdes Escudero Ruiz de Lacanal, M. José Martínez Aedo, Juan Pedro López Sigueros y Antonio Jurado Ortiz
Hospital General Carlos Haya, Málaga.
Introducción:El Síndrome de McCune-Albright (SMA) se describe como un entidad esporádica, caracterizada por presencia de displasia fibrosa poliostótica, lesiones cutáneas hiperpigmentadas y alteraciones endocrinológicas, la más frecuente de las cuales es la pubertad precoz independiente de gonadotropinas.
Caso clínico:Niña de 1,10/12 años que presenta desde hace 4 meses episodios de metrorragia de 1 o 2 días de duración y telarquia bilateral.
Exploración física:Peso: 13 kg; Longitud: 80,8 cm.
Buen estado general. Normoconformada. ACR: normal. Abdomen: blando y depresible, sin masas ni megalias. Telarquia bilateral III con pigmentación areolar. No adrenarquia. Genitourinario femenino normal. Máculas hiperpigmentadas irregulares que ocupan parte de espalda y hombros. No deformidades óseas.
Pruebas complementarias:FSH: 0,1 mU/ml; LH: 0 mU/ml; PRL: 40 ng/ml; Estradiol: 63 pg/ml; Cortisol: 170 ng/ml; ACTH: 38 pg/ml; TSH: 3,20 mcU/ml; T4L: 11,7 pmol/l; PTH: 16 pg/ml; Ecografía pélvica (coincidiendo con metrorragia): hiperplasia endometrial a nivel de fundus. Quiste ovárico derecho de 4,7 x3,2 x 2,6 cm; Mapa óseo: normal; Edad ósea: 1,6/12 para EC: 1,10/12; RMN Hipotálamo-Hipófisis con contraste: normal; Estudio genético en biposia de piel: pendiente de resultado.
Evolución:Ante los hallazgos clíncos, analíticos y ecográficos compatibles con pubertad precoz periférica, se inició tratamiento con Tamoxifen (10 mg/día), con lo que se consiguió la regresión de la telarquia, pero persistiendo episodios de metrorragia., por lo que a los 3 años de edad se inicia tratamiento con Anastrazol (1 mg/día), pero continúa presentando quistes ováricos funcionantes con metrorragia a pesar de tratamiento frenador.
Conclusiones:El diagnóstico del SMA es clínico, cuando aparecen al menos dos de los síntomas de la tríada clásica, aunque recientemente se sugiere que se puede realizar con uno cualquiera de los signos de este amplio síndrome, sobre todo, si se puede demostrar la mutación en los tejidos afectados.
P304SINDROME DE TURNER Y DUCTOPENIA BILIAR
Natalia Ramos Sánchez, Ángel Carrillo Herranz, Inmaculada Sánchez Pérez, Luz Golmayo Gaztelu, Ana Coca Pérez, Emma Lara Orejas, M. del Milagro Alonso Blanco y Cristina Camarero Salcés
Hospital Ramón y Cajal, Madrid y Universidad de Alcalá de Henares, Madrid.
Introducción:En el S. de Turner (ST) se han observado alteraciones hepáticas con un sustrato anatomo-patológico variado. Describimos una paciente con ST, elevación persistente y asintomática de los enzimas hepáticos y ductopenia biliar leve en la biopsia hepática.
Caso clínico:Paciente de 18 años de edad con ST, tratada con GH humana (desde los 8 hasta los 16 años), estrógenos equinos conjugados (12-16 años) y etinil estradiol y gestodeno (16-18 años). Desde los 11 años de edad, previamente a la administración de estrógenos, presenta una elevación asintomática de ASAT y ALAT inicialmente y posteriormente también de gamma-GT; en varias ocasiones se detectaron anticuerpos antinucleares (1/160). La ecografía hepática fue normal. La biopsia hepática mostró una arquitectura conservada. El 25 % de los espacios porta presentaba estructuras vasculares aisladas sin conducto biliar acompañante. Se suspendió temporalmente el tratamiento hormonal y se administró ácido ursodeoxicólico, observándose 4 meses después normalización de gamma-GT y GOT con mínima elevación de GPT (50 U/l).
Comentarios:La elevación asintomática de enzimas hepáticos en pacientes con ST ocurre con frecuencia variable en diferentes series (20-80 %) y aumenta con la edad. La presencia de ductopenia biliar leve no se ha documentado previamente. El curso no progresivo de las alteraciones hepáticas con nula expresión clínica, ductopenia leve y ausencia de fibrosis en la biopsia hepática de esta paciente descartan los síndromes colestásicos crónicos y la hepatitis autoinmune. Es improbable que la terapia estrogénica sea responsable de las alteraciones observadas dado el comienzo previo a su administración, sin embargo no podemos excluir definitivamente la participación de estos fármacos en el mantenimiento de la lesión. La asociación de ANA y la ductopenia biliar observada en esta paciente, podría sugerir la naturaleza autoinmune de la lesión biliar.
Conclusión:Las alteraciones hepáticas en pacientes con ST no son infrecuentes por lo que es aconsejable la vigilancia periódica con pruebas de funcionalismo hepático. La biopsia hepática está indicada cuando se detecten alteraciones enzimáticas persistentes. Las pacientes con ductopenia biliar leve, pueden beneficiarse del tratamiento con ácido ursodeoxicólico.
P305PSEUDOHIPOPARATIROIDISMO ASINTOMATICO COMO DIAGNOSTICO CASUAL
M.ª Elisa Corrales del Río, M. Pilar Gutiérrez Díez, Florencio Jiménez, Elena García-Zarza Martínez, Marta Ruiz, Cristina Ferrero Martín y David Montes Bentura
Hospital Universitario de Getafe.
Introducción:El pseudohipoparatiroidismo (PHP) es una entidad poco frecuente en Pediatría, descrito por Albright (1942). Se caracteriza por: hipocalcemia, hiperfosfatemia con PTH elevada y función renal normal. Se clasifica en tipo I (no aumenta AMPc urinario tras administrar PTH exógena, test de Ellsworth-Howard negativo) y tipo II (test positivo, aumenta AMPc). El tipo Ia se asocia a resistencia a otras hormonas (TSH, ADH, glucagón...), porque se debe a la actividad disminuida de la proteína Gs. Esta proteína tiene la función de transducir la señal desde el receptor para la PTH activando la adenilciclasa y produciendo un aumento del AMPc. En el tipo Ib la actividad de las proteínas Gs es normal. El fenotipo Albright (talla baja, obesidad, retraso mental, calcificaciones intracraneales y de partes blandas, acortamiento de metacarpianos y metatarsianos...) suele ir asociado a la forma Ia. El tratamiento consiste en suplementos de calcio y vitamina D; su objetivo es la normalización de la calcemia y de las cifras de PTH evitando la osteopenia.
Caso clínico:Niña de 11 años controlada en Alergología Infantil por asma por ejercicio, hallándose casualmente hipocalcemia (6,9) e hiperfosforemia (7). Antecedentes: enuresis primaria (varios antecedentes familiares). No ha presentado crisis secundarias a hipocalcemia. Madre hipotiroidea (desconoce la causa) en tratamiento sustitutivo. Diversos antecedentes familiares de neoplasias en distintas localizaciones. Exploración: Ec 11,5a, Et 10,9a, peso 44,3 kg (P75-95), talla 140 cm (DS 0,8, P20), IMC 22,6, desarrollo puberal P1S1A1 (estadio I de Tanner), TA 116/74, FC 85, cociente intelectual normal.
Pruebas complementarias:1. Bioquímica: normal salvo Ca 6,9; Ca++ 4,13; P 7; PTH 577.2. Función renal, TSH, calcitonina, osteocalcina: normales. 4. CT de cráneo: normal. 5. Serie ósea: normal. 6. Cariotipo 46XX. 7. AMPc en plasma tras 2 horas de ayuno: 41,8 nmol/l (9,6-26,8). AMPc en orina normal.
Diagnóstico:PHP, posiblemente Ib, pendiente de completar estudio (prueba de Ellsworth-Howard y actividad de Gs alfa).
Conclusiones:1. La ausencia de fenotipo OHA en casos de hipocalcemia con hiperfosforemia no descarta PHP, por lo que hay que determinar PTH. 2. La no disponibilidad de PTH exógena y estudio de la actividad de las proteínas Gs dificultan la clasificación.
P306HIPOTIROIDISMO ADQUIRIDO: ASSOCIAÇÃO CASUAL COM OSTEOGÉNESE IMPERFEITA TIPO 3 OU SECUNDARIO AO TRATAMENTO COM PAMIDRONATO
Jorge Sales Marques
Centro Hospitalar de Vila Nova de Gaia, Portugal.
Lactente de 16 meses de idade, sexo feminino, foi - lhe diagnosticado no período neonatal osteogénese imperfeita tipo 3, por apresentar fractura bilateral do fémur e escleróticas azuis. Nasceu de parto eutócico, 39 semanas de gestação. Peso ao nascer - 3160 g (percentil 25-50), estatura - 45, 5 cm (percentil 5) e perímetro craneano - 35 cm (percentil 75). Rastreio de hipotiroidismo congénito negativo. Pais jovens e não consanguíneos. Ao 5 mês de vida, inicia tratamento com pamidronato, na dose de 0,5 mg/kg com ciclos de 3 dias, repetidos cada 6 semanas, conforme o protocolo de tratamento preconizado por Ploktin H e col. em 2000, em crianças com osteogénesis imperfeita tipos 3 e 4, com idades inferiores a 3 anos. Desde então, fez 10 ciclos de tratamento com pamidronato, sempre sem intercorrências e com melhoria da densidade óssea, redução do fósforo e da fosfatase alcalina no sangue e aumento da paratormona plasmática, cálcio e fósforo urinário. A qualidade de vida da criança melhorou de forma extraordinária, a deformidade óssea reduziu e fez apenas uma pequena fractura do pé durante estes meses. Actualmente, caminha sem apoio. Durante o último ciclo, foi efectuado estudo da função tiroideia que revelou o valor de TSH aumentado, com Ft4 normal Foi reconfirmado o aumento numa segunda colheita de sangue. Os anticorpos antitiroideus estavam normais. Iniciou tratamento com tiroxina, com normalização da função tiroideia após um mês. A associação de hipotiroidismo com osteogénese imperfeita não tem sido descrita. Os efeitos secundários da utilização do pamidronato são conhecidos, mas não relacionados com hipotiroidismo adquirido. Qual a etiologia?
P307COLESTASE, HIPOGLICEMIA E MICROPÉNIS EM LACTENTE DE 4 MESES
Jorge Sales Marques, Teresa Caldeira, Carla Brandão,
Susana Pereira y Eduarda Marques
Centro Hospitalar de Vila Nova de Gaia, Portugal.
Lactente de 4 meses de idade, sexo masculino, recorre ao SU, por quadro de prostração. Ao exame físico encontrava - se ictérico, hipotónico, com micropénis e hipoglicemia. Tinha crescido 6 cm em 3 meses. Nasceu de cesariana às 39 semanas de gestação. Gravidez vigiada, pais jovens, não consanguíneos. Peso ao nascer - 3525 g, estatura - 47 cm. Apgar 9/10. Foi internada em neonatologia ao 3 dia de vida por hipotonia, cianose, hipoglicemia e hiperbilirrubinémia. Detectado infecção urinária por E. coli, com ecografia renopélvica normal. Alta medicado com trimetropin.
Foram efectuados exames complementares de diagnóstico que revelaram alterados para: hemoglobina-8,5 g/dl, TGO-56 U/l, TGP-110 U/l, gama GT-454 U/dl, fosfatase alcalina-547 U/dl, bilirrubina total-7,4 mg/dl e directa-6, 9 mg/dl, cortisol-0, 24 μg/dl (6,2-19,4) T4-5, 0 μg/dl (6,5-17,5), FSH- < 0,10 m UI/ml (1,5-12,4), LH- < 0,10 m UI/ml (1,7-8,6), prolactina-3,93 ng/ml (4,1-18,4), insulina- < 0, 20 mU/ml (3,0-17,0), ressonância magnética cerebra - hipoplasia da hipófise. Entre outros exames, o rastreio de doenças infecciosas e metabólicas foi normal. Apesar de não ter sido feito prova de estimulação de hormona de crescimento por a quantidade de sangue necessária para o estudo ser grande (4 ml por tempo), após ter iniciado tratamento com hormona de crescimento, tiroxina e hidrocortisona, às 48 horas as glicemias normalizaram. Foi feita prova terapêutica com retirada de hormona de crescimento, tendo após dois dias voltado a fazer hipoglicemia. A função hepática normalizou após um mês. Em 3 semanas, o bebé cresceu 3 cm. Os dados apontam para um caso clínico de panhipopituitarismo.
O panhipopituitarismo é uma patologia grave e requer diagnóstico e tratamento atempado para evitar sequelas. Nos rapazes, o diagnóstico é mais precoce pela associação de micropénis e hipoglicemia. É rara a apresentação clínica com icterícia colestática.
A colestase é secundária a défice de hormona de crescimento e/ou cortisol, produzindo - se uma imaturidade na síntese e transporte de sais biliares. Normaliza em média 6 a 7 semanas após início da terapêutica hormonal
P308
AVALIAÇÃO CLINICA E PREVISÃO DA ESTATURA FINAL EM CRIANÇAS COM S. DE TURNER
Jorge Sales Marques, Rosa Campos, Sonia Aires y José Soares
Centro Hospitalar de Vila Nova de Gaia, Portugal, Hospital São Sebastião, Santa Maria da Feira (Portugal) y Instituto de Genética Médica Jacinto Magalhães, Porto (Portugal).
Objectivo:Revisão de 7 casos de crianças com S. de Turner, com relevância sobre idade do diagnóstico, fenótipo, doenças associadas e eficiência do tratamento com hormona de crescimento em relação a previsão da estatura final.
Material e métodos:Foram revistas 7 casos de crianças com S. Turner seguidas na consulta de endocrinologia pediátrica do Centro Hospitalar de Vila Nova de Gaia desde 1999 e avaliadas os seguintes itens; idade da criança na altura do diagnóstico, fenótipo, cariótipo, doenças associadas, tratamento, objectivo parenteral e previsão da estatura final antes e após o início do tratamento com hormona de crescimento. O cariótipo foi realizado a partir de cultura de linfócitos sanguíneos pelo Centro de Genética Clínica em dois casos e pelo Instituto de Genética Médica nos restantes casos. O estudo da função tiroideia e anticorpos antitiroideus foi efectuado respectivamente no Laboratório de Endocrinologia e Imunologia do Centro Hospitalar de Vila Nova de Gaia.
Resultados/conclusões:1. A idade média do diagnóstico foi tardia - 7, 5 anos. 2. Em relação ao fenótipo, há a destacar que em apenas um caso (14 %) tinha pterigium colli e hipertrofia do clitóris e também em um caso (14 %), a forma de apresentação foi linfedema das mãos e dos pés.3. O cariótipo 45 XO foi o resultado mais frequente (57 %). 4. A tiroidite linfocitária foi frequente (71 %), tendo necessidade de fazer tratamento com tiroxina em dois dos casos afectados (40 %).5. A altura final sem tratamento seria em média de 142, 2 cm, longe da média do objectivo parenteral de 163, 4 cm.6. Após o início do tratamento com hormona de crescimento, a previsão da altura final passou a ser de 150, 0 cm, ou seja um ganho de 7, 8 cm.7. É fundamental que em todas as crianças do sexo feminino com baixa estatura, sem uma explicação plausível para a sua situação, o cariótipo seja incluído como meio auxiliar de diagnóstico, atendendo que quanto mais precoce for o tratamento com hormona de crescimento, melhor será o prognóstico em relação a estatura final.
P309SITUACION INTERSEXUAL Y CANCER DE MAMA EN UNA FAMILIA CON MUTACION EN EL GEN DEL RECEPTOR DE ANDROGENOS
Marta López Capapé, Margarita Revenga Parra, Esmeralda Colino Alcol, M. del Carmen Miranda Cid, Ángel Carrillo Herranz, M. del Milagro Alonso Blanco y Raquel Barrio Castellanos
Hospital Ramón y Cajal, Madrid y Universidad de Alcalá de Henares, Madrid.
Introducción:Las mutaciones en el gen del receptor de andrógenos (AR), localizado en el brazo largo del cromosoma X, dan lugar a un amplio espectro de alteraciones en la diferenciación sexual masculina, desde una feminización completa hasta infertilidad en pacientes con virilización adecuada.
Presentamos una familia con situación intersexual y carcinoma de mama en varios miembros de la misma con resistencia parcial a andrógenos.
Caso clínico:Varón de 19 meses con situación intersexual nacido de embarazo, parto y período neonatal normales sin episodios de deshidratación. Talla y peso en P50. A.F: padres sanos. Hipospadias, criptorquidia, micropene y esterilidad en los varones de 4 generaciones y cáncer de mama en 2 tíos maternos. EF: hipospadias perineal, escroto poco diferenciado con aspecto de labios mayores, pene interescrotal de 22 mm con hendidura ventral, teste derecho no palpable e izquierdo en parte alta de escroto. Resto sin hallazgos.
Cariotipo:46 XY. A los 2 meses: testosterona basal: 138 ng/dl, FSH: 0,3 ng/ml y LH: 1,7 ng/ml. A los 19 meses: Test GnRH LH basal/pico: 1,7/11,5 mUI/ml. FSH b/p: 1,3/9,5 mUI/ml. Test HCG: testosterona b/p: 4/313 ng/dl, DHTb/p: < 0,05/0,2 ng/ml. Relación Testosterona/DHT: 16. 17OHP: 0,12 ng/ml, DHEA-S: 35,9 ng/dl, androstendiona: < 10 ng/ml.
Biopsia testicular: cambios mínimos, no disgenesia testicular.
Estudio molecular del gen AR: mutación Ala 597 Thr, en la región de unión del receptor al ADN.
Evolución:Orquidopexia a los 9 años. Múltiples intervenciones para reconstrucción uretral. Inicio puberal a los 12 años con evolución normal. Edad actual 16 años. Testes finales de 8 ml y pene de 7,5 cm. Virilización normal. No eyaculaciones.
Conclusiones:1. La mutación Ala 597 Thr en nuestra familia presenta homogeneidad fenotípica.2. La existencia de cáncer de mama en 2 varones afectos sugiere la posible asociación entre éste y alteraciones en el gen del AR o genes adyacentes.
P310
SINDROME DE ADIPSIA HIPERNATREMIA
Marta López Capapé, Luz Golmayo Gaztelu, Natalia Ramos Sánchez, M. Nieves Gallego Cobos, Gustavo Lorenzo Sanz, M. del Milagro Alonso Blanco y Raquel Barrio Castellanos
Hospital Ramón y Cajal, Madrid y Universidad de Alcalá de Henares, Madrid.
Introducción:El Síndrome de Adipsia Hipernatremia (SAH), entidad rara en la edad pediátrica, se caracteriza por deshidratación hipertónica crónica o recurrente con adipsia/hipodipsia. Es secundaria a alteraciones de los osmorreceptores hipotalámicos de la sed y con frecuencia de los de ADH. Presentamos un caso de SAH con adipsia sin alteración en los osmorreceptores de ADH en un niño sin evidencia de lesión hipotalámica.
Caso clínico:Varón de 12 años con astenia y pérdida de peso de 7 kg en 3 meses asociado a disminución de la ingesta de sólidos y líquidos; sin cefalea ni alteraciones del sueño ni termorregulación. AF y AP sin interés. Exploración inicial: deshidratación de 2.º grado. Talla: 150cm (P50-75). Peso: 26,8 kg (1,7 DS). IMC: 11,91 (2,52 DS) A1P1 Testes 4 ml. Fondo de ojo y exploración neurológica normal. Al ingreso Nas: 162 mmol/l, Creat: 0,89 mg/dl, Urea: 72, BUN: 33,6, Osms: 335 mM/kg. Densidad en orina ≥ 1.030. Estudio hormonal: prolactinabasal: 10,1 ng/ml; TSH: 1,85 μU/ml, T4L: 1,16 ng/ml, Test Synacthen de 1 μg: Cortisolbasal/pico: 16,8/22 μg/dl; Test de Clonidina: GHbasal/pico: 0,18/2,8 ng/ml; Test de HI: GHbasal/pico: 0,11/3,6 ng/ml. Test de infusión salina hipertónica: respuesta de ADH normal pero adipsia mantenida (tabla). Estudio de imagen: RMN intracraneal normal. Gammagrafia con tecnecio y serie ósea normales. Valoración psicológica: normal.

Evolución:Tras rehidratación se pauta dieta normal e ingesta hídrica de 1,5 litros al día con buen cumplimiento. A los 2 meses Talla: 151,2 cm. Peso: 33,5 kg (+6,7 kg). IMC: 14,7 (1,7DS). Velocidad de crecimiento normal. Asintomático.
Conclusiones:1. En nuestro paciente existe afectación selectiva de los osmorreceptores de la sed con normofunción de la ADH.2. La ausencia de respuesta de GH a estímulos ha sido referida en el SAH idiopático en 4 casos pediátricos (aunque asociado a obesidad) y con velocidad de crecimiento normal.3. Es obligado descartar patología hipotalámica orgánica de manera evolutiva.
P311SUPRESION DEL EJE SUPRARRENAL EN NIÑOS EN TRATAMIENTO CRONICO CON ESTEROIDES INHALADOS
Almudena del Pino de la Fuente, Ana M. Cordón Martínez, M.ª José García Arias, Silvia Ortega Pérez, Emilio José García García, Fco. Javier Pérez Frías, Juan Pedro López Sigueros y Antonio Jurado Ortiz
Hospital Materno Infantil, Málaga y Hospital General Carlos Haya, Málaga.
Introducción:Los corticoides inhalados tienen una indicación bien establecida en la prevención y el tratamiento de la hiperreactividad bronquial y el asma, pero los estudios sobre sus efectos en niños son escasos. El aumento de su uso prolongado conlleva la posibilidad de efectos sistémicos, de los cuales, la supresión del eje suprarrenal es uno de los más importantes. Presentamos dos casos de supresión del eje suprarrenal en niños en tratamiento prolongado con esteroides inhalados.
Caso 1:Niño de 18 meses, diagnosticado de hiperreactividad bronquial y en tratamiento con altas dosis de fluticasona inhalada (1000 μg/día) desde los 9 meses de edad. Un mes antes de su ingreso había recibido tratamiento con prednisona oral comenzando con 1 mg/kg/día, y reduciendo progresivamente la dosis hasta un total de 10 días de tratamiento. Acudió al Serviccio de Urgencias por un cuadro de vómitos, deshidratación leve y obnubilación. Se detectó hipoglucemia (36 mg/dl) y se trató con rehidratación intravenosa. Fue dado de alta a las 36 horas. Posteriormente se recibieron niveles plasmáticos de cortisol de 37 ng/ml (rango normal: 130-260) y ACTH de 6 pcg/ml (rango normal: 5-60). Dos meses después, en el curso de una infección de vías aéreas superiores, reingresó por vómitos y decaimiento y requirió esteroides parenterales. La dosis de fluticasona fue reducida gradualmente hasta 500 mg/día.
Caso 2:Niño de 9 años, diagnosticado de hiperreactividad bronquial, en tratamiento con fluticasona inhalada (500 μg/día) durante 19 meses, y posteriormente, 250 μg/día durante los últimos 7 meses. En una analítica de control se detectó supresión del eje adrenal: cortisol: 53 ng/ml y ACTH: 27 pcg/ml. La dosis de esteroides fue reducida de manera gradual. Actualmente tiene tratamiento con budesonida inhalada (320 μg/día) y los niveles hormonales han mejorado (cortisol: 90 ng/ml; ACTH: 35 pg/ml),
Comentario:Los paciente en tratamiento crónico con esteroides inhalados pueden presentar supresión del eje suprarrenal, en ocasiones sin sintomatología asociada. Destacamos la importancia del control del eje suprarrenal en estos pacientes, no sólo en los casos de dosis altas (más de 400 μg/día de fluticasona), sino también con dosis más bajas.
P312
HIPOTIROIDISMO CONGÉNITO
Carmen Fuentes Gutiérrez, José Antonio López Medina, Montserrat de Felipe Jiménez-Casquet, José Luis Barrionuevo Porras, Rosa Espigares Martín, M. Rosario Benavides Román, Purificación Cárdenas Guerrero y Luis Ortega Martos
Hospital Virgen de las Nieves, Granada.
Introducción:El programa de detección precoz del hipotiroidismo congénito, que se inicia en España en 1978 y está implantado en 1982 en todas las comunidades autónomas, ha permitido el inicio del tratamiento sustitutivo en fases cada vez más precoces de la etapa neonatal, permitiendo un desarrollo físico y cerebral normal en estos niños. En España se estima una incidencia de 1/2000 RN. En Andalucía se han registrado 474 casos en los últimos 14 años. Hemos realizado una revisión de la casuística de nuestro hospital en los últimos 16 años.
Material y métodos:Estudio retrospectivo de niños con diagnóstico de hipotiroidismo congénito detectados por cribado neonatal entre 1986-2002 (N = 66), con análisis descriptivo de variables: n.º de casos por período, sexo y relación v/m, presencia de síntomas/signos al diagnóstico, etiología, edad de inicio del tratamiento, realización de test mental, presencia de cromosomopatías, procedencia.
Resultados:Se diagnosticaron 66 nuevos casos: 74,3 % de mujeres y 25,7 % de varones, todos con hipotiroidismo primario. Etiología: 48,5 % tiroides sublingual, 25,7 % atireosis, 25,7 % dishormonogénesis. Procedencia: Almería 24,2 %, Jaén 33,3 %, Granada 42,5 %. Signos físicos más frecuentes: fontanela posterior de 5 mm: 44 %, macroglosia 25 %, hernia umbilical 25 %. Edad de inicio del tratamiento: 21 días (1986-1990), 15 días (1991-1994), 23 días (1995-1998), 12 días (1999-2002). Cromosomopatías: 4,5 % (3 casos: 1 s. de Klinefelter, 1 trisomía parcial 1, 1 s. 22q11) (catch 22). Test mental realizado: 58 %.
Conclusiones:Los programas de detección precoz de hipotiroidismo congénito han permitido un acortamiento progresivo del inicio del tratamiento sustitutivo, que ee de unos 12 días en nuetra zona. En nuestra revisión comprobamos un claro predominio femenino en esta patología, así como la presencia de signos inespecíficos al diagnóstico (fontanela posterior mayor de 5 mm, macroglosia y hernia umbilical). Es destacable la presencia de signos de un 4,5 % de cromosomopatías (el hipotiroidismo congénito se asocia en un 10 % a otras alteraciones congénitas, frente al 3 % en la población general). Los test mentales no se realizaron en la totalidad de los pacientes.
GASTROENTEROLOGIA
P313
ELASTASA PANCREATICA 1 FECAL EN NIÑOS AFECTOS DE DIABETES TIPO 1
Libia Quero Acosta, Federico Argüelles Martín, Francisco Javier Arroyo Díez, Diego de la Cruz, Beatriz Espín Jaime, Joaquín Mateos Cañas y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Introducción y objetivos:Las técnicas utilizadas para valorar la secreción exocrina de pacientes diabéticos en los años 80 eran muy invasivas o tenían un valor predictivo muy bajo. Actualmente disponemos de una prueba novedosa no invasiva o indirecta: la elastasa pancreática 1 fecal (E1). Dicha prueba tiene una sensibilidad y una especificidad de 93 %. En diabéticos tipo 1, se ha descrito tradicionalmente una función exocrina conservada, sin embargo estudios realizados en los últimos tres años evidencian hasta un 30 % de alteración en esta función en este grupo de pacientes, sobre todo en pacientes con evolución prolongada, superior a diez años. Basado en esto nos propusimos valorar función exocrina pancreática en pacientes diabéticos tipo 1 con menos de 10 años de diagnóstico y correlacionar los niveles de E1 con valores de hemoglobina glicosilada y tiempo de evolución.
Material y métodos:Selección al azar de 31 niños diabéticos tipo 1. Criterio de inclusión menos de 10 años de evolución. Recolección de datos por una encuesta tipo test más una muestra de heces por paciente. Se determinó la concentración de E1 empleando el método de laboratorio consistente en un sándwich ELISA con dos anticuerpos monoclonales altamente específicos de la elastasa pancreática humana.
Resultados:Ningún paciente presentó valores de E1 por debajo de 200 μg/g de heces (punto de corte diagnóstico de insuficiencia pancreática). Al dividir la población en dos grupos; basándose en valores cercanos a 200 y valores superiores a 400 μg/g de heces se evidenciaron dos grupos. Grupo 1: 27 niños (87 %) con valores de E1 superiores a 400 μg/g heces, edad promedio: 11,48 ± 4,8 años, tiempo de evolución: 5,19 ± 2,2 años y valores de hemoglobina glicosilada de 8,3 ± 4,2 %. Grupo 2: 4 niños (13 %) con valores de E1 entre 200 y 230 μg/g de heces: edad promedio 13,7 ± 2,1 años, tiempo de evolución 7,2 ± 0,9 años, hemoglobina glicosilada 7,23 ± 2,7 años. Al aplicar chi cuadrado existe significación estadística entre las diferencias observadas entre las edades promedio y el tiempo de evolución.
Conclusión:Ningún niño tuvo el diagnóstico de insuficiencia pancreática exocrina,pero cuatro niños (13 %) presentaron valores cercanos a este punto de corte. La edad promedio de este grupo de pacientes aunado al tiempo de evolución son factores pronósticos a tomar en cuenta y es probable que se deba practicar determinaciones rutinarias de E1 a estos niños para verificar si progresan a una insuficiencia pancreática exocrina.
P314ENFERMEDAD CELIACA: NUEVA FORMA DE PRESENTACION
Manuel Haro Gómez, María José Villalobos Linares, M. del Carmen Vega Castaño, Orlando Farfán, Filiberto Ramírez Gurruchaga y Federico Argüelles Martín
Hospital Universitario Virgen Macarena, Sevilla.
Introducción y objetivos:En los últimos años se ha comprobado que la enfermedad celíaca tiene una alta prevalencia en nuestro medio. Este hecho probablemente no se deba a un aumento en la incidencia de la enfermedad, sino a la demostración de nuevas formas clínicas por métodos diagnósticos más innovadores. El objetivo de nuestro estudio es comparar los síntomas clínicos que presentan al diagnóstico los pacientes entre 1980-1994 (grupo A) y 1995-2001 (grupo B).
Material y método:Se realiza un estudio descriptivo retrospectivo en una muestra de 53 historias escogidas aleatoriamente de los niños diagnosticados de enfermedad celíaca en nuestro servicio desde 1980 hasta 2001, revisando sus datos clínicos. El criterio diagnóstico utilizado es tener al menos una biopsia y serología compatibles.
Resultado:Al grupo A pertenecen 18 pacientes (35 %) y al grupo B 34 (65 %). Los síntomas y signos que padecen los pacientes del grupo A con respecto al B con diferencia estadísticamente significativa (d.e.s) son fallo de medro (A75-B45 %), anorexia (A63-B37 %), distensión abdominal (A64-B48 %), desnutrición (A44-B15 %), malabsorción (A44-B15 %), intolerancia a la lactosa (A56-B21 %). Los síntomas y signos de A y B sin d.e.s son diarrea (70 %), vómito (40 %), dolor abdominal (15 %), irritabilidad (32 %), heces malolientes (27 %), apetito excesivo (2 %), astenia (10 %), palidez (14 %), atrofia muscular (10 %), edema (4 %), peso < P3 (50 %). La edad media de comienzo de los síntomas en el grupo A es 19 meses y en el B 18 meses y la edad media del diagnóstico es 35 meses en A y B sin d.e.s. La edad media de introducción del gluten en A es 6 meses y en B es 7 meses sin d.e.s. En la analítica encontramos anemia (56 %), ferropenia (58 %), hipertransaminasemia (26 %), hipoproteinemia (12 %) sin d.e.s. La evolución clínica tras el diagnóstico es buena en el grupo A en un 56 % y en el B en un 94 % con d.e.s.
Comentario:Los niños del grupo A presentan a su diagnóstico un deterioro clínico mayor que los del B, es decir, ahora se están diagnosticando niños con menor afectación del estado general. No hemos podido demostrar que esto se deba a la precocidad en su diagnóstico, ni en el comienzo de los síntomas, ni en la precocidad de la ingesta del gluten en el grupo A respecto al B. La mejor evolución en el grupo B probablemente se debe al mejor control médico y al mejor conocimiento de las dietas exentas de gluten en los últimos años.
P315
ALERGIA A LA PROTEINA DE LA LECHE DE VACA (APLV) MEDIADA POR IGE: FACTORES PRONOSTICOS Y EVOLUCION
Orlando Farfán, Federico Argüelles Martín, Libia Quero Acosta, Filiberto Ramírez Gurruchaga e Isabel Gil
Hospital Universitario Virgen Macarena, Sevilla.
Introducción:La APLV es la reacción adversa a alimentos más frecuente en el primer año de vida.
Objetivos:Describir la evolución de la APLV mediada por IgE en relación con variables no descritas extensamente en la literatura.
Material y métodos:Estudio retrospectivo, descriptivo de 83 niños con diagnóstico de APLV en los últimos 6 años. Se midieron porcentajes, chi cuadrado de los resultados y comparación de medias (test de Wilcoxon).
Resultados:En la población estudiada la edad de comienzo más temprana (antes de los 6 meses) no se correlacionó con una mayor predisposición a padecer otra alergia alimentaria o a una evolución más prolongada de la enfermedad; por el contrario, una edad de comienzo más temprana se asoció con una menor edad de negativización (p = 0,05) La presencia de enfermedad atópica en un familiar de primer grado no predijo una edad de comienzo más temprana para la APLV. La presencia de otra alergia alimentaria se observó en un 22 % de la muestra, siendo el huevo el alimento más frecuentemente implicado La edad de negativización tuvo una media de 25,9 meses. Se buscó relacionar edad, fórmula de reemplazo utilizada y presencia de otra alergia alimentaria con esta variable de negativización. No se observaron diferencias significativas entre varones y mujeres, aunque se observó una tendencia en los primeros a negativizar antes que las segundas (24,3 vs 29,7, p = 0,9). En cambio, sí se observó una edad de negativización significativamente más tardía en aquellos que presentaban otra alergia alimentaria o habían consumido fórmula de soja con respecto a aquellos que no presentaban estas características (34,6 vs 24,5 meses, y 34,5 vs 19,9 meses, respectivamente; p = 0,007 y 0,002, respectivamente).
Conclusión:La población estudiada muestra características de evolución diferentes para algunos factores relatados en la literatura. Los diferencias halladas en cuanto al tipo de fórmula utilizada constituyen un hallazgo de importancia que debería ser confirmado por estudios prospectivos de casos seleccionados.
P316ENFERMEDAD CELIACA FAMILIAR O DETECCION DE ENFERMEDAD CELIACA SILENTE (OLIGOSINTOMATICA?) EN FAMILIARES DE UN CASO INDICE
Ana Fernández Romero, Pedro Terol Barrera, Federico Argüelles Martín, Filiberto Ramírez Gurruchaga y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Antecedentes y objetivo:Se compara a la enfermedad celiaca con un iceberg, dada la mayor proporción de casos silentes u oligosintomáticos en relación a los de presentación típica. Están demostrados el mayor riesgo de enfermedad en familiares de enfermos celiacos así como la asociación de determinados HLA con esta patología. Un medio de detectar pacientes de riesgo es el estudio de familiares en primer grado de celiacos diagnosticados.
Métodos:Tipificación del HLA y determinación de anticuerpos antigliadina y antiendomisio, así como biopsia yeyunal (en los casos con serología positiva y/o presencia de haplotipos de riesgo) en familiares en primer grado de niña afecta de celiaquía.
Resultados:Niña de 2 años con anticuerpos antiendomisio positivos y clínica de diarrea frecuente y ligera afectación de la curva de peso. Es diagnosticada de enfermedad celiaca al presentar atrofia vellositaria subtotal en la biopsia yeyunal obtenida por sonda de Watson-Crosby. Se estudia a sus 2 hermanas (asintomáticas?) presentando ambas anticuerpos antiendomisio positivos. El padre, que presenta una artritis migratoria no filiada de varios años de evolución, tiene anticuerpos de enfermedad celiaca negativos. Se realiza estudio de HLA en los 5 miembros de la familia para detectar haplotipos de riesgo (incluyendo a la madre, que está asintomática), encontrándose positividad para A1 Cw7 B8 (HLA clase I) y para DRB1*03 DQA1*0501 DQB1*0201 (HLA clase II) en homocigosis en el padre y en heterocigosis en las 3 hijas. Las biopsias yeyunales fueron diagnósticas de enfermedad celiaca en el padre y en las 3 hijas.
Conclusiones:1. Dada la alta frecuencia de formas silentes u oligosintomáticas de enfermedad celiaca es muy importante detectar los pacientes de riesgo, siendo necesario realizar cribado en familiares de enfermos celiacos 2. Más del 95 % de celiacos comparten el haplotipo de complejo mayor de histocompatibilidad (HLA) DQ 2 (concretamente DQB1*02[01-02], DQA1*0501) o DQ8. El haplotipo HLA-DRB1*0301 también está asociado con un riesgo incrementado de enfermedad celiaca. 3. La prevalencia de enfermedad celiaca entre familiares en primer grado de enfermos celiacos oscila entre un 2 y un 15 %.
P317 CONTRIBUCION DE LA CAPSULOENDOSCOPIA PARA EL ESTUDIO DE LA HEMORRAGIA INTESTINAL EN EL NIÑO
Estefanía Quintela Molinillo, Libia Quero Acosta, Orlando Farfán, Filiberto Ramírez Gurruchaga, Federico Argüelles Arias, Federico Argüelles Martín y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Introducción:La hemorragia digestiva es la segunda causa de sangrado agudo en la infancia tras la traumática. La etiología es muy diferente a la de adulto. Su presentación clínica puede orientar hacia el origen, aunque se ve influenciado por otros muchos factores: tiempo de evacuación, cuantía del sangrado... En el intestino delgado se localiza el 5 %. Hasta hace poco su exploración se limitaba a métodos radiológicos, y en muchos se requería la cirugía. Con la enteroscopia esto mejora, aunque presenta limitaciónes en cuanto al área visualizada y además es molesta para los niños. Actualmente con la capsuloendoscopia, técnica inocua y específica para la exploración del intestino delgado, estas limitaciones han disminuido.
Nuestro caso:Varón de 7 años que consulta en el Servicio de Urgencias Pediátricas de nuestro Hospital por rectorragia indolora de escasa cuantía de 2 años de evolución. Carece de antecedentes personales y familiares de interés. Se realiza exploración física, con atención al área abdominal y anorrectal, no objetivándose nada que puediera orientar hacia su etiología. Por la presentación clínica, la edad del paciente y la carencia de otros datos relevantes, se emite el juicio clínico de rectorragia leve. ¿Fisura anal? ¿Pólipos intestinales? ¿Enfermedad inflamatoria? Para completar su estudio se solicita: colonoscopia, objetivándose lesiones compatibles con Hiperplasia fonicular linfoide. En Ileón, a unos 30 cm de la Válvula Ileocecal se localiza lesión hemangimatosa, friable y se intenta cauterizar con Argón Láser resultando imposible debido a su tamaño. Se realiza estudios con glóbulos rojos marcados en tres fases y cortes tomográficos con el objetivo de valorar de forma menos invasiva el número y extensión de la lesión. Con la ecografía Doopler Abdominal se valorara los grandes vasos abdominales. Por la localización de la lesión se decide realizar estudio endoscópico con cápsula para visualizar todo el intestino delgado, constatándose una única lesión angiomatosa en ileón.
Conclusión:La capsuloendoscopia es un método adecuado para la valoración de la mucosa del intestino delgado, ya que permite visualizar todo esta área de manera dinámica, en tiempo real. Es una técnica no invasiva con la que no se necesita irradiar al paciente. Actualmente este método es empleado escasamente en niños, y queda por resolver la preparación del paciente para dicho estudio.
P318GASTROENTERITIS AGUDA COMO CAUSA DE INGRESO HOSPITALARIO
Mercedes Loscertales Abril, Cayetano Cintado Bueno, María José Martínez Roda, Fernando Ferreira Pérez, Eduardo Vigil Martín e Ignacio Gómez de Terreros y Sánchez
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivos:La gastroenteritis aguda sigue siendo una de las causas más frecuentes de ingreso hospitalario en los países de nuestro entorno, y dan lugar a elevado consumo de recursos. El objetivo de este trabajo ha sido estudiar las gastroenteritis agudas ingresadas en el Hospital Infantil Universitario "Virgen del Rocío" durante los dos últimos años (2001-2002), analizando las estancias, en función de la edad, la etiología y la existencia de patología aguda asociada.
Métodos:Se analizan 919 registros de alta del Conjunto mínimo de bases de datos (CMBA), seleccionados por el diagnóstico principal y los dos primeros secundarios.
Los registros se agruparon por grupos de edades y los resultados del coprocultivo (positivo o negativo). Los estudios estadísticos se han realizado con paquete SPSS.
Resultados:Las gastroenteritis aguda representan el 9,4 % de los ingresos urgentes al año, de ellos 524 varones y 395 hembras. El 36,7 % eran menores de seis mesesy el 61 % menores de 2 años. Los Grupos relacionados por el Diagnostico (GRD) más frecuentes, fueron el 776 (42,9 %) y el 777 (41,6 %) (p < 0001). La estancia media (EM) observada fue de 6,8 y la EM depurada de 5,54. Los enteropatógenos aislados fueron: Salmonella 10,8 %, Rotavirus 9,6 %, Campilobacter yeyuni 4,7 %, Giardia Lamblia 0,8 %. En 667 niños (grupo A) el coprocultivo fue negativo, con códigos CIE92000 558,9 y 009,1 (71 % y 3 %). La EM fue significativamente más elevada p < 0001 en aquellos pacientes en los que se aisló algún germen en coprocultivo, con una mayor diferencia cuando son menores de 24 meses.
Al analizar el Grupo A hemos podido constatar que 283 niños, cursaron con patología aguda asociada (Bronquitis y bronquilitis 20,5 %, catarros de vías altas 25,3 %, infecciones de vías urinarias 13,8 %), y 27,3 % eran menores de seis meses. De los 394 niños que no presentaban patología ni aguda ni crónica asociada, 25 % presentaron acidosis y/o hiponatremia y el 26,6 % eran mayores de 5 años.
En este grupo, la EM más elevada, se correlacionó significativamente (p < 001) con la menor edad y la presencia de patología aguda asociada. Sin embargo la suma de estancias fue similar en ambos subgrupos.
Conclusiones:Las gastroenteritis agudas no tipificadas, sin patología aguda asociada, ni signos analíticos de deshidratación, constituyen actualmente en nuestro medio una causa importante de ingreso hospitalario y consumo de recursos.
P319SINDROME DEL NEVUS AZUL DE CONSISTENCIA DE GOMA O "BLUE RUBBER BLEB NAEVUS"
Laura Cabanes Colliga, Amparo Villaverde Rodríguez, Andrés Bodas Pineda, Elena del Castillo Navío, Juan José Borraz Torca y Carlos Maluenda Carrillo
Hospital Clínico San Carlos, Madrid.
Introducción:Presentamos el caso de un paciente varón, diagnosticado hace 3 años de Angiomatosis gastrointestinal múltiple y en el que el hallazgo reciente, con 11 años, de varias lesiones vasculares cutáneas, nos debe hacer replantear el diagnóstico.
Caso clínico:El motivo de consulta inicial fue un cuadro de vómitos postprandiales, dolor abdominal epigástrico y deposiciones negruzcas de un mes de evolución. Además aportaba hemograma realizado desde atención primaria con anemia importante.
Como único antecedente personal destacaba Linfangioma quístico cervical.
A la exploración física destacaba palidez cutáneomucosa, sin lesiones angiomatosas, y soplo cardiaco, con peso y talla dentro de percentiles normales.
En las pruebas complementarias se confirmó intensa anemia ferropénica, se descartaron coagulopatías y mediante pancolonoscopia y endoscopia oral se detectaron múltiples lesiones angiomatosas en estómago, duodeno postbulbar y cólon distal que se fueron coagulando con láser progresivamente.
Ante la persistencia de sangrado activo, se realizó enteroscopia hasta asas de yeuno medio y arteriografía, sin hallarse nuevas lesiones.
Se descartaron también lesiones vasculares a otros niveles, propias de algunos síndromes neurocutáneos.
Sin embargo la detección reciente de varios nevus azules en la planta del pie, nos hace replantear el diagnóstico como Síndrome del nevus azul.
Conclusiones:El Síndrome del nevus azul de consistencia de goma o "Blue rubber bleb naevus" es una entidad infrecuente, a tener en cuenta en caso de asociación de tumores vasculares cutáneos-digestivos y anemia crónica ferropénica.
La evolución es crónica y su gravedad y pronóstico dependen del desarrollo de complicaciones hemorrágicas.
P320RECTORRAGIA NEONATAL PRECOZ DE ORIGEN INFECCIOSO
Alfonso Peña Valenceja, Alicia Sánchez Mínguez, Helena Pérez Gutiérrez, Carolina López García, Marta Isabel Carrascal Arranz y Alfredo Blanco del Val
Hospital Universitario del Río Hortega, Valladolid.
Caso clínico:Recién nacido de 40 horas de vida. Comienza con febrícula y deposiciones líquidas con sangre roja y mucosidad. Peso actual: 3.420 kg (3.580 kg al nacimiento). Al nacer profilaxis ocular y antihemorrágica. Vacunado de primera dosis de vacuna VHB. Inició diuresis y meconio en las primeras 24 h de vida. Primeras deposiciones de aspecto y consistencia normal. Madre de 27 años. Sana. Grupo O, Rh positivo. No abortos ni hijos previos. Embarazo controlado normal. Serologías de infección connatal negativas. Parto eutócico. Presentación cefálica. Bolsa rota de 5 horas. Líquido amniótico claro. Apgar 9/10. Rea I.
Anamnesis:No ingesta de fármacos, no estimulación rectal. No sonda oro/nasogástrica. No vómitos ni reflujo. Lactancia materna exclusiva. Leves grietas en el pezón.
Exploración física:No presencia de otros puntos de sangrado. Abdomen normal, algo distendido. No aspecto quirúrgico. No presencia de fisuras perianales. No se objetivan hemangiomas.
Pruebas Complementarias: Hemograma: leucocitos 11.800 (N:54 %; L:33 %; M:8 %). Hb:14,9 g/dl. Htco:45 %. Plaq: 85.000 (agregados). Coagulación, PCR, Gases, Perfil hepato-renal normales. Rx tórax y Eco Abdominal normales. Se recogen cultivos para bacterias y virus: hemo, copro y urocultivo.
Evolución:Se coloca una sonda nasogástrica (con aspirado gástrico negativo), sueroterapia, y se deja el paciente a dieta absoluta. A las 6h del ingreso sufre un empeoramiento hemodinámico que se remonta con un bolo de suero salino fisiológico iv, a 20cc/kg, repitiendose las pruebas de imagen: Eco Abdominal con signos de colitis. Se inicia antibioterapia con Ampicilina y Gentamicina. A las 48 del ingreso se recibe un coprocultivo positivo para Campylobacter, sustituyendo la antibioterapia inicial por Eritromicina. La evolución fue favorable, reintroduciendo la alimentación oral sin problemas, siendo el paciente dado de alta a los 7 días.
Conclusión:La hemorragia digestiva baja en el período neonatal precoz es una entidad poco frecuente, en un alto porcentaje de etiología desconocida, y de resolución espontánea en la mayoría de los casos. La etiología infecciosa no debe pasarnos desapercibida, debido a las visitas, cada vez más frecuentes, de los diferentes servicios de maternidad de nuestros hospitales.
P321IMPORTANCIA DEL SCREENING DE PATOLOGIA HEPATICA EN UNA CONSULTA DE CENTRO ESPECIALIZADO
Fernando Ferreira Pérez, Cayetano Cintado Bueno, María José Martínez Roda, Mercedes Loscertales Abril e Ignacio Gómez de Terreros y Sánchez
Hospital Universitario Virgen del Rocío, Sevilla.
Objetivo:Evaluar la importancia de la realización de pruebas de screening para intentar esclarecer la etiología en los hallazgos de afectación hepática.
Material y métodos:Se analizan 100 casos de niños remitidos a una consulta especializada de Hospital, en los dos últimos años, por hallazgos durante una exploración rutinaria de hepatomegalia,y/o aumento de transaminasas o ictericia de aparición brusca.
En dicha consulta se realizó exploración clínica, hemograma, VSG, PCR, enzimograma hepático, estudio de coagulación, metabolismo del hierro, proteinograma, inmunoglobulinas y serología de hepatitis A, B, C, EBV, CMV y toxoplasma. En caso de que estas analíticas no nos aportaran datos se pasa a un segundo escalón analizando ceruloplasmina, a1 antitripsina, ecografía abdominal y anticuerpos de enfermedad celíaca.
Resultados:Del total de los 100 casos, en 90 de ellos se llego a un diagnóstico. En 31 casos mononucleosis aislada, y en seis de ellos asociándas a un síndrome de Gilbert, un síndrome de Gianotti Crosti, en 2, a hepatitis A y en otros 2, a CMV. Hepatitis A tenian 20 niños, uno de ellos asociado a síndrome de Gilbert. En 3 que comprobó la existencia de hepatitis B activa, una de las cuales asociada a infección por CMV. Otros 2 niños cursaban con hepatitis C. Entre los restantes diagnosticos destacan 3 hepatitis por CMV,. 2 síndrome de Gilbert aislados, 1 caso de leishmaniasis, 1 quiste quiste de colédoco, malformación vascular y colestasis. En 19 casos, a pesar de constatarse la elevación de transaminasas sostenida no pudo ser filiada la lesion hepatica.
Conclusiones:Destacar por un lado, la importancia de un screening en los hallazgos rutinarios de afectación hepática dado que, en nuestra serie, el 3 % presentó hepatitis B y el 2 % hepatitis C y en todos fue posible el tratamiento. El hecho de que en un grupo de niños nos encontremos ante un 5 % con patología potencialmente grave, pero a su vez tratable, justifica en gran medida la realización de pruebas de screenig cuando en una exploración rutinaria se encuentren los signos clínicos y/o analíticos antes descritos.
P322 ESTUDIO CLINICO-EPIDEMIOLOGICO DE SALMONELOSIS EN EL HOSPITAL CLINICO SAN CARLOS ENTRE 1997-2002
Elena del Castillo Navío, Juan José Borraz Torca, Olga Pérez Rodríguez, Laura Cabanes Colliga, Carlos Maluenda Carrillo y Andrés Bodas Pineda
Hospital Clínico San Carlos, Madrid.
Objetivos:La incidencia de infección por Salmonella ha crecido considerablemente durante la última década a pesar de la mejoría general de las condiciones sociosanitarias. Revisamos la población de pacientes ingresados en nuestro servicio por salmonelosis durante los 5 últimos años para describir la situación en nuestro medio respecto a esta patología.
Material y métodos:Se realiza un análisis retrospectivo de la historia clínica de 73 pacientes de edades comprendidas entre 6 meses y 15 años ingresados que presentaron salmonelosis en cualquiera de sus formas clínicas y que fueron confirmadas con cultivo entre 1997 y 2002. Se analizan la distribución por edades, las manifestaciones clínicas, las alteraciones analíticas que presentaron, el género de salmonella identificado y la terapia realizada.
Resultados:La edad media de presentación del proceso fue de 5,3 años, con un aumento de la incidencia en menores de 3 años (31 %). Encontramos un 13,7 % pacientes con patología de base y un 17,8 % con antibioterapia (atb) previa. El 96 % cursó con gastroenteritis de las cuales el 21,4 % se debían a toxiinfección alimentaria constatadas epidemiológicamente. Los serotipos aislados fueronS. enteritidis (64,4 %),S. derby (9,6 %),S. stanley (27,4 %),S. typhimurium (1,4 %),S. sp (17,8 %). Presentaron bacteriemia sin enterocolitis el 4,1 %. El 90 % presentaron fiebre, 4,1 % dudosos peritonismo, signos meníngeos o confusión con LCR normal, 2,7 % eritema nodoso y artralgias, exantema máculo papuloso o estreñimiento. En el laboratorio se objetivó: leucocitosis (54,7 %) con neutrofilia (35,6 %), leucopenia (2,7 %), leucocituria con urocultivo negativo (8,2 %), microhematuria (2,7 %) e hiponatremia (27 %). Se administró antibiótico al 5,7 % de las gastroenteritis. Se indicó la recogida de un coprocultivo de control al mes en el 15,7 %.
Conclusiones:Durante los 5 años hemos observado que los pacientes menores de 3 años, con alguna enfermedad de base o los que recibieron antibioterapia previa representan el grupo más numeroso. El serotipo predominante esS. enteritidis. La forma clínica de presentación típica es la gastroenteritis aguda, con leucocitosis, neutrofilia, hiponatremia y en un alto porcentaje leucocituria. Recomendamos realizar coprocultivo de control en un mes ya que lo consideramos importante por su importante valor epidemiológico y de Salud Pública.
P323DIARREA INTRATABLE EN UN LACTANTE DE 4 MESES
José M. Iglesias Meleiro, Pilar A. Crespo Suárez, Carmen Curros Novo, Montserrat López Franco, Elena V. Rodrigo Sáez y Manuel Fernández Sanmartín
Hospital Clínico Universitario. Complejo Hospitalario Universitario, Santiago de Compostela.
Objetivo:Descripción del caso de un lactante de 4 meses con diarrea intratable, las posibilidades diagnósticas barajadas, su diagnóstico definitivo y el tratamiento aplicado.
Material, métodos y resultados: Paciente de 4 meses de edad que ingresa en nuestro Centro por presenta un cuadro de diarrea de 5 días de evolución con acidosis metabólica e intolerancia a disacaridos; tras recuperación inicial con fluidoterapia intravenosa y posteriormente dieta exenta de lactosa, presenta repetidas recaidas con despeños diarréicos y graves descompensaciones metabólicas, asi como fracaso a la introducción de fórmulas semi y elementales, realizando intolerancia a monosacáridos y necesitando nutrición mediante alimentación parenteral central completa durante 20 días; entre los estudios complementarios realizados destacan: coprocultivos seriados (negativos), determinación de Ig E total (normal) e Ig(s) específicas para lecha de vaca (negativas), catecolaminas en orina, cloro en sudor y electrolitos en heces (normales). En la biopsia de colon se destaca la presencia de un denso infiltrado inflamatorio constituido por eosinófilos a nivel de la mucosa y muscular de la mucosa, llegándose al diagnóstico etiológico de colitis eosinofílica.
Conclusiones:Dentro de la posible etiología de la diarrea crónica en el lactante ha de considerarse la gastroenterocolitis eosinofílica, una entidad poco frecuente (1/100.000), de fisiopatología desconocida, definida por la aparición en la mucosa intestinal de infiltrado inflamatorio de predominio eosinófilo, localizado preferentemente en mucosa gástrica y duodenal, aunque también se puede presentar, como en nuestro caso, con afectación exclusiva a nivel del colon.
El tratamiento de esta entidad ha de individualizarse en función del grado de afectación, tanto a nivel clínico como histológico, estando descrita buena respuesta, en la mayoría de los casos, a la administración de corticoides, en ciclos cortos, sistémicos y/o en forma de enemas, antihistamínicos e inhibidores de la degranulación del mastocito, especialmente cuando existe eosinofilia o aumento importante de los niveles de IgE sérica. También se han descrito casos de intolerancia a las fórmulas hidrolizadas, como en nuestro paciente, necesitando recurrir a alimentación enteral a base de aminoácidos libres.
P324SINDROME DE INTESTINO CORTO POSTCIRUGIA CON TORPIDA EVOLUCION
M.ª José García Arias, Javier Blasco Alonso, María González López, Silvia Ortega Pérez, Almudena del Pino de la Fuente, Carmen Serrano Recio, Leticia Olivares Sánchez, Luis del Río Mapelli, Carlos Sierra Salinas y Antonio Jurado Ortiz
Hospital General Carlos Haya, Málaga.
Introducción:El síndrome de intestino corto tiene múltiples causas, siendo de gran importancia el secundario a cirugía resectiva, por sus secuelas y mala evolución, en ocasiones asociándose a otras patologías.
Caso clínico:RNPT (34+6sem, 2.880 g) varón, diagnosticado prenatalmente de hidrops fetal, con doble atresia intestinal en yeyuno e ileon intervenidas al 2.º día de vida, realizándose anastomosis término-terminal. Al 5.º día postoperatorio presentó un cuadro de distensión abdominal sugestivo de obstrucción intestinal alta que precisó reintervención quirúrgica, confirmándose la existencia de una dehiscencia de anastomosis. Posteriormente precisó nutriciónparenteral exclusiva durante 19 días presentando tránsito intestinal enlentecido con marcada distensión abdominal. Se inició NEDC con fórmula elemental, persitiendo cierta distensión abdominal, con mejoría progresiva de la motilidad intestinal. Secundariamente a la nutrición parenteral prolongada y al "intestino corto", desarrolló colestasis con cifras de Bilirrubina total de hasta 7 mg/dl y elevación de transaminasas (GGT: 155; FA: 369; GOT: 73; GPT: 50).
Tras un mes de la última intervención, presentó deterioro brusco con datos radiológicos de neumatosis intestinal sin perforación, requiriendo reposo digestivo y reinicio de nutrición parenteral. La mala evolución en el postoperatorio del paciente nos hizo sospechar la existencia de una insuficiencia pancreátrica exocrina asociada, presentado unas cifras de quimiotripsina en heces no detectables y de elastasa pancreática de 7,5 μg/g de heces. Se confirmó una Fibrosis Quística como causa, ratificándose con estudio genético concluyente (homocigosis DF508-DF508). Desde entonces sigue tratamiento sustitutivo con enzimas pancreáticas y fórmula semielemental, manteniéndose asintomático desde el punto de vista respiratorio y con mejor tolerancia digestiva.
Conclusiones:Una de las causas frecuentes de hidrops fetal es la patología obstructiva intestinal. Ante una mala evolución postquirúrgica intestinal se debe plantear el diagnóstico de insuficiencia pancreática exocrina, siendo lafibrosis quística una de las más frecuentes. Las secuelas de un síndrome de intestino corto se deben sobre todo a colestasis y alteraciones del tránsito gastro-intestinal, teniendo su indicación principal las fórmulas elementales o semielementales.
INFECTOLOGIA
P325SEPSIS NEUMOCOCICA RECURRENTE EN NIÑA CON ASPLENIA Y DÉFICIT DE C3 E IGG
M. Ángeles Murillo Pozo, Antonio Vázquez Florido, Cristina Montero Valladares, Víctor Manuel Navas López, Alberto Varona García, Juan David González Rodríguez, Juan Antonio Soult Rubio, Miguel Muñoz Sáez, José Domingo López Castilla y Mercedes Carranza Conde
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivo:La sepsis bacteriana es la infección más grave que el niño puede adquirir en la comunidad. Existen factores que predisponen a padecer infección grave por neumococo, como las inmunodeficiencias.
Presentamos un caso de sepsis recurrente por neumococo, en niña con asplenia y deficit de C3 e IgG.
Observación clínica:Se trata de una niña de 13 meses de edad, sin antecedentes personales de interés, que ingresa en la Unidad de Cuidados Intensivos Pediátricos (UCIP) por cuadro de fiebre alta, decaimiento, exantema petequial generalizado, coloración pálido-cianótica, relleno capilar enlentecido y afectación del nivel de conciencia; precisando reposición de volemia, fármacos inotrópicos y vasoactivos, hemoderivados y antibioterapia. Evoluciona favorablemente, siendo alta de UCIP a los 6 días de ingreso. En hemocultivo se aislóStreptococcus pneumoniae, serotipo 6B.Dos meses y medio después, reingresa en UCIP en situación de extrema gravedad, por nuevo cuadro de sepsis severa, shock séptico y fracaso multiorgánico, con inestabilidad hemodinámica, insuficiencia renal, fallo hematológico y síndrome de distress respiratorio agudo, precisando reposición de volemia, fármacos inotrópicos y vasoactivos, hemoderivados, antibioterapia, ventilación mecánica y hemodiálisis. Se mantiene varios días en situación de extrema gravedad, mejorando posteriormente y siendo alta de UCIP a los 15 días de ingreso. En hemocultivo se identificóStreptococcus pneumoniae, serotipo 6B.Los exámenes complementarios realizados demostraron asplenia y déficit de C3 e IgG específica frente a neumococo.
Comentarios: 1. Streptococcus pneumoniae puede originar infecciones invasoras muy graves, como la sepsis, con shock séptico y fracaso multiorgánico. 2. Los factores que predisponen a padecer enfermedad neumocócica invasora no suelen ser diagnosticados previamente y pueden dar lugar a cuadros de repetición.3. Es preciso realizar estudios detallados en todo niño que padece enfermedad invasora grave, para detectar esos factores de riesgo. 4. La vacuna conjugada heptavalente es el método más eficaz de prevención de esta grave enfermedad.
P326APLICACION DE TÉCNICAS DE DETECCION RAPIDA DEL ANTIGENO DE STREPTOCOCCUS PYOGENES EN LA URGENCIA HOSPITALARIA
Itziar Marsinyach Ros, Aida de la Huerga, Paz Chimenti Camacho, Piedad Isabel Dobon Westphal, M. Concepción Míguez Navarro, Carmen Gutiérrez, Kay Boris Brandstrup Azuero, Cristina Serrano Loredo, Carlos Merello Godino y Paula Vázquez López
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción:El Streptococcus pyogenes(SBHGA) es responsable del 15-30 % de las faringoamigdalitis en la edad pediátrica. Es la única indicación de tratamiento antibiótico fundamentalmente para prevenir sus posibles complicaciones supurativas y no supurativas. Planteamos el uso de test de detección rápida del antígeno del SBHGA en las urgencias hospitalarias con el fin de evitar la antibioterapia empírica innecesaria.
Material y métodos:Se realiza un estudio prospectivo en pacientes pediátricos que acudieron a la urgencia del hospital entre los meses de octubre y febrero con clínica sugestiva de faringoamigdalitis estreptocócica y se les realizó una prueba de detección rápida del antígeno del SBHGA (StrepA Abbott Testpack®plus™). Se recogió muestra de exudado faríngeo para cultivo de confirmación. Para cada paciente se cumplimentó un cuestionario clínico-epidemiológico; se utilizó la escala estandarizada de Wald para estimar la probabilidad clínica de amigdalitis de origen estreptocócico. Se analizó la sensibilidad del microorganismo a los antibióticos de uso más frecuente y las pautas de antibioterapia prescritas.
Resultados:Se recogió una muestra de 241 pacientes (47,3 % niñas y 52,4 % niños) con edades comprendidas entre los 9 meses y los 15 años (media de edad 5 años). 69 pacientes (28 %) tuvieron un test rápido positivo de los cuales 66 % presentaron un cuadro de comienzo agudo, el 46,3 % odinofagia, el 71 % fiebre por encima de 38,5 °C y el 28 % adenitis submaxilar. Respecto a la correlación clínico-microbiológica (score de Wald) tuvieron test rápido (+) el 23,5 % de los que presentaron puntuación 1-2, el 25 % con puntuación 3-4 y el 52 % con puntuación 5-6. Hemos obtenido para el test una sensibilidad del 89 %, especificidad del 94 %, VPP 89 % y VPN 95 %. Se pautó tratamiento antibiótico al 30 % de los pacientes estudiados (58 % recibieron Penicilina V). Se encontró una resistencia a eritromicina del 25 %.
Conclusiones:1. El uso de técnicas de detección rápida del antígeno de SBHGA permite disminuir el uso indiscriminado de antibióticos en la faringoamigdalitis compensando su coste económico.2. Los parámetros de validación del test nos permiten equipararlo al cultivo como método diagnóstico, por lo que podría plantearse prescindir de la confirmación microbiológica en determinados casos.3. Al igual que en otros estudios hemos objetivado una pobre correlación clínico-microbiológica por lo que estas escalas sólo deberían tener un valor orientativo.
P327
DOENÇA DE RITTER EM CRIANÇA DE 1 ANO
Filipa Vasconcelos Espada, Georgeta Oliveira, Sofía Aroso, Ana Paula Aguiar y Eduarda Cruz
Hospital Pedro Hispano, Matosinhos (Portugal).
Introdução:O diagnóstico diferencial entre Síndroma de Steven Johnson/Síndroma de Lyell e Síndroma da pele escaldada estafilocócica (anteriormente designado por Doença de Ritter) pode ser difícil devido às manifestações clínicas semelhantes, porém é essencial para a instituição de uma terapêutica adequada e eficaz.
Caso clínico:Os autores apresentam o caso clínico de um menino de 1 ano, transferido por suspeita de Síndroma de Steven-Johnson.
Tratava-se de uma criança, sem antecedentes patológicos relevantes, que recorreu ao Serviço de Urgência por aparecimento de edemas palpebrais, hiperémia conjuntival, irritabilidade, exantema eritematoso disperso, bolhas e descamação cutânea da região pré-auricular direita. O quadro clínico foi interpretado como Síndroma de Steven-Jonhson pelo que fez administração parentérica de prednisolona, sendo, posteriormente, transferido para o Hospital Pedro Hispano.
Na admissão apresentava-se apirético, hemodinamicamente estável, queixoso, com rinorreia purulenta, exantema escarlatiniforme disperso e áreas bolhosas com descamação e sinal de Nikolsky positivo, na face e tronco.
Durante as 12h de observação houve progressão das lesões descamativas com atingimento de 29 % da superfície corporal.
Foi colocada a hipótese de diagnóstico de Síndroma de pele escaldada estafilocócica.
Analiticamente apresentava leucocitose com neutrofilia, PCR negativa, VS normal, albumina no limite inferior da normalidade, CK e LDH ligeiramente elevadas.
Foi instituída flucloxacilina sistémica, fluidoterapia, segundo fórmula de Parkland para queimados, analgesia e isolamento com medidas rigorosas de assepsia.
Manteve-se sempre apirético, hemodinamicamente estável, sem infecção secundária das lesões. A partir de D3 de internamento iniciou estabilização do exantema escarlatiniforme descamativo evoluindo para cicatrização.
No exame cultural da nasofarínge foi isolada uma estirpe deStaphilococcus aureus resistente a penicilina, confirmando diagnóstico de Síndroma da pele escaldada estafilocócica. Teve alta, após completar 10 dias de antibioterapia endovenosa.
P328MIOSITIS INFECCIOSAS
Dolores Falcón Neyra, M. Soledad Camacho Lovillo, Ana Olivar Gallardo, Segundo López Ros, Ana M. Reina González, Cristina Montero Valladares, Juan Antonio León Leal y Juan Navarro González
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción:La miositis se caracteriza por inflamación muscular con degeneración de fibras musculares. La causa infecciosa más frecuente de la polimiositis es viral, produciéndose una reacción antigénica cruzada de anticuerpos antivirales contra las células musculares. Los virus más frecuentes son Coxsackie, VEB y virus influenza. La clínica comprende debilidad muscular, mialgia selectiva, ausencia de signos inflamatorios y fiebre. El diagnóstico es clínico y se apoya en el aumento de las enzimas musculares. Se trata de procesos benignos y autolimitados. El tratamiento se basa en reposo y AINES orales.
Objetivos:Hemos querido hacer una revisión retrospectiva de esta patología en nuestro medio contrastando los datos con lo descrito en la bibliografía.
Material y métodos:Obtención de datos a partir de la revisión de historias clínicas y bibliografía actualizada.
Resultados:Hemos revisados los casos ingresados en nuestro centro en los últimos 7 años. El 65 % de los casos son hombres. El 75 % de los niños son mayores de 5 años y el 50 % mayor de 8 años. Observamos mayor incidencia en los meses de primavera (60 % de la muestra). La sintomatología debut en la mayoría de los casos fue mialgia selectiva, debilidad muscular y fiebre en el curso o convalescencia de una infección banal. La localización más frecuente fue en grupo gemelar. En el 100 % de los casos hubo un aumento de la enzima creatinkinasa. El tiempo de evolución fue de 5,2 días. El tratamiento se basó en el uso de AINES orales y reposo. En ningún caso se observaron secuelas o recidivas a corto plazo.
Conclusión:Tanto la epidemiología como la forma de presentación y evolución clínica concuerdan con la bibliografía revisada. Concluimos que la miositis en nuestro medio es un cuadro clínico benigno y autolimitado que no precisa hospitalización si existe seguimiento por parte del pediatra de medicina primaria.
P329 REPERCUSION EN UN HOSPITAL TERCIARIO DE UN NUEVO PROTOCOLO PARA EL MANEJO DE LOS PACIENTES CON MENINGITIS
Antonio Pérez Martínez, Mercedes de la Torre Espí y Juan Carlos Molina Cabañero
Hospital del Niño Jesús, Madrid.
Antecedentes:En enero del años 2002 se instauró en el servicio de urgencias de nuestro hospital un protocolo para el manejo de los casos de meningitis, basándonos en el clásico trabajo de Thomé y Boyer y nuestra propia experiencia, publicada recientemente en la literatura.
Objetivos:Evaluar la validez del protocolo para diferenciar la etiología de las meningitis, así como valorar el impacto, la repercusión y el seguimiento por parte de los profesionales de nuestro centro.
Pacientes y método:Se han revisado retrospectivamente los casos diagnosticados de meningitis en nuestro hospital durante el año 2002. Se excluyeron a los pacientes que presentaban alguna de las siguientes características: neonato, sepsis, sospecha de tuberculosis, portadores de válvula de derivación craneal, cirugía craneal reciente o tratamiento con antibiótico en las últimas 48 horas. A todos los pacientes incluidos en el estudio se les aplicó la escala de valoración de Boyer modificada, el tratamiento antibiótico pautado y los días de hospitalización.
Resultados:De los 90 casos encontrados, sólo 55 formaron parte de este estudio. En 50 pacientes el cultivo fue estéril. En este grupo la escala de Boyer fue ≤ 3. Recibieron antibiótico 9 pacientes con cultivos estériles. La mediana de ingreso en este grupo de pacientes fue de 4 días (rango 3-11). En 5 pacientes el cultivo fue positivo (4 meningococos B y 1 neumococo). Este grupo de pacientes presentó al diagnóstico una puntuación en la escala de Boyer ≥ 5 y todos recibieron tratamiento antibiótico desde el inicio. La mediana de ingreso fue de 9 días (rango 7-14).
Conclusiones:La escala de valoración de Boyer modificada es útil para discriminar la etiología bacteriana o viral de las meningitis, mejorando las indicaciones de antibioticoterapia y disminuyendo los días de hospitalización.
P330ENFERMEDAD INVASIVA POR S. AUREUS EN PACIENTE INMUNOCOMPETENTE
M. Pilar Ranchal Pérez, David Moreno Pérez, Francisco Jesús García Martín, Yolanda María Chica Fuentes, Olga M. Escobosa Sánchez, Lourdes Escudero Ruiz de Lacanal, Carmen Martínez Ferriz y Antonio Jurado Ortiz
Hospital Materno Infantil, Málaga.
La sepsis por S. aureus es una entidad más frecuente en lactantes y especialmente en aquellos con alguna enfermedad de base.
Presentamos el caso de una enfermedad invasiva por S. aureus con focos localizados en tejidos blandos, hueso y pulmón.
Caso clínico:Niña de 18m previamente sana que ingresa en hospital comarcal para estudio de convulsión febril atípica y sospecha de cuadro viral con ecografía de cráneo, EEG y LCR normales. Como único hallazgo analítico presentó leucopenia (2.600, N = 1.000/mm3).
Durante su estancia hospitalaria mantuvo fiebre y sufrió deterioro clínico trasladándose a nuestro centro donde ingresa en UCIP por estado tóxico severo sin repercusión hemodinámica y con importante afectación cutánea consistente en tumefacciones induradas dolorosas sin calor ni eritema en línea axilar, costado y cadera derechos, ambos muslos y pies. Además presentaba una lesión equimótica de 1 cm en mano izquierda, una gran adenopatía submaxilar izquierda dura y dolorosa, hepatomegalia de 5 cm e impotencia funcional de la cadera derecha.
Presentó leucocitosis con neutrofilia (34.690, 80 %N), anemia y trombopenia, así como coagulopatía, hipertransaminasemia y elevación de la CPK.
Se instauró tratamiento empírico con vancomocina y clindamicina a pesar de lo cual tuvo hemocultivos persistentemente positivos para S. aureus hasta en tres ocasiones, y desarrolló focos infecciosos a nivel respiratorio (neumonía cavitada con neumotórax recidivante), óseo (osteomielitis de cadera y metáfisis proximal de húmero derechos) y nuevas lesiones cutáneas (paniculitis y abscesificación).
Dada la gravedad del cuadro se sospechó un déficit inmunitario por lo que se estudiaron subpoblaciones linfocitarias, inmunoglobulinas, moléculas de adhesión (CD11 y CD18) y la función fagocítica del neutrófilo, que resultaron normales. En el seguimiento posterior durante año y medio la paciente ha continuado con analíticas normales y sin infecciones sugerentes de inmunodeficiencia.
Conclusiones:La enfermedad invasiva por S. aureus es un cuadro potencialmente grave pero excepcional en niños inmunocompetentes, por lo que es obligado realizar un estudio inmunitario.
P331PITIRIASIS LIQUENOIDE Y VARIOLIFORME AGUDA
Elena Lucas Sáez, M. Elena del Prado Sanz, Mercedes Gracia Casanova, Margarita Navarro Lucía, Jorge Pons García, M. Teresa Urgel Gómez, Santiago Gallego Vela y Pilar Arnauda Espatolero
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción:La Pitiriasis Liquenoide y Varioliforme Aguda (PLEVA) es una dermatosis papulosa con características clínicas típicas y de aparición brusca cuya etiología se desconoce. La hipótesis más aceptada es un tipo de reacción de hipersensibilidad frente a un agente infeccioso, apoyada por el modo de presentación clínica en forma de brotes epidémicos.
Caso clínico:Presentamos el caso de una niña de 10 años que acudió a Urgencias por presentar lesiones cutáneas pruriginosas de 20 días de evolución. A la exploración se observaron pápulas eritemato-edematosas, sobreelevadas, polimorfas, de entre 0,3-0,5 cm de diámetro, algunas de ellas con costra hemorrágica en su centro. Se distribuían predominantemente y de forma simétrica en tronco, cara y extremidades, respetando palmas y plantas. No se observaron lesiones en mucosa oral ni genital. El resto de exploración fue normal, a excepción de una faringe hiperémica con amígdalas hipertróficas y congestivas, así como adenopatías laterocervicales palpables.
Valorada por Dermatología, y con diagnóstico clínico compatible con PLEVA, se decidió ingreso en Pediatría para estudio y tratamiento. Los datos analíticos así como los cultivos microbiológicos no evidenciaron causa infecciosa.
Se realizó biopsia cutánea con diagnóstico histopatológico de PLEVA.
Instauramos tratamiento antibiótico con eritromicina 500 mg/8 h, que por intolerancia gástrica hubo de ser sustituido por claritromicina 250 mg/12 h. Tópicamente se trató con baños diarios emolientes de avena e hidratación corporal abundante. La evolución de las lesiones agudas fue favorable, no observándose nuevos brotes en sucesivas revisiones, quedando solamente máculas hipocrómicas residuales.
Conclusiones:Dentro de la Pitiriasis Liquenoide, la forma aguda o PLEVA es más frecuente en la infacia, mientras que la crónica predomina en adultos.
En el caso que presentamos, el proceso faríngeo podría ser el responsable del brote cutáneo, apoyando este hecho la buena respuesta al tratamiento antibiótico, a pesar de que el frotis faríngeo resultara negativo.
Entre los distintos tratamiento empleados en edades infantiles figuran los macrólidos vía oral y los corticoides y emolientes tópicos. Otras alternativas como la fotoquimioterapia y el metrotexate, rara vez se justifican en edades pediátricas.
P332TUBERCULOSIS MULTIRRESISTENTE: A PROPOSITO DE UN CASO
Estíbaliz Onís González e Itziar Pocheville Gurutzeta
Hospital de Cruces, Baracaldo.
Introducción:La emergencia de cepas de Mycobacterium tuberculosis multirresistentes representa un problema mundial. Aproximadamente 2 billones de personas están infectadas y cada año ocurren 8,8 millones de casos nuevos. La resistencia se adquiere con la infección inicial o se desarrolla durante el tratamiento, bien por mal cumplimiento o por regímenes de tratamiento inadecuados.
Caso clínico:Niña de 6 años referida desde Guinea por tuberculosis (TBC) ganglionar diseminada de 2 años de evolución que fue tratada con isoniacida (INH), pirazinamida (PZ) y rifampicina (RIF) sin resultado satisfactorio. Ingresó en otro centro hospitalario español donde se confirmó el diagnóstico y se objetivaron absceso de psoas izquierdo y mal de Pott a nivel L1-L2 que precisaron drenaje y artrodesis. Se aislaron bacilos tuberculosos resistentes a hidrazida por lo que se instauró nueva pauta antifímica con Rifampicina + Etambutol + Pirazinamida + Amicacina. 12 meses después consulta en nuestro centro por síndrome febril, dolor dorsal y aumento de tamaño de las adenopatías y se confirma progresión de la enfermedad ganglionar con reaparición del absceso de psoas. En el estudio de sensibilidad a fármacos antituberculosos se objetiva resistencia a INH, RIF y PZ por lo que inicia nueva terapia con Etambutol + PAS + Ciprofloxacino + Estreptomicina. Así mismo se realiza estudio inmunológico que detecta defecto micobactericida en la vía IFNγ -IL2-receptores por lo que se añade IFNγ al tratamiento. Actualmente ha cumplido 12 meses de tratamiento con evolución satisfactoria y ausencia de recaidas.
Comentarios:La incidencia de TBC resistente a fármacos ha aumentado en los últimos años y es más frecuente en niños nacidos en el extranjero (Asia, África y Ámerica latina) y tratados previamente por TBC. El régimen inicial de tratamiento debe incluir al menos 4 fármacos, de los que al menos dos deben ser bactericidas, hasta que se disponga de los resultados de pruebas de sensibilidad y no están indicados regímenes de corta duración.
P333SEPSIS FULMINANTE POR NEUMOCOCO: SINDROME DE WATERHOUSE-FRIEDERICHSEN
Alberto Varona García, José Carlos Flores González, Cristina Montero Valladares, Lidia M. Santalo González, Antonio Vázquez Florido, Víctor Manuel Navas López, Elia Sánchez Valderrábanos, Miguel Muñoz Sáez, Juan Antonio Soult Rubio y José Domingo López Castilla
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivo:La sepsis bacteriana es una infección grave que pone en peligro la vida del niño. Su forma de presentación más severa es la sepsis fulminante, con síndrome de Waterhouse-Friederichsen.
Presentamos un caso de sepsis fulminante por neumococo, con necrosis suprarrenal demostrada en el estudio necrópsico o síndrome de Waterhouse-Friederichsen.
Observación clínica:Se trata de una niña de 2 años de edad, con antecedentes personales de sepsis-meningitis por meningococo B en período neonatal, que acude a Urgencias por cuadro de fiebre alta de 12 horas de evolución. A su ingreso presenta mal estado general, coloración pálido-cianótica, petequias generalizadas, pulsos filiformes, relleno capilar muy enlentecido y afectación del nivel de conciencia con escasa respuesta a estímulos. Ingresa en la Unidad de Cuidados Intensivos Pediátricos en situación de extrema gravedad, precisando reposición de volemia, fármacos inotrópicos y vasoactivos, ventilación mecánica, hemoderivados, antibioterapia y corticoterapia; a pesar de lo cual no se consigue restablecer la estabilidad hemodinámica, siendo exitus a las 5 horas de su ingreso. En hemocultivo, realizado a su ingreso, se aislóStreptococcus pneumoniae. El estudio postmortem reveló necrosis suprarrenal bilateral. En los cultivos postmortem, de sangre y líquido cefalo-raquídeo, se identificóStreptococcus pneumoniae.
Comentarios:La sepsis bacteriana y, sobre todo, el síndrome de Waterhouse-Friederichsen han sido cuadros clínicos asociados casi exclusivamente a la infección invasora por Neisseria meningitidis. Sin embargo, en los últimos meses hemos observado casos de sepsis y shock séptico de etiología neumocócica.
El caso presentado es un cuadro clínico excepcional, ya que no existen en la literatura científica casos de síndrome de Waterhouse-Friederichsen en el curso de sepsis por neumococo.
Por tanto, ante estos cuadros clínicos debemos sospechar, no sólo una etiología meningocócica, sino también porStreptococcus pneumoniae.
Debemos insistir en que, actualmente, existe un medio eficaz para prevenir esta grave enfermedad, como es la vacuna conjugada neumocócica heptavalente.
P334EPIDEMIOLOGIA DE LAS SEPSIS Y MENINGITIS BACTERIANAS: 1993-2003
Juan Antonio Soult Rubio, Cristina Montero Valladares, Víctor Manuel Navas López, Antonio Vázquez Florido, Alberto Varona García, Juan David González Rodríguez, Miguel Muñoz Sáez, Julio Salvador Parrilla Parrilla, José Domingo López Castilla y Elia Sánchez Valderrábanos
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivo:Las sepsis y meningitis bacterianas son infecciones graves. La incidencia de los distintos gérmenes está sufriendo grandes variaciones, algunas influenciadas por el uso de las vacunas conjugadas. Nuestro objetivo es determinar los cambios en la epidemiología de las sepsis y meningitis bacterianas en nuestro medio.
Métodos:Estudio prospectivo de los casos de sepsis y meningitis bacterianas diagnosticados desde enero de 1993 a febrero de 2003, inclusive.
Resultados:Entre 1993-1997 el germen más frecuente fue meningococo B, desde 1997 a 2000 fue meningococo C, en 2001 de nuevo meningococo B y en los últimos meses de 2002 y primeros de 2003 ha sido neumococo. Algunos cambios han sido determinados por el uso de vacunas conjugadas; así, desde que en 1994 se comercializa la vacuna conjugada frente a Hib, la incidencia de este germen descendió progresivamente y desde que en enero de 1998 se introdujo en el calendario vacunal sólo se han observado 2 casos: uno en 1999 y otro en 2001, en niños no vacunados. También, desde que en julio de 2000 se inicia la inmunización sistemática con la vacuna conjugada frente a meningococo C, el número de casos descendió de forma espectacular, registrándose sólo 3 casos en 2001, 1 en 2002 y 1 en los meses de 2003. La incidencia de meningococo B ha permanecido estable durante el período estudiado, registrándose entre 10 y 15 casos por año. Sin embargo, la incidencia de neumococo ha sufrido un aumento muy significativo a partir de 2001 y sobre todo en los últimos meses de 2002 y primeros de 2003, en los cuales ha pasado a ocupar el primer lugar, por delante de meningoco B.
Conclusiones:1. La vacunación generalizada frente aNeisseria meningitidis C yHaemophilus influenzae b ha demostrado ser un medio de prevención eficaz.2. Aún se observan casos por meningococo C, por lo que debería generalizarse la vacunación al grupo de población (10-18 años) no vacunado.3. El aumento de incidencia y la morbi-mortalidad de las meningitis y sepsis neumocócicas exige que se tomen medidas para prevenir la enfermedad.4. El uso generalizado de la vacuna conjugada neumocócica heptavalente es el método de prevención más eficaz.
P335PROTEINA C REACTIVA EN LA INFANCIA: SIGNIFICADO DIAGNOSTICO Y EVOLUTIVO DE VALORES SUPERIORES A 100 MG/L
Ana Cristina Rodríguez Dehli, Rosa P. Arias Llorente, M. Nuria Fernández González, José David Herrero Morin, Santiago Jiménez Treviño, Paula Touza Pol, Gil Daniel Coto Cotallo y José Blas López Sastre
Hospital Central de Asturias, Oviedo.
Introducción:Niveles elevados de proteína C reactiva (PCR) se relacionan con procesos potencialmente severos. Este estudio pretende describir qué patologías se asociaron más frecuentemente con niveles de PCR en suero superiores a 100 mg/l en pacientes pediátricos, así como el significado pronóstico de los mismos.
Métodos:De las 2957 determinaciones de PCR realizadas en pacientes pediatricos a lo largo del año 2002 en nuestro hospital, se revisaron epidemiológica y clínicamente aquellas superiores a 100 mg/l en los diferentes rangos de edad.
Resultados:Se encontraron niveles de PCR superiores a 100 mg/l en el 4,53 % de las determinaciones, con ratio varón/mujer de 1,3/1. Los pacientes se clasificaron en grupos de edad: neonatos (12,7 %), lactantes (42,5 %), preescolares (2-5 años, 23 %) y escolares (6-13 años, 21,8 %). Las patologías más frecuentemente encontradas fueron sepsis nosocomiales en neonatos (72,7 %), sepsis en lactantes (64,8 %), y neumonías en preescolares (35 %) y en escolares (26 %). Se encontraron niveles medios de leucocitos en sangre de 18963 y 17560 e índices cayados/segmentados medios de 0,11 y 0,1 en neonatos y no neonatos respectivamente. Se empleó antibioterapia en el 94,2 %, con una duración media de 16 días, siendo el antibiótico más usado la teicoplanina en neonatos y la cefotaxima en el resto. Un 37,9 % de los pacientes precisaron ingreso en unidades de cuidados intensivos; el 36 % de los pacientes neonatos y el 42,1 % de los restantes, siendo exitus por el proceso asociado a elevación de PCR un 1,1 % de los casos.
Conclusiones:Los resultados sugieren que niveles de PCR superiores a 100 mg/l en edad pediátrica se asocian mayoritariamente a sepsis en neonatos y lactantes, y a neumonía en preescolares y escolares, acompañándose de alteración de otros parámetros analíticos. Nuestros pacientes con frecuencia precisaron tratamiento antibiótico prolongado e ingreso en unidades de cuidados intensivos, siendo exitus un 1,1 % de los mismos.
P336TUBERCULOSIS INFANTIL DE PRESENTACION MULTIORGANICA EN NIÑA INMUNOCOMPETENTE
M. del Carmen Martín Ruiz, Eulogio Agulla Rodiño, Luis Zarallo Cortés y Julián Vaquerizo Madrid
Hospital Universitario Infanta Cristina, Badajoz.
Introducción:Presentamos un caso de TBC con infiltrado pulmonar, meningitis y espondilodiscitis de evolución muy desfavorable, en una niña sin antecedentes de enfermedad previa de interés ni trastornos de la inmunidad.
Caso clínico:Niña de 2 años que ingresa con coma neurológico agudo e hidrocefalia tetraventricular. Dos semanas antes fiebre, vómitos y cefalea. Aparición posterior de deterioro neurológico progresivo.
Antecedentes personales:Contacto íntimo con familiar afecto de TBC bacilífera. PPD en la niña en ese momento negativo aunque se inició quimioprofilaxis con isoniacida a dosis habituales.
Exploración:Coma profundo, respuesta en extensión a estímulos dolorosos, pupilas isocóricas poco reactivas, hipotonía, reflejos plantares en extensión, no signos meníngeos.
Exámenes complementarios:PPD(+). LCR y de drenaje ventricular: pleocitosis mononuclear, hipoglucorraquia, proteinorraquia, se aisla Mycobacteria Complex. EEG: afectación cerebral difusa, asimetría interhemisférica, anomalías epileptiformes, paroxismos generalizados. Pruebas de imagen: condensación LSD; destrucción espacio intervertebral D8-D9 con fusión y lesión de cuerpos vertebrales, masa paravertebral derecha; hidrocefalia y atrofia cerebral de los núcleos de la base, lóbulos temporales y frontales. Potenciales evocados visuales ausentes. Estudio serológico diverso negativo.
Evolución:SIADH, hepatitis tóxica, sepsis, otitis, epilepsia, amaurosis.
Tratamiento:Drenaje ventricular externo, derivación ventrículo peritoneal, tuberculostáticos (sensibilidad al antibiograma), corticoides, antibioterapia de amplio espectro, plaquetas, anticonvulsivantes, gastrostomia, rehabilitación.
Discusión:La forma de presentación multiorgánica de la TBC infantil es infrecuente en la actualidad.
Se confirma la importancia de un diagnóstico y tratamiento precoz como prevención de secuelas importantes.
Nuestro caso plantea la necesidad de un despistaje diagnóstico más exhaustivo en caso de foco de contagio actual a pesar del PPD negativo.
P337INFECÇÃO PELO VIH NA CRIANÇA
M. Ana G. del Castillo y Laura Marques
Hospital de Crianças Maria Pia, Porto (Portugal).
Antecedentes:A infecção pelo Vírus da Imunodeficiência Humana na criança é uma situação grave, de mau prognóstico e difícil tratamento. Actualmente é possível prevenir a transmissão vertical na quase totalidade dos casos através da terapêutica antiretrovírica adequada à grávida associada a cesariana electiva, profilaxia peri-parto e ao recém-nascido e evicção da amamentação.
Objectivo:Caracterizar os casos de infecção ou risco de infecção pelo Vírus da Imunodeficiência Humana (VIH) seguidos na Consulta de Imunodeficiências.
Material e métodos:Analise retrospectiva dos casos com infecção ou risco de infecção VIH seguidos na Consulta de Imunodeficiências do nosso hospital desde 1994.
Resultados:Nestes 8 anos foram acompanhadas 66 crianças. Foi confirmada a infecção em 20 (18 pelo VIH1 e 2 pelo VIH2). Adquiriram a infecção por via vertical 17 delas, 1 por via sexual e 2 por via transfusional (infectadas pelo VIH 2).
Seis crianças foram diagnosticadas entre os 4 e as 8 semanas, 11 após os 4 meses de idade (nas quais a seropositividade materna era desconhecida), 1 aos 13 anos (transmissão sexual) e duas aos 7 e 15 anos (via transfusional). Oito crianças encontravam-se no estadio N1 na data do diagnóstico, uma no estadio N2, A1, B1 e A3 respectivamente e dois no B3. Cinco crianças apresentaram critérios de Sida.
A maioria das crianças efectuaram terapêutica antiretrovírica tripla. Nove tiveram uma boa resposta à terapêutica tanto do ponto de vista clínico como analítico e cinco tiveram vírus resistentes. Duas crianças faleceram. Uma, aos 9,5 anos com critérios de Sida e imunodepressão severa, apresentava leishmaníase sistémica resistente à terapêutica e Tuberculose disseminada. A outra criança faleceu aos 2,5 anos, também com critérios de Sida, por uma sepsis fulminante. Três crianças foram transferidas para serviços de outros hospitais (por idade ou por mudança de residência).
Conclusões:Em Portugal tem sido implementado o rastreio universal de infecção VIH à grávida. No entanto os resultados não são aínda satifatórios, como é patente nesta casuística em que a seropositividade materna era desconhecida em 11 crianças que foram infectadas e cujo diagnóstico foi tardío. É necessario melhorar esta situação no sentido de prevenir eficazmente a transmissão vertical desta infecção.
P338INFECCION POR CITOMEGALOVIRUS EN NIÑOS MENORES DE 18 MESES
Zuriñe Martínez de Compañón Martínez de Marigorta, Victoria Trenchs Sáinzde la Maza, Laia Alsina Manrique de Lara, Carlos Mainou Cid, Teresa Juncosa Morros y Carmen Muñoz Almagro
Hospital San Joan de Deu, Barcelona.
Introducción:La infección por citomegalovirus (CMV) es muy prevalente en nuestro medio, pasando muchas veces desapercibida. En los primeros meses de vida la infección congénita (IC) toma mayor protagonismo por las secuelas que puede presentar. Así mismo, el gran polimorfismo clínico de la infección adquirida (IA) justifica su inclusión en el diagnóstico diferencial de sintomatología muy diversa.
Objetivos:1. Describir diferentes formas de presentación de infección por CMV, haciendo hincapié en la IC y la IA.2. Valorar el uso de la técnica de PCR para su diagnóstico.
Métodos:Estudio retrospectivo (1997-2002) de niños menores de 18meses con serologías y determinación PCR positivas para CMV.
Resultados:Se recogen 20 casos, edad media de 5,6 meses en la 1.ª valoración, 12 son niñas. Las serologías a CMV se solicitan en 11 casos por sospecha clínica de IC (2 seroconversiones maternas en el embarazo, 1 gestación de riesgo y 8 con estigmas en la exploración física al nacer), en 3 niños asintomáticos en estudio de adopción, en 5 casos por síntomas infecciosos (1 gastroenteritis, 1 adenitis, 1 síndrome mononucleosiforme, 1 meningoencefalitis y 1 síndrome febril con leucopenia) y en 1 niño estudiado por Síndrome de West. En todos hay un resultado de PCR en plasma, linfocitos u orina positivo. Una de las sospechas de IC se confirma ante positividad de PCR en linfocitos al 3.º día de vida. Se realiza la determinación de PCR en sangre de talón en 3 casos, todas ellas negativas; uno es una sospecha clínica de IC, que se descarta. Las secuelas de las IC incluyen hipoacusia, retraso psicomotor, coriorretinitis, alteraciones hepáticas e inmunitarias. Entre las IA 3 casos permanecen asintomáticos y 1 es éxitus.
Comentarios:1. A pesar de la diferencia clínica entre IC e IA es importante contar con la técnica de PCR para llegar a un diagnóstico cierto, sobre todo en caso de duda en infecciones perinatales.2. Dado su gran polimorfismo clínico debería prodigarse más el uso de técnicas diagnósticas para su detección.3. No hay que olvidar la potencial gravedad de la IA por CMV, principalmente en déficits inmunitarios.
P339TROMBOSIS VENOSA PROFUNDA EN UN CASO DE VARICELA Y SHOCK TOXICO ESTREPTOCOCICO
José Manuel Jiménez Hinojosa, José María Lloreda García, Antonio Madrid Madrid, Yolanda María Chica Fuentes, Olga M. Escobosa Sánchez, M. Pilar Ranchal Pérez, Lourdes Escudero Ruiz de Lacanal, David Moreno Pérez, Francisco Jesús García Martín y Antonio Jurado Ortiz
Hospital Materno Infantil, Málaga y Hospital General Carlos Haya, Málaga.
Introducción:La infección por el virus varicela-zóster se ha relacionado con la aparición de diversos tipos de vasculopatías, entre ellas la trombosis venosa profunda, que parece ser debida a lesión del endotelio por factores no bien conocidos desencadenados por la infección viral. La aparición del síndrome de shock tóxico estreptocócico (SSTS) también se ha descrito ligada al cuadro de varicela, entre otras situaciones.
Caso clínico:Niña de 4 años de edad que en el curso de una varicela de 4 días de evolución presenta empeoramiento del estado general, fiebre de hasta 39 °C, y dolor en miembro inferior izquierdo coincidiendo con la aparición de un exantema eritematoso micro-papuloso generalizado, con algunas petequias puntiformes disemidadas. A la exploración presentaba mediano estado general. La TA era de 100/80 mmHg y la FC de 180 lpm. Llamaba la atención la presencia a nivel de pierna izquierda de tumefacción y calor local, con dolor a la palpación en dicha extremidad y en hemiabdomen homolateral. A nivel de la piel destacaba el exantema escarlatiniforme previamente descrito y lesiones típicas de varicela. Resto de exploración normal. En las pruebas complementarias destacaron: leucopenia (2.850 leucocitos/mm3); trombopenia (19.800 plaq./mm3); urea y creatinina en plasma elevadas (123 y 1,5 mg/dl respectivamente); TPTA alargado (52 seg.). Se realizó eco-doppler de miembro inferior izquierdo detectándose obstrucción de vena femoral y poplítea con buena permeabilidad del sistema venoso superficial, catalogándose de trombosis venosa profunda y tratada con reposo y anticoagulantes. Durante su evolución presentó otros datos sugestivos de SSTS (hipotensión, oliguria, aumento de transaminasas). Recibió tratamiento con penicilina G y clindamicina i.v. previa toma de hemocultivo donde se aisló Streptococo pyogenes. La paciente recibió además las medidas de soporte habituales teniendo una buena evolución clínica.
Comentarios:Llama al atención la concurrencia de dos complicaciones potencialmente graves y poco frecuentes de la varicela en una niña previamente sana: la trombosis venosa y el SSTS. El aumento en la incidencia de estas complicaciones, unido a la probada efectividad de la vacunación, hace que deba plantearse la posibilidad de incluir la vacuna antivaricela en el calendario actual.
P340INFARTO CEREBRAL CON AFECTACION DE GANGLIOS BASALES Y LOBULO TEMPORO-OCCIPITAL EN UN CASO DE VARICELA
M. Pilar González Santiago, Ana M. Carrasco Torres, Pilar Sevilla Ramos, Margarita I. Cebrero García, Álvaro Lassaletta Atienza y José Enrique García de Frías
Hospital Príncipe de Asturias, Alcalá de Henares.
Introducción:La enfermedad cerebrovascular en la infancia es poco frecuente, representando la infección por varicela un factor de riesgo importante (55 % en infartos de ganglios basales en niños son atribuidos a la varicela como causa).
Caso clínico:Niña de 9 años, sin antecedentes de interés, que acude por un episodio de cefalea súbita derecha, confusión y discurso inapropiado sin aparente desconexión; posteriormente observan hemiparesia izquierda. Afebril. Doce días antes había iniciado exantema febril compatible con varicela.
Exploración:Consciente, orientada, tendencia a la somnolencia. Respuestas verbales adecuadas. Hemiparesia izquierda. Parálisis facial central izquierda. Lesiones compatibles con varicela en fase de costra. Resto de exploración normal. Se realizó analítica y pruebas complementarias encaminadas a descartar otras etiologías que fueron normales. EEG: trazado de base lento con descargas en forma de punta-onda frontotemporal derecha. TAC craneal normal. RNM craneal: lesiones compatibles con infarto isquémico de los ganglios basales. Angiorresonancia: asimetría en el calibre de la carótida interna y la cerebral media, estando esta arrosariada y con menos ramas terminales.
Durante el ingreso recibió Aciclovir iv, corticoterapia y Aspirina a los 2 dias. Presentó febrícula esporádica; situación basal normal salvo leve hemiparesia izquierda y parálisis facial central izquierda. El 9.º día de ingreso se observó un empeoramiento con cefalea, vómitos y somnolencia. Se realiza una 2.ª RMN donde se aprecia un amplio infarto en hemisferio derecho. Se traslada a UCIP. Angiografía cerebral: obstrucción del sifón carotídeo derecho con buena circulación colateral. Se añade a la Aspirina, Clopidogrel y Nimodipino con mejoría de los síntomas. La paciente presenta actualmente leve hemiparesia con cierta distonia.
Comentario:El infarto cerebral atribuido a varicela es poco frecuente siendo sin embargo, la varicela una etiología importante en el accidente isquémico cerebral en niños. Suele producirse semanas después del exantema. La localización más frecuente es en ganglios basales siendo la evolución favorable. Se han descrito pocos casos en los que se produzca reinfarto. En nuestra paciente la progresión del infarto o reinfarto a pesar del tratamiento justificó la asociación del 2.º antiagregante.
P341TUBERCULOSIS PULMONAR: UNA CAUSA OLVIDADA DE MASA PULMONAR
Africa Jordán Jiménez, Francisco Hernández Oliveros, Fernando Baquero Artigao, M. Isabel de José Gómez, Francisco Martínez Cortés, Clementina Borque Andrés, Fernando del Castillo Martín, Susana Rivas Vila, Luis Felipe Ávila Ramírez y María Jesús García de Miguel
Hospital Universitario La Paz, Madrid.
Introducción:La tuberculosis pulmonar primaria, en lactantes y niños, suele ser asintomática y con escasas manifestaciones radiológicas. Una forma de presentación poco frecuente es la de imagen de masa pulmonar. El diagnóstico diferencial en estos casos es difícil e incluye malformaciones pulmonares y tumores propios de la infancia.
Pacientes:
Caso 1:Niña de 9 meses diagnosticada de malformación adenomatoide quística. En la biopsia intraoperatoria se observaron granulomas tuberculoides. En el examen definitivo de la pieza quirúrgica se apreció afectación miliar de diafragma, pleura y LID. El Mantoux preoperatorio fue negativo, positivizándose con posterioridad (10 mm). El cultivo de mycobacterias en líquido pleural y jugo gástrico fue negativo. Tras lobectomía inferior derecha recibió tratamiento con tres fármacos tuberculostáticos durante 6 meses.
Caso 2:Niña de 4 meses con diagnóstico de teratoma tímico. Durante la cirugía se encontró una masa pulmonar en LSD cuyo estudio intraoperatorio objetivó la presencia de granulomas tuberculoides con intensa destrucción tisular. Debido al grado de afectación se realizó lobectomía. El Mantoux fue positivo (10 mm). En el cultivo del líquido pleural se aisló M. Tuberculosis. Presentó empiema pleural y fístula broncocutánea durante el postoperatorio, ambas complicaciones respondieron al drenaje torácico y antibioterapia. Durante seis meses recibió tratamiento tuberculostático.
Conclusiones: 1.La TBC pulmonar debe considerarse en el diagnóstico diferencial en niños que presentan masa pulmonar. 2. La sospecha radiológica más frecuente es malfomación adenomatoidea quística y teratoma tímico. 3. El tratamiento mediante lobectomía está indicado cuando existe destrucción tisular irreversible. 4. Las principales complicaciones postquirúrgicas son el empiema y la fístula broncopleural.
P342REVISION DE LOS CASOS DE PALUDISMO EN NUESTRO MEDIO
Elena Montesinos Sanchís, Sandra Solaz Barrios, José Fernando Mata Bernáldez, José Tacons Mateu, Carmen Lecuona López, Concepción Tomás Rates, José Ardit Lucas y Vicente Álvarez Ángel
Hospital General Universitario de Valencia.
Introducción:Como consecuencia del aumento de la población inmigrante en nuestro país, nos encontramos con un aumento de patologías que estamos poco habituados a manejar, entre ellas el paludismo.
Método:Revisión retrospectiva de los casos de paludismo pediátrico de nuestro hospital en los últimos dos años.
Resultados:Se diagnosticaron ocho casos, todos ellos procedentes de Guinea Ecuatorial y de raza negra. Tres casos nacidos en España habían viajado a dicho país el mes previo al diagnóstico, llevando una profilaxis correcta uno de ellos. En cinco casos existía otro episodio de paludismo previo.
P. falciparum fue el responsable de seis casos y P. vivax en los otros dos. La forma de presentación constante en todos ellos fue la fiebre, seguida de hepatoesplenomegalia en seis pacientes. En la analítica siete pacientes cursaron con anemia (dos de ellos precisando transfusión), seis con trombopenia, y como otros hallazgos encontramos leucopenia y linfocitosis.
El diagnóstico se llevó a cabo con gota gruesa para determinar la presencia de parásitos, y con frotis para el diagnóstico de especie y cuantificación de la parasitación. Ningún caso presentó criterios clínicos de gravedad.
Se trataron con Sulfato de Quinina (7 días) más clindamicina (5 días) los menores de 8 años y Sulfato de Quinina más doxicilina (7 días) los mayores de dicha edad. En los dos casos parasitados por P. vivax se empleó el fosfato de primaquina para la prevención de recidivas. La efectividad del tratamiento se comprobó al finalizar éste con la toma de una gota gruesa que fue negativa en todos ellos, no encontrando recidivas hasta el momento.
Como curiosidad encontramos parasitación en heces en cuatro de los casos y escabiosis en otro.
Conclusiones:Destacar la importancia de la sospecha clínica en todo paciente con fiebre procedente de una zona endémica de paludismo, siendo importante destacar el país de procedencia para el tratamiento, dado el aumento de las resistencias a la cloroquina. Además muchos de estos casos pueden presentar otras parasitaciones asociadas.
P343ABSCESO PERIAMIGDALINO POR ENTEROBACTER CLOACAE Y STREPTOCOCCUS MILLERI EN LACTANTE DE 4 MESES
José María Lloreda García, José Manuel Jiménez Hinojosa, Gemma Colomé Rivero, Salvador Ariza Aranda, Pascual Caballero Fernández, David Moreno Pérez, Isabel Durán Hidalgo, Francisco Jesús García Martín y Antonio Jurado Ortiz
Hospital General Carlos Haya, Málaga.
Introducción:El absceso periamigdalino es una patología poco frecuente en la infancia y excepcional por debajo del año de edad. Presentamos un caso en un paciente de 4 meses con un absceso polimicrobiano y bacteriemia
Caso clínico:Lactante de 4 23/30 meses que ingresó por presentar cuadro de 15 días de evolución de fiebre diaria, máximo 40°, rechazo del alimento, con llanto intenso e hiperextensión del cuello con las tomas, y tumoración submaxilar izquierda dolorosa. Recibió tratamiento domiciliario con amoxicilina durante 5 días, sin mejoría. Destacó en la exploración un moderado estado general, irritabilidad y adenopatía submaxilar izquierda de 2-3 cm de diámetro, dura y dolorosa a la palpación. Hiperemia amigdalar y faríngea, con desplazamiento del velo del paladar hacia abajo y abombamiento de amígdala izquierda, desplazada hacia la línea media. Hemograma: leucocitos 29.070 (74 % neutrófilos; 15 % linfocitos), PCR: 146,7 mg/l. TAC orofaríngeo: masa quística de 3 * 2,5 cm en región amigdalar izquierda que comprime y desplaza la luz, compatible con absceso. Se drenó y se inició empíricamente tratamiento con amoxicilina-clavulánico vía intravenosa. Tras recibir cultivos (Enterobacter cloacae, resistente a amoxicilina-clavulánico y Streptococcus milleri, resistente a ampicilina y cefotaxima, en cultivo del absceso y Streptococcus milleri en hemocultivo) se instauró tratamiento con meropenem vía intravenosa durante 9 días. Dado que la literatura describe casos de abscesos múltiples por Streptococcus milleri, se descartó esta posibilidad mediante estudio ecográfico. Se realizó un estudio inmunológico (inmunoglobulinas, capacidad oxidativa de los neutrófilos y subpoblaciones linfocitarias) con valores normales para su edad.
Comentarios:El absceso periamigdalino es una patología inusual en lactantes y debe distinguirse del absceso retrofaríngeo y otros procesos contiguos. La bacteriemia por Streptococcus milleri se asocia a otros abscesos, especialmente del sistema nervioso central y abdominales. Dada la precocidad del caso, se investigó un trastorno inmunitario de base.
P344SIFILIS CONGÉNITA EN EMBARAZO CONTROLADO
Alejandra López Guinea, Julia Cano Fernández, Blanca Espinola Docio, Enma de la Torre Montes de Neira, Gladys Yep Chullen y José Manuel Melendi Crespo
Hospital del Niño Jesús, Madrid y Universidad Autónoma de Madrid.
Introducción:La sífilis congénita es una enfermedad poco prevalente en nuestro medio, cuyo despistaje se incluye en el control serológico de la embarazada en la primera visita prenatal. Su clínica multisistémica raramente afecta de forma grave al riñón.
Objetivos:Describir un caso frecuente de sífilis congénita tras embarazo controlado y serología negativa en el primer trimestre que se manifestó con grave afectación renal.
Caso clínico:Lactante de dos meses y medio con hematuria macroscópica de tres días de evolución, sin otros síntomas. Antecedentes: padres ecuatorianos, embarazo controlado en España con serologías negativas en el primer trimestre, parto hospitalario a término. Peso al nacimiento 2480 g y período neonatal inmediato normal. A los quince días de vida, erupción de máculas eritematosa en palmas y plantas. Exploración: palidez de piel y mucosas con intensa descamación cutánea en manos y pies, hepatoesplenomegalia, edemas generalizados. Análisis de sangre: anemia, trombopenia, leucocitosis con intensa desviación izquierda y elevación de reactantes de fase aguda e Ig M. Hipoproteinemia, hipoalbuminemia, hipertrigliceridemia y LDH elevada. Análisis de orina: hematuria macroscópica y proteinuria franca. Ecografía abdominal: nefromegalia bilateral. Función renal y tensiones arteriales normales. Evolución: al cuarto día se pide serología para sífilis y se encuentra RPR positivo a título 1/32, TPHA y FTA-Abs positivos. Se instaura tratamiento con Penicilina G sódica intravenosa durante 14 días y la paciente mejora clínica y analíticamente. Repetida la serología materna, muestra signos de infección reciente por Treponema Pallidum.
Conclusiones:Se destaca la importancia de realizar un diagnostico deferencial completo ante cualquier cuadro clínico, sin obviar parología poco frecuente. Importancia de efectuar el adecuado despistaje de infecciones connatales durante la gestación
P345SEGUIMIENTO DE LA PREVENCION EN LA TRANSMISION VERTICAL DE HIJOS DE MADRE VIH EN EL HOSPITAL UNIVERSITARIO "VIRGEN DE LA ARRIXACA" DE MURCIA
Carlos Pérez Cánovas, Juan José Quesada López, Arancha Escribano Muñoz, Juana M. Espín López, Lluis Marín Vives y Juan José Agüera Arenas
Hospital Universitario Virgen de la Arrixaca, El Palmar.
Introducción:La introducción del Protocolo ACTG 076 a partir de 1995 basado en el uso de Zidovudina (AZT) a partir de la semana 14 de gestación, durante el parto y en el recién nacido hasta la sexta semana de vida se ha demostrado claramente eficaz disminuyendo la incidencia de transmisión vertical en un 66 %. Si a esto le asociamos la realización de cesárea electiva y evitación de maniobras invasivas la incidencia desciende hasta el 2 %.
Metodología:Se realiza un estudio retrospectivo descriptivo de los hijos de madres VIH + nacidos en el Hospital Virgen de la Arrixaca de Murcia entre los años 1995 y 2002, ambos inclusive. Se analiza la administración de AZT u otros antirretrovirales durante la gestación, parto y primeras seis semanas de vida del recién nacido, así como la realización de cesárea electiva. Se determina la posibilidad de infección mediante resultados de PCR y cultivo del VIH. Así mismo se recogen datos analíticos durante el seguimiento de los niños y la edad en la que se produce la desaparición de anticuerpos frente a VIH en niños no infectados hasta el año 2001.
Resultados:Se han incluido un total de 87 niños hijos de madres VIH + durante el tiempo que transcurre el estudio de los cuales en 59 (67 %) se siguió el protocolo mientras que en 28 (33 %) no se hizo. No se produjo ningún caso de infección en los niños entre los que se siguió el protocolo. De los no protocolizados hubo 6 infectados (21 %) frente a 22 no infectados (79 %). La edad media en la que se objetivó la negativización de anticuerpos frente a VIH en los no infectados fue a los 14 meses. No se encontró ninguna alteración en hemogramas seriados que haya obligado a la supresión de tratamiento antirretroviral, así como tampoco han aparecido datos de toxicidad mitocondrial o acidosis láctica.
Conclusiones:1. El seguimiento del protocolo ACTG 076 unido a cesárea electiva parece ser eficaz en nuestro medio.2. La incidencia de transmisión vertical en hijos de madre VIH + que no siguieron protocolo se asemeja a la incidencia descrita en países europeos.3. No encontramos anomalías en hemogramas seriados durante el seguimiento que hayan obligado a la supresión del tratamiento antirretroviral.
P346VIRUS DE EPSTEIN BARR: AGENTE ETIOPATOGÉNICO DE NEOPLASIAS
Salvador Ariza Aranda, Pascual Caballero Fernández, Esther García Requena, Rafael Vera Medialdea, Antonio Herrero Hernández, Francisco Jesús García Martín y Antonio Jurado Ortiz
Hospital General Carlos Haya, Málaga y Hospital Materno Infantil, Málaga.
Introducción:El VEB posee un poder oncógenico conocido. Presentamos dos casos, en los que se constata: Carcinoma (ca.) Nasofaringeo en niño de 11 años procedente de Marruecos y linfoma angiocéntrico en niña de 6 años.
Casos clínicos:
Caso 1:Niño de 11 años que desde hace 4 meses presenta tumoración cervical izquierda, con aumento de tamaño progresivo, anorexia y perdida de peso. Además, refiere epixtasis recidivantes, cefalea hemicraneal e hipoacusia derecha. A la exploración destaca: adenopatias laterocervicales izquierdas que forman un paquete (3-4 cm), duras, adheridas, no dolorosas a palpación; ORL: amígdala derecha hipertrófica. Serologia: VEB: Ig G-VCA (630); Ig M-VCA (). EBNA Ig G+. EBEA Ig G 160. ECO y TC región cervical: masa faríngea que afecta a la totalidad del anillo de Waldeyer, con grandes adenopatias laterocervicales y ocupación de celdillas etmoidales derechas. AP de biopsia cervical: Ca. Nasofaringeo indiferenciado y PCR a VEB+. Se inicia tratamiento quimioterápico según protocolo NPC-2003-GPOH, con posterior radioterapia.
Caso 2:Niña de 6 años ingresada por pancitopenia en cuadro de fiebre y odinofagia de 10 dias de evolución. Presenta afectación general, adenopatias cervicales rodaderas, hipermia amigdalar y hepatoesplenomegalia. Serologia: VEB: Ig G-VCA 620; Ig M-VCA 0,227; PCR en suero a VEB: +. Triglicéridos 308 mg/dl. Ferritina: 1.432 ng/ml. Biopsia de MO: hemofagocitosis. TC de tórax: múltiples nódulos pulmonares de 2-5 mm. EcoDoppler abdomen: nódulos hipoecogénicos en hígado, bazo y riñones. Biopsia pulmonar: linfoma angiocéntrico. Recibe quimioterapia con CVP y CHOPP. Fallece por insuficiencia respiratoria secundaria a necrosis pulmonar y diátesis hemorrágica.
Comentarios:El VEB posee un importante papel en la etiopatogenia de neoplasias, parece tener una relevancia especial su capacidad de latencia, gracias a proteinas (EBNA-1 y LMPA1), con poder oncogénico per se. Se postula la existencia de variantes geneticas, aún no definidas, que faciliten dicha oncogénesis.
En nuestro medio, el ca. Nasofaringeo indiferenciado tiene incidencia < 1 %; sin embargo, en zonas del Magreb (nuestro primer caso), Alaska y Asia puede ser de un 10 %. Cerca del 100 % de estos tumores se relacionan con VEB. La serologia anti-VCA se utiliza para valorar la efectividad del tratamiento o recidivas tumorales.
P347SINDROME DE GUILLÉN BARRÉ: REVISION DE CASOS CLINICOS
Manuel Haro Gómez, M. del Carmen Vega Castaño, María José Villalobos Linares, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Objetivo:Aportar nuestra experiencia en los aspectos clínicos, diagnósticos y terapéuticos del síndrome Guillén Barré y contrastarla con los datos existentes en la literatura.
Material y método:Se realiza un estudio descriptivo retrospectivo mediante la revisión de historias clínicas de niños de < 14 años ingresados con el diagnóstico de sd. Guillén Barré desde 1989 hasta 2002. Los criterios diagnósticos utilizados son presencia de debilidad progresiva de más de un miembro, arreflexia, LCR con disociación albúmino-citológica y pleocitosis de < 10 células/mmc.
Resultados:Durante el período de estudio ingresaron en nuestro servicio 8 niños (5 varones y 3 hembras) de edades comprendidas entre 4 y 11 años. Todos se manifiestan con debilidad, arreflexia, mialgias de EEII con progresión ascendente y simétrica. Las parestesias aparecen en 3 niños, ataxia en 6, oftalmoplejía en 2, meningismo en 4, disfagia en 1, sialorrea en 1, llanto débil en 1, disfonía en 1, rinolalia en 1, hipertensión en 1, sudoración en 1. El LCR presenta disociación albúmino-citológica con PANDY + en todos los casos. Todos los ENG tienen un patrón con polineuritis desmielinizante/axonal distal/proximal. Los estudios serológicos son positivos a CMV, VRS, Parainfluenzae 1-2-3, Micoplasma en 1 niño; Coxiella Burnetti en 1 niño; VRS y CMV en 1 niño; CMV en 1 niño; CMV y Influenzae A-B en 1 niño. Se trató con gammaglobulina (IgG) iv a 4 niños entre el año 1996-2002 (3 con evolución favorable (e.f), 2 niños con IgG más corticoides entre 1991-1995 (1 con e.f) y 2 niños sin tratamiento entre 1989-1994 (1 con e.f). En la evolución de la enfermedad 7 niños consiguen una deambulación normal en los 3 meses siguientes, 1 niño tarda 12 meses y 2 niños aún presentan arreflexia-hiporreflexia 1-2 años posteriores.
Comentario:Entre las causas del sd Guillén Barré se baraja la autoinmune y la infecciosa. Destacamos que en nuestra revisión hemos obtenido un número importante de casos en los que se encuentran un gérmen que podría estar relacionado con el desarrollo de la enfermedad. El tratamiento utilizado ha dependido del año del diagnóstico y no se ha podido comprobar una mejor evolución clínica de unas alternativas terapéuticas respecto a las otras. Según la literatura la IgG induce mejoría evidente y los corticoides no tienen efecto.
P348ARTRITIS SÉPTICA POR NUEVO PATOGENO: KINGELLA KINGAE
M. del Carmen Vega Castaño, Mercedes López-Campos Bodineau, Concepción Hidalgo Figueroa, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Objetivo:Presentación de un caso de artritis séptica por Kingella kingae por la rareza del germen y la escasa bibliografía con descripción clínica en nuestro país.
Material y método:Anamnesis: niña de 17 meses que consulta por fiebre de dos días de evolución. Dos días antes del inicio del cuadro presentó un episodio de dolor y claudicación del miembro inferior izquierdo. Exploración: peso: 11.700 g (P75), longitud: 83,5 cm (P97), tumefacción de rodilla izquierda, dolorosa a la movilización y a la palpación, peloteo articular, surco rotuliano borrado. Rinorrea blanquecina posterior. Pruebas complementarias: hemograma: leucocitosis (13.600) con un 53,8 % de segmentados. PCR: 43,9 mg/l. Rx de rodillas: aumento de partes blandas sin alteración ósea. Ecografía de rodillas: presencia de líquido intraarticular. Artrocentesis: se obtiene líquido turbio con incontables PMN, y se aísla un germen Gram negativo identificado como K. kingae. Gammagrafía ósea: aumento de la vascularización a ése nivel sin afectación ósea. Tto: férula posterior inguinopedica. Cefotaxima y vancomicina durante 6 días para cubrir los gérmenes más frecuentes de artritis séptica en esta edad; tras el resultado del cultivo se suspendió vancomicina y se continuó con cefotaxima i.v. durante 7 días más. Al alta prosiguió con Tto oral con amoxicilina-clavulánico durante 2 semanas más. Evolución: favorable hasta la curación.
Comentarios:Kingella kingae es un cocobacilo gramnegativo excepcional como agente patógeno que emerge como causa de infecciones en la edad pediátrica apareciendo el 90 % de los casos en menores de 2 años. Forma parte de la flora de vía respiratoria alta y en la mayoría de los casos existía clínica de vías aéreas superiores los días previos. Se ha identificado como etiología de bacteriemia oculta, endocarditis, infecciones de vías respiratorias bajas, del SNC y osteoarticulares, por lo que hay que resaltar el interés de su búsqueda sistemática en menores de 5 años con clínica osteoarticular insidiosa con fiebre. El diagnóstico debe realizarse mediante artrocentesis y cultivo del material en frasco de hemocultivo. Evolución favorable con tratamiento i.v. con b -lactámicos durante un mínimo de 7 días seguido de tratamiento oral hasta completar 4-6 semanas.
P349OTOMASTOIDITIS
Cristina Flor Parra, Susana Cora López, María Jesús Balboa Vega, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Justificación:La mastoiditis como complicación más frecuente de la otitis media aguda pasó a ser con la introducción de los antibióticos una complicación excepcional, pero últimamente se está observando un incremento de la misma.
Objetivos:Evidenciar el aumento de frecuencia de mastoiditis agudas diagnosticadas en nuestro hospital en los últimos años.
Material y métodos:Revisamos los casos de otomastoiditis, entre el 1/1/95 y el 31/12/02, analizando edad, sexo, motivo de ingreso, antecedentes, epidemiología, clínica, pruebas complementarias, tratamiento y evolución.
Resultados:Estudiamos 23 niños, con edad media de 3,7 años. La media de mastoiditis aguda por año fue de 2,8 casos, encontrándose 16 (69,5 %) en los últimos 3. Distribución por edades: 2 casos (8,7 %) en menores de 1 año, 6 (26,1 %) entre 1 y 3 años y 15 (65,2 %) en mayores de 3. Distribución por sexo: 10 mujeres (43,4 %) y 13 hombres (56,5 %). Antecedentes: 8 casos (37,4 %) con OMA previa, 1 (4,3 %) otitis media crónica, 1 (4,3 %) otitis media supurada, 2 (8,6 %) varicela en el mes previo y 2 (8,6 %) faringoamigdalitis de repetición. La mayor incidencia se dio en invierno (10 casos, 43,4 %). Los síntomas más frecuentes fueron: tumefacción retroauricular (20 casos, 60,8 %), fiebre (14 casos, 60,8 %) y dolor en la región mastoidea (13 casos, 56,5 %). Pruebas complementarias: la cifra media de leucocitos totales fue de 17246/ul, la VSG media, de 40,95 mm 1.ª hora, y el valor medio de PCR fue de 42,13 mg/l. Se realizó TC en 20 casos (86,9 %): en 2 no se encontraron hallazgos significativos; en 18 (78,2 %) ocupación de celdillas mastoideas, lesiones de osteitis en 10 (43,4 %), absceso subperióstico en 2 (8,6 %) y complicaciones intracraneales en 2 (8,6 %). Se tomó muestra de exudado ótico en 11 casos (47,8 %), con cultivo negativo en 6 (26,1 %) y positivo en los restantes, aislándose P. Aeruginosa en 3 (13,1 %), S. Pneumoniae en 1 (4,3 %) y C. Albicans en 1 (4,3 %). Todos recibieron tratamiento con betalactámicos i.v. entre 7 y 10 días, y 6 (26 %) requieron tratamiento quirúrgico, siendo la evolución favorable en el 100 %. Estancia media:9,6 días.
Comentarios:Los casos de mastoiditis en nuestro centro han aumentado de forma significativa en los últimos años. Se da con mayor frecuencia en mayores de 3 años. Los aislamientos de gérmenes en cultivos, escasos. El 26 % requirió intervención quirúrgica y la evolución fue favorable en el 100 % de los casos.
P350PALUDISMO. UNA ENFERMEDAD EMERGENTE
Susana Cora López, Cristina Flor Parra, Cristina Hidalgo Figuerosa, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Antecedentes y objetivo:El paludismo, erradicado en nuestro país en 1964, está resurgiendo debido al aumento de la emigración desde países endémicos en los últimos años. Esto hace que su conocimiento sea cada día más necesario.
Métodos:Revisamos los 2 primeros casos de paludismo diagnosticados en nuestro hospital, en enero-03, analizando edad, sexo, procedencia, antecedentes, clinica, pruebas complementarias, tratamiento y evolución.
Resultados:Fueron dos hermanas de origen guineano nacidas en España de 3 (caso 1) y 6 (caso 2) años de edad, que habían regresado de un viaje a Guinea Ecuatorial 9 días antes del inicio del cuadro, sin realizar profilaxis para paludismo. Los datos fueron:Clínica:ambas presentaron fiebre de 3 y 4 días de evolución respectivamente, junto con vómitos y diarrea en el caso 2. P. Complementarias: ambas presentaron: anemia microcítica e hipocroma, elevación de reticulocitos, leucocitosis con neutrofilia y trombopenia; LDH, D-dímero, VSG y PCR elevadas; frotis de sangre periférica: microcitosis y parásitos intraeritrocitarios, granulación tóxica de neutrófilos, linfocitos activados y vacuolización de monocitos y disminución cuantitativa de serie plaquetaria; aumento de IgG en estudio de inmunidad humoral; trofozoitos de Plasmodium Falciparum en gota gruesa. Además, el caso 1 presentó: aumento de la hemoglobina fetal, descenso de albúmina y elevación de alfa-1 y gamma globulinas, quistes de Giardia Lamblia y antígenos de adenovirus en heces. El caso 2 presentó a su vez, descenso de albúmina y elevación de alfa 1 y alfa 2 globulinas y elevación de urobilinógeno en orina, siendo el resto de pruebas normales en ambas. Tratamiento: mefloquina (2 dosis con intervalo de 10 horas de 10 mg/kg/dosis v.o.) y Doxiciclina (5 mg/kg/día v.o. cada 12 horas, 8 días). Además el caso 1 recibió Metronidazol (30 mg/kg/día v.o. en 4 dosis al día, 7 días). Evolución: favorable en ambas, remitiendo la fiebre a las 24 horas del ingreso, sin volver a presentar vómitos el caso 2. Estancia hospitalaria: 9 días.
Conclusiones:Es importante considerar esta patología en niños procedentes de países endémicos con fiebre, sobre todo en aquellos nacidos en España que viajan a esas zonas, dado que en ellos es mayor el riesgo de gravedad. La oleada de emigración desde áreas endémicas últimamente, está motivando un aumento en incidencia del paludismo, lo que hace cada día más necesario el estudio y manejo de la misma por parte de los pediatras.
P351SIFILIS CONGÉNITA: OLVIDADA PERO NO DESAPARECIDA
Mercedes López-Campos Bodineau, M. del Carmen Vega Castaño, Pedro Terol Barrera, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Justificación:La sífilis es un problema cuya frecuencia esta aumentando en los países desarrollados.
Objetivos:Poner en evidencia la importancia del diagnóstico y tratamiento precoz en el embarazo y en el RN. Material y método: revisión retrospectiva de las historias de niños con diagnóstico de sífilis congénita en nuestro hospital desde Enero de 1995 hasta Diciembre de 2002. Se revisaron la edad, motivo de ingreso, antecedentes personales y familiares, exploración clínica, serología, exámenes complementarios, tratamiento y evolución.
Resultados:El grupo estudiado esta formado por 6 pacientes con una edad media de 14 días en el momento del diagnostico. Los motivos de ingreso fueron sintomatología clínica en 2 casos (33 %), distocia social en 3 casos (50 %), serología positiva para sífilis en screening neonatal en 1 caso (17 %).Antecedentes personales: embarazo controlado en el 50 % casos y en uno de ellos (17 %) serología luética positiva al 8.º mes de embarazo. Al nacimiento 3 pacientes tenían peso inferior al P3 (50 %), en 3 casos apareció en la evolución síndrome de abstinencia, el porcentaje con antecedentes maternos de ADVP fue del 67 % (4 casos), serología VHC positiva 17 % (1 caso) y serología VIH positiva 17 % (1 caso). Los síntomas más prevalentes fueron retraso estaturoponderal en 5 casos (83 %), palidez cutáneomucosa en 2 casos (33 %), pseudoparálisis de Parrot y hepatomegalia en 2 casos (33 %). Ausencia de clínica en 33 %. El porcentaje de pacientes con serología para sífilis positiva fue del 100 % y con serología para VHC del 33 % (2 casos). Datos analíticos de interés: anemia normocítica normocrómica junto con elevación de transaminasas en 2 pacientes; en LCR bioquímica anormal en 3 casos y serología para sífilis positiva en 1 caso. Rx de huesos largos: periostitis en 2 casos. Tratamiento: penicilina G parenteral durante una media de 15 días. En dos casos transfusión de concentrado de hematíes (33 %). Evolución favorable en el 100 % casos. Comentarios: dos terceras partes de los nacidos infectados de sífilis no tiene síntomas la nacer, de ahí la importancia de realizar en toda mujer un cribado con prueba no treponémica en la 1.º visita prenatal. La penicilina G parenteral es el único tratamiento eficaz documentado. El régimen de penicilina debe ser realizado sin interrupciones durante 10 días consecutivos.
P352CELULITIS CERVICOTORACICA Y MONONUCLEOSIS INFECCIOSA
Laura Castells Vilella, M.ª Socorro Uriz Urzainqui, Violeta Tenorio Romojaro, Raquel Dora Young Cecilia, M. Consuelo Sánchez Garre, M. Rosa Martorell Albareda e Inma Romero Novo
Hospital Mutua de Terrassa.
Introducción:Las infecciones cutáneas bacterianas son muy frecuentes en niños, siendo el estreptococo b -hemolítico del grupo A y el estafilococo aureus los principales responsables. La diferenciación de las dermohipodermitis como la erisipela, la celulitis y la fascitis, es básicamente clínica y en ocasiones difícil. El tratamiento incluye una cobertura antibiótica amplia y, en los casos graves, desbridamiento quirúrgico para disminuir su morbimortalidad.
Aportamos el caso de un niño de 15 meses con síndrome mononucleósico que presenta concomitantemente celulitis cérvico-torácica.
Caso clínico:Niño de 15 meses, sano, que inicia 4 días antes cuadro compatible clínica y analíticamente con mononucleosis infecciosa. Ingresa por presentar afectación marcada del estado general, importantes adenopatías laterocervicales y placa inflamatoria en zona cervical anterior. A la exploración destaca la sensación de enfermedad grave, palidez cutánea, exantema morbiliforme y extensa placa inflamatoria mal definida, de 26 * 17 cm, desde maxilar inferior hasta región torácica anterosuperior, paquetes adenopáticos laterocervicales, y hepatomegalia de 3 cm. La analítica muestra leucocitosis con desviación izquierda y PCR de 417 mg/l, iniciándose tratamiento con Amoxicilina-clavulánico. Persiste con picos febriles y rápida extensión de la placa inflamatoria. Se practica TAC que evidencia importante infiltración del tejido subcutáneo, próximo a la fascia muscular, en zonas submaxilar, cervical anterior y torácica alta. Ante la importante celulitis, riesgo fascitis necrotizante, y al observar panadizo ungueal en 2.º dedo de la mano derecha, se cambia antibioticoterapia por Cloxacilina y Clindamicina. La evolución es favorable, con mejoría clínica progresiva, remisión de la fiebre, formándose absceso submentoniano que se desbrida y cultiva. El hemocultivo y cultivo del absceso fueron negativos, creciendo S. Aureus en el cultivo ungueal, lo que sugiere que el panadizo fuera la puerta de entrada de la infección cutánea.
Comentarios:La alteración de la barrera cutáneo-mucosa, la neutropenia, la disfagocitosis y las alteraciones de la inmunidad predisponen a infecciones bacterianas. Algunos autores sugieren la asociación entre la infección por el virus Epstein Barr y una susceptibilidad aumentada a padecer infecciones bacterianas en niños sanos, como sería la referida en nuestro caso.
P353OSTEOMIELITIS TUBERCULOSA: PRESENTACION DE UN CASO
M. del Carmen Mosquera Pérez, Nieves Balado Insunza, Miriam García García, Martín Mosteiro Cerviño, Domingo González Lestón, José Luis Chamorro Martín, José Antonio Couceiro Gianzo y Jesús Antelo Cortizas
Complejo Hospitalario Xeral-Cíes, Vigo.
Introducción:La afectación osteoarticular por tuberculosis representa del 1 al 3 % de los casos de enfermedad tuberculosa. Las zonas corporales más afectadas son las vértebras, la cadera, la rodilla y los pies.
Caso clínico:Niño de 2 años y medio de edad que ingresa por presentar cojera de 2 meses de evolución y que en los últimos días presenta absceso a nivel de rodilla izquierda. A la exploración física: buen estado general, temperatura 37 °C, tumefacción y calor en rodilla izquierda. Resto de exploración física dentro de la normalidad.
Hemograma: leucocitos 14750/mm3 (Neut. 47 %, linf. 45 %, mono. 6 %, eosin. 1 %). Hb.11,4g/dl. VSG 52 mm/h. Proteína C Reactiva 17 mg/l. ASLO negativo. Inmunoglobulinas normales. HIV negativo. Bioquímica normal. Mantoux 27 x 21 mm. Radiografía de tórax normal. Radiografía de rodilla izquierda: lesión osteolítica en metáfisis y epífisis proximal de tibia. RM de rodilla izquierda concordante con la radiología, mostrando extensión y componente de absceso en partes blandas. Gammagrafía ósea: sugestiva de osteomielitis, fase de secuestro. Anatomía patológica de la lesión: granulomatosis epitelioide necrotizante. BAAR y amplificación ARN de jugo gástrico negativos. BAAR, amplificación ARN y Lowenstein de la lesión, positivo para Mycobacterium tuberculosis. Cultivo de material de absceso negativo. Estudio familiar de TB: negativo.
A su ingreso se procede al drenaje, limpieza y toma de muestras del absceso. Se inicia tratamiento antibiótico empírico. La evolución no favorable junto con Mantoux y pruebas específicas de TB positivas, confirman el diagnóstico de osteomielitis tuberculosa, iniciándose tratamiento específico.
Conclusiones:El diagnóstico de la osteomielitis tuberculosa suele ser difícil, lo que explica que normalmente éste sea tardío. Para realizarlo se requiere la sospecha clínica basada en la evolución subaguda con mala respuesta al tratamiento empírico habitual. El Mantoux y la baciloscopia de diversas muestras son un factor primordial en la orientación diagnóstica que se confirma con la anatomía patológica y el cultivo del bacilo de muestras del foco de osteomielitis.
P354MENINGITIS NEUMOCOCICA. REVISION DE LOS ULTIMOS 10 AÑOS
Patricia Esparza Paz, Nere Arostegi Kareaga, Alberto González García, Ramón M. Gaztañaga Expósito, Ángeles M. Ruiz Benito, Esperanza Pérez Ruiz y Francisco Javier Mintegui Aramburu
Hospital Donostia, San Sebastián.
Objetivo:Describir las características epidemilógicas, clínicas y secuelas de las meningitis neumocócicas ingresadas en los últimos 10 años en el Servicio de Pediatría del Hospital Donostia.
Material y métodos:Estudio retrospectivo por revisión de historias clínicas (según CIE-9-MC.320.1) desde Enero 1993-Enero 2003 de meningitis neumocócicas diagnosticadas mediante el aislamiento deStreptococcus Pneumoniae en LCR. Dos pacientes habían recibido vacunación incompleta antineumocócica. Se han analizado las complicaciones durante el ingreso y las observadas en los controles posteriores.
Resultados:Se han revisado 16 pacientes con 19 episodios de meningitis neumocócicas. Predominio de varones del 62,5 % (10 casos). La edad media fue 18 meses (rango 3 meses-5 años); representando el 56 % los < 12 meses y el 81 % los < 24 meses. En los dos únicos casos mayores de 2 años se objetivó por recidiva de nueos episodios la presencia de fístula de LCR. Las estaciones de mayor incidencia fueron invierno-primavera. Los signos y síntomas más frecuentes al ingreso fueron fiebre (100 %), afectación del estado general (80 %), rigidez de nuca o abombamiento de la fontalela anterior (80 %), vómitos (64 %) y convulsión (7 %). Presentaron hemocultivo positivo el 78 %. La distribución de los serotipos fue la siguiente: 18C (3 casos), 19F (2 casos), 14 (2 casos), 6B (2 casos), 4,11,15, 23F,24,25 y 38. Los serotipos de los vacunados no estaban incluidos en la vacuna heptavalente. El 33,3 % de las cepas fueron resistentes a la penicilina y el 7,7 % a la cefotaxima. Durante el ingreso 3 pacientes presentaron colección subdural debutando 2 de ellos con crisis convulsiva. La estancia media fue de 18,8 días (rango 11-56 días). En nuestra serie no se ha registrado ningún fallecimiento y la morbilidad ha sido: hidrocefalia que requirió shunt ventrículo-peritoneal, hipoacusia neurosensorial bilateral con implante coclear en oído izquierdo y paresia espástica de extremidades inferiores; las complicaciones representan, por tanto, el 18,7 % de todos los casos.
Conclusiones:1. Las meningitis neumocócicas son causa de elevada morbi-mortalidad en la edad pediátrica.2. Su incidencia es mayor en lactantes menores de 2 años. 3. La vacuna conjugada habría sido eficaz en el 70,5 % de los casos.
P355INFECCION POR MICOBACTERIAS NO TUBERCULOSAS EN CASUISTICA DE UN HOSPITAL TERCIARIO 1996-2001
Ana M. Sánchez Torres, Cristina Ots Ruiz, Ruth Díez Dorado, Luis Martín Jiménez, Julio García Rodríguez y Fernando del Castillo Martín
Hospital Universitario La Paz, Madrid.
Antecedentes:Las Micobacterias no tuberculosas (MNT) son un grupo de patógenos ambientales cuya forma clínica más frecuente en la edad pediátrica es la linfadenitis cervical. El manejo terapéutico es complejo,combinando tratamiento médico (resistencias farmacológicas) y quirúrgico.
Objetivo:Revisión de los casos de infección por MNT,con diagnóstico microbiológico confirmado durante un período de 5 años (1996-2001) en un hospital terciario.
Métodos:Estudio retrospectivo de pacientes con cultivos positivos o identificación mediante técnicas genéticas en distintos tipos de muestras orgánicas.
Resultados:Se analizaron 15 casos con edades entre 2 y 20 años, 2 fueron colonizaciones. De los 13 casos de infección 9 (69,2 %) presentaban linfadenitis subaguda, 1 afectación pulmonar (fibrosis quística), 1 peritonitis (IRC en diálisis peritoneal), 1 colitis y 1 infección diseminada (SIDA). En las linfadenitis subagudas el rango de edad osciló entre 1 y 5 años,el agente etiológico predominante (66,6 %) fue M. avium complex (MAC). El diagnóstico se realizó mediante histología y cultivo del material obtenido por PAAF. El tratamiento fue médico,quirúrgico o combinado. Se obtuvieron los mejores resultados con la terapia combinada (menor evolución, tasa de recidiva y queloides). En los casos de infección pulmonar, peritonitis y colitis se aisló MAC en aspirado bronquial, líquido peritoneal y biopsia intestinal; en la enfermedad diseminada se aislóM. xenopi en sangre. En estos casos se utilizaron tratamientos médicos de pauta múltiple (incluyendo Claritromicina/Azitromicina y Etambutol) con lenta evolución favorable y resolución del cuadro en los tres primeros en 1,12 y 6 meses respectivamente. En la enfermedad diseminada la paciente fallece a los 3 meses del inicio del tratamiento.
Conclusión:La infección por MNT es poco frecuente en edad pediátrica. Su manifestación más usual es la linfadenitis subaguda en niños entre 1-5 años por MAC. El tratamiento de elección es combinado,médico con un abordaje quirúrgico precoz. En pacientes inmunodeprimidos ante cuadros clínicos con un alto índice de sospecha deberá instaurarse un tratamiento médico precoz con una pauta múltiple que incluya Claritromicina o Azitromicina y Etambutol.
P356ADENOMATOSIS QUISTICA PULMONAR TIPO I. PRESENTACION DE UN CASO
Blanca Lodoso Torrecilla, Ana Gloria Andrés Andrés, Cristina Verdú Sánchez, Fernando del Castillo Martín, Francisco Hernández Oliveros, Luis Lassaleta Garbayo, M. Isabel de José Gómez, Clementina Borque Andrés, Francisco Martínez Cortés y María Jesús García de Miguel
Hospital Universitario La Paz, Madrid.
Introducción:La adenomatosis quística pulmonar es una enfermedad congénita infrecuente. Se caracteriza por una proliferación alveolar anómala,que conlleva un atrapamiento aéreo. La clínica está determinada por la compresión de tejido sano o por sobreinfección.
Caso clínico:Niña de 2 años sin antecedentes de interés remitida desde su centro de referencia con el diagnóstico de neumonía necrotizante por neumococo resistente a penicilina, con neumatocele y derrame pleural de evolución tórpida. Exploración al ingreso: REG, palidez cutaneo-mucosa, AP: hipoventilación en hemitorax derecho con crepitantes en base; moco abundante en cavum. Analítica al ingreso: Hb:7,3, Hto: 23,3 %, leucocitos: 17.100 (N70 %, L20 %, M5 %, E1 %), plaquetas: 1.574 x 10e3, VSG:134, BQ: normal. Rx tórax y TC torácico al ingreso: condensación en lóbulo superior derecho con bullas apicales y neumonía necrotizante en lóbulo inferior derecho con hidroneumotorax en el que predomina componente aéreo. Mejoría clínica y radiológica tras tratamiento antibiótico y toracoscopia con drenaje pleural.
Tras 4 meses asintomática, comienza con cuadro de dificultad respiratoria precisando de nuevo ingreso; Rx de tórax: bulla que ocupa todo el pulmón derecho. TC torácico: imagen multiquística en lóbulo superior derecho compatible con neumatocele postneumónico vs adenomatosis quística pulmonar. Se realiza lobectomía superior derecha, confirmándose por anatomía patológica el diagnóstico de adenomatosis quística tipo I.
Conclusiones: 1.La adenomatosis quística pulmonar es una enfermedad, generalmente, de diagnóstico prenatal por ecografia, o neonatal por distress respiratorio. Hay que sospecharla también, ante neumonías con neumatoceles de evolución tórpida en niños más mayores. 2. Es importante hacer un diagnóstico y tratamiento quirúrgico precoces para evitar complicaciones. 3. El interés de este caso radica en el debut tardío, permaneciendo asintomática hasta la sobreinfección de los quistes.
P357ADENOPATIA BRAQUIAL Y GRANULOMAS ESPLÉNICOS EN PACIENTE DE 11 AÑOS: ENFERMEDAD POR ARAÑAZO DE GATO
Javier Blumenfeld Olivares, Miguel Ángel Roa Francia, Marta Villares Alonso, Marta Ortega Molina, Giangaspro y Juan Arnáez Solís
Complejo Hospitalario de Móstoles, y Hospital 12 de Octubre, Madrid.
Introducción:La enfermedad por arañazo de gato; causada porBartonella Henselae, siendo los gatos el vector de transmisión; suele producir una linfadenopatía crónica, también se han descrito síndrome oculoglandular de Parinaud, encefalitis, y síntomas generales. Esta misma bacteria es responsable de manifestaciones graves como angiomatosis bacilar y pielosis hepatis en inmunodeprimidos.
Caso clínico:Paciente de once años que acude a urgencias por tumefaccíon dolorosa en brazo derecho, que abarca desde el tercio inferior del brazo hasta el codo que ha ido aumentando progresivamente en últimos seis días, acompañado de fiebre (39°) en últimas cuarenta y ocho horas. Además presenta lasiones papulo-eritematosas en dorso de la misma mano.Antecedentes: dueña de un gato. Resto sin interés. Exploración física: lesión indurada con signos inflamatorios en región interna de brazo derecho con celulitis circundante. Adenopatía axilar derecha móvil y dolorosa. Pápulas eritematosas ovaladas en el dorso del primer dedo de mano izquierda. Resto sin interés. Pruebas complementarias: hemograma, Hitachi, Inmunoglobulinas, ANAs y FR sin alteraciones. Rx de codo y de tórax sin hallazgos. Ecografía braquial: múltiples imágenes nodulares sólidas de ecogenicidad heterogénea en relación con paquete vascular medial del codo, todas estas con vascularización prominente (visto con eco-doppler).Evolución: al ingreso comienza tratamiento antibiótico empírico por celulitis circundante, se produce mejoría clínica por lo que se da de alta, reingresa a los pocos dias por aumento de la tumoración y de los signos inflamatorios. Se realiza biopsia lesional, ante la sospecha de patología tumoral y ecografía abdominal. En la biopsia se distingen granulomas necrotizantes en los ganglios biopsiados y en la ecografia se distinguen varias lesiones ocupantes de espacio en bazo. Finalmente la serologia para Bartonella Henselae (Ig M) es positiva. Iniciamos tratamiento con claritromicina oral con notable mejoría en pocos días tanto de la adenopatía braquial como de los posibles granulomas esplénicos.
Juicio clínico:Enfermedad por arañazo de gato.
Conclusiones:Los granulomas esplénicos y hepáticos pueden presentarse en el contexto de una enfermedad por arañazo de gato.
P358UTILIDAD DE LA ESTERASA LEUCOCITARIA EN EL DIAGNOSTICO DE SOSPECHA DE INFECCION DEL TRACTO URINARIO EN EL LACTANTE FEBRIL
Marta San Román Muñoz, M.ª José Lozano de la Torre, José Lorenzo Guerra Díez, Laura Buesa Casajús, Cristina Álvarez Álvarez, Hortensia Vallverdú Torón, Marco Uyaguari Quezada, Elena Pérez Gil, Vicente Madrigal Díez y Ángel Pérez Puente
Hospital Universitario Marqués de Valdecilla, Santander y Universidad de Cantabria, Santander.
Antecedentes:La infección del tracto urinario (ITU) es la causa más frecuente de infección bacteriana en el lactante con fiebre. El diagnóstico de sospecha se establece habitualmente por la presencia de leucocitos y/o gérmenes mediante el análisis de las tiras reactivas que detectan la esterasa leucocitaria y la presencia de nitritos respectivamente. La leucocitosis y la elevación de la PCR orientan hacia una pielonefritis. El diagnóstico de definitivo de ITU se basa en el resultado del cultivo de orina. La confirmación diagnóstica se obtiene mediante el urocultivo.
Objetivo:Establecer la utilidad de los distintos parámetros clínico-analíticos en el diagnóstico de sospecha de ITU.
Métodos:En 20 lactantes menores de un año con ITU por E. coli, ingresados en nuestro servicio durante cinco meses (septiembre de 2002 a enero de 2003), se analizaron al ingreso las manifestaciones clínicas y los resultados analíticos (tira reactiva y sedimento de orina recogida mediante bolsa estéril, hemograma y PCR).
Resultados:La edadmedia fue de 6 meses, siendo 10 varones y 10 mujeres. Lafiebre sin foco aparente estaba presente en todos los pacientes. Otrossíntomas asociados fueron rechazo de tomas (35 %) e irritabilidad (30 %).
Laleucocituria determinada por tira reactiva se detectó en todos los casos, incluso en lactantes con menos de 3 horas de evolución de fiebre. Losnitritos sólo fueron positivos en 7 pacientes (35 %). Se constatóleucocitosis superior a 15.000 en el 100 % de los niños con más de 24 horas de fiebre. LaPCR fue > a 30 mg/l en todos los lactantes con fiebre de más 3 horas de evolución.
Conclusiones: 1.La presencia de leucocituriadetectada por tira reactiva, asociada o no a nitraturia, en un lactante con fiebre es un método rápido y fiable para establecer el diagnóstico de sospecha de ITU en Urgencias y en Atención Primaria.2. La leucocituria detectada por dicho método es un hallazgo precoz ya que se detecta en todos los lactantes independientemente del tiempo de evolución de fiebre.3. La elevación de la PCR por encima de 30 mg/l en el lactante febril con sospecha de ITU, es también un dato precoz y constante (aún en ausencia de leucocitosis) en los casos de fiebre de más de 3 horas de evolución.
P359ARTRITIS SÉPTICA. REVISION DE LOS ULTIMOS 6 AÑOS: ¿"CLUSTER" EN EL 2002?
Beatriz Solís Gómez, Fidel Gallinas Victoriano, Mercedes Herranz Aguirre, Valentín Baranda Areta, Serafín García Mata, Luis Torroba Álvarez, Natividad Viguria Sánchez y Enrique Bernaola Iturbe
Hospital Virgen del Camino, Pamplona.
Antecedentes y objetivos:El manejo de las artritis sépticas requiere la colaboración entre pediatras y traumatólogos. El objetivo es revisar las artritis sépticas en nuestro Servicio, al observar un aumento de la incidencia el último año.
Material y métodos:Se revisaron las historias de los niños diagnosticados de artritis séptica en los últimos 6 años, entendiendo por artritis séptica aquellas con un líquido sinovial purulento, cultivo positivo, o sospecha clínica y pruebas de imagen compatibles. Se analizaron: edad, sexo, localización, historia previa, síntomas locales, estudios de imagen, analíticos y microbiológicos de sangre y de líquido sinovial, días de fiebre, duración del ingreso y del tratamiento antibiótico.
Resultados:Desde 01/01/1996 a 31/12/2002 se registraron un total de 22 casos, 12 (54,5 %) en el último año. Un total de 12 niños y 10 niñas (edad media: 4 años). Localización: 8 rodilla, 8 cadera (6 en el último año), 2 codo, 2 tobillo y 2 hombro. Del total, 9 referían en la anamnesis infecciones de vías respiratorias, 3 traumatismos, 2 lesiones cutáneas adyacentes y 1 sepsis meningocócica C. Los síntomas locales más frecuentes fueron: dolor, tumefacción y calor. La Rx y la ecografía fueron las pruebas de imagen más solicitadas. Un 33 % de los hemocultivos fueron positivos (7), aislándose: 3S. aureus, 2S. viridans y 2 bacterias Gram (). De los negativos, 5 habían recibido antibióticos previamente (35,7 %). Se realizaron 14 artrocentesis, obteniendose en 13 líquido purulento y aislándo gémenes en 6 casos (42,9 %): 2 S. aureus, 1S. viridans, 1Staphilococcus simulans, 1S. epidermidis y 1 bacteria Gram (). La PCR media fue 6,48, la VSG 64 y número de leucocitos 14.023. La duración media de la fiebre fue de 3,78 días y del ingreso 8,7 días. Se trataron con betalactámicos, asociados o no a aminoglucósidos, durante una media de 25 días, precisando 2 drenaje quirúrquico.
Conclusiones: 1.Hemos observado un aumento en la incidencia de artritis sépticas en el último año, especialmente de las localizadas en cadera.2. El gérmen más frecuente en el hemocultivo y líquido sinovial es elS. aureus.3. La rentabilidad obtenida de los estudios microbiológicos (hemocultivos y cultivos de líquido sinovial) es similar a la referida en la literatura.
NEFROLOGIA
P360 MANEJO QUIRURGICO DE LA INSUFICIENCIA RENAL EN LA HIDRONEFROSIS NEONATAL
Raquel Amo Rodríguez, M. Dolores Rodrigo Jiménez, Rosa Romero Ruiz, Carmen Clavero, Sonia Yeste, Juan Bregante, Jaime Mulet y Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca.
Introducción:La displasia renal multiquística es la causa más frecuente de enfermedad quística en el recién nacido. El riñón multiquístico se caracteriza por ausencia de función renal, pudiendo asociarse a anomalías en el riñón contralteral.
Caso clínico:RN mujer con detección prenatal de dilatación renal bilateral en la 28.ª semana de gestación, persistiendo la dilatación en los controles ecográficos prenatales. Parto vaginal espontáneo. A las 36 horas de vida presenta una Creatinina sérica (Crs) de 1,7 mg/dl y un GFR de 3,9 ml/mto/1,73 m2. En el control Ecográfico postnatal muestra dilatación de ambas unidades renales y la presencia de una dilatación ureteral derecha. Presentando ureterohidronefrosis derecha masiva e hidronefrosis izquierda asociada a insuficiencia renal se coloca nefrostomía bilateral al 3.er día de vida comprobándose que el riñón izquierdo está constituido por un riñón displásico multiquístico. Tras 48 horas de nefrostomía la función renal mejora Crs: 1,3, GFR: 16,17 ml/mto/1,73 m. A los 9 días se realiza estudio Renográfico con DTPA y DMSA objetivándose un Riñón Izquierdo afucionante y un Riñón Derecho con deformidad de contornos e incapacidad para eliminar el contraste. En la CUMS se aprecia ausencia de reflujo vesico-ureteral. A las 3 semanas de vida se retira la nefrostomía derecha y se realiza Renograma diurético en el que se objetiva parénquima renal derecho funcionante con dilatación no obstructiva. Al 2.º mes de vida presenta empeoramiento de la función renal con renograma diurético: patrón obstructivo pieloureteral. Ante el diagnóstico de obstrucción pielo-ureteral secundaria a angulación del megaureter derecho se realiza rectificación ureteral derecha y ureteroneocistostomía por vía lumbar en procedimiento único. La evolución postoperatoria fue favorable mejorando la captación y la eliminación en el renograma diurético de control.
Discusión:La displasia renal multiquística unilateral asociada a patología renal contralateral severa pueden presentar insuficiencia renal en el período neonatal que precisa de un tratamiento médico-quirúrgico agresivo para preservar la función renal. El reimplante y la rectificación ureteral en un procedimiento quirúrgico único proporciona una adecuada eliminación evitando las secuelas de los procedimientos dobles (pieloplastia y ureteroneocistostomía) y evita las prolongadas cateterizaciones con posible riesgo de infección de orina.
P361ACIDOSIS TUBULAR RENAL DISTAL
M. del Carmen Fons Estupiñá, Margarita Vázquez Olivares, Jesús Fleta Zaragozano, César Loris Pablo, Josefa López Moreno y José Luis Olivares López
Hospital Clínico Universitario Lozano Blesa, Zaragoza y Hospital Miguel Servet, Zaragoza.
Introducción y objetivos:La acidosis tubular renal (ATR) es una causa común de acidosis metabólica, causada por un defecto en la reabsorción tubular renal de bicarbonato y en la excreción urinaria de ión hidrógeno. En la ATR distal (Tipo I) la excreción urinaria de bicarbonato es escasa y no sobrepasa el 5 % de la cantidad filtrada.
Caso clínico:Niña de 7 meses de edad de origen marroquí que presenta anorexia, irritabilidad, vómitos aislados en algunas tomas y retraso de crecimiento. Exploración física: peso, 4,5 kg (< P3). Longitud, 60 cm (< P3). PC, 40 cm (< P3). Fontanela anterior 5 * 4 cm. Rosario costal. Soplo sistólico I/VI. Hipotonía generalizada. Resto sin alteraciones.
Pruebas complementarias: Hemograma: Hb, 12,3 g/dl, Ht, 38,4 %. Iones: Na+143 mEq/l. K+4,1. Cl 121. Gasometría: pH 7,25, pO2 100, pCO2 20,1, Sat O2 96,9, CO3H 9, Ex. Base 18. Acidez titulable: 0. Amonio: 23 uEqxmx 1,73 m2. Anión GAP en sangre 13. Anión GAP en orina + 40. Ph urinario mínimo alcanzado en situación de acidosis metabólica (pH 7,3; bicarbonato plasma 14): 7,7. Fracción excreción de bicarbonato: inferior a 5 ml.M.100 ml. Cociente calcio creatinina urinario 0,7. Urocultivo: Streptococo agalactiae. Ecografía renal: imágenes hiperecógenas a nivel papilar sugestivas de nefrocalcinosis. Potenciales evocados: normales. Evolución y tratamiento: tras la admisnistración de dosis variables de bicarbonato vía oral, se consiguió la reducción de la calciuria (cociente calcio/creatinina urinario de 0,17) y de la acidosis.
Comentarios:La acidosis tubular renal tiene una incidencia de 1/10.000. La forma habitual en el niño tiene un carácter primario o idiopático aunque existen casos asociados a otras enfermedades renales, sistémicas o secundarios a la administración de fármacos y tóxicos. Siempre se debe investigar la posible asociación con sordera nerviosa. El pronóstico es bueno si el diagnóstico es precoz y se previene la aparición de nefrocalcinosis con la administración de bicarbonato.
P362FENOMENO DE NUTCRACKER
Isabel Santos Ruiz, Emilia Hidalgo Barquero del Rosal, José M. García Blanco y P. Rincón Rodero
Hospital Universitario Infanta Cristina, Badajoz.
Introducción:El fenómeno de Nutcracker o "cascanueces" es una rara anomalía vascular, caracterizada por disminución del ángulo formado entre aorta y arteria mesentérica superior, comprimiendo entre ambas la vena renal izqda (VRI), lo que ocasiona a veces sintomatología diversa (proteinuria, hematuria,...), como consecuencia del desarrollo de hipertensión en la VRI.
Caso clínico:Niña de 11 años, sin antecedentes personales de interés, que ingresa por hematuria macroscópica de 2 semanas de evolución, sin otros síntomas. No antecedente de traumatismo, ni de proceso infeccioso previo o concomitante. Como antecedentes familiares destaca la presencia de varios miembros con historia de cólicos nefríticos y una hermana con duplicidad pieloureteral. La exploración física es normal salvo hábito asténico e hiperlordosis lumbar. Entre las pruebas complementarias solo destaca la presencia de hematuria con leve proteinuria, siendo el estudio inmunológico, metabolismo fosfocálcico y el estudio de función renal con calciuria, citraturia, oxaluria y aminoaciduria normales, así como las pruebas de imagen (eco abdominal, pielografía IV y cistouretrografía miccional). Tras el alta, persisten los episodios de hematuria macroscópica intermitente, más frecuentes durante horario escolar, por lo que se completa estudio con eco-doppler, que muestra flujo VRI en límite superior y angio-TAC abdominal, observánsose escasa distancia entre arteria mesentérica superior y VRI, que hace que ésta se vea comprimida a dicho nivel, hallazgo que es confirmado con angio-RMN. Estos datos sugieren una compresión intermitente de la VRI, en relación con hiperlordosis lumbar constitucional, exacerbada durante el horario escolar (sedestación). Se remite a Traumatología y Rehabilitación para corregir su hiperlordosis (con corsé y ejercicios para reforzar musculatura lumbar). Actualmente permanece asintomática.
Discusión:Este fenómeno debe incluirse en el diagnóstico diferencial de hematurias y proteinurias no filiadas en el que todos los estudios de función renal y urológicos son normales. En su diagnóstico es de gran utilidad el angio-TAC helicoidal, que permite la reconstrucción de imágenes tridimensionales. El tratamiento debe ser precoz, para evitar posibles complicaciones (TVR). Existen diversos métodos quirurgicos, aunque a veces puede ser suficiente con un tratamiento rehabilitador, como en nuestro caso.
P363HIPERTENSION RENOVASCULAR
Nuria Cortés, Eva M. Pacheco Navas, Ramón Vilalta Casas, Manuel Matas, Ángel Vila López y Lluis M. Callis Bracons
Hospital Vall D'Hebrón, Barcelona.
Objetivos:Valorar la respuesta al tratamiento quirúrgico en pacientes afectos de hipertensión arterial (HTA) renovascular en la edad pediátrica.
Métodos:Revisión de una muestra de 7 pacientes afectos de HTA renovascular no controlada mediante tratamiento médico que precisaron intervención quirúrgica entre los años 1997 y 2002.
Resultados:De los 7 pacientes 5 eran varones y 2 hembras. La edad del diagnóstico era en 3 casos inferior a los 4 años de edad y en el resto mayor de 10 años. El signo guía que motivó el estudio fue en todos ellos HTA confirmada mediante holter de 24 horas, en 5 de ellos detectada casualmente en control rutinario y en 2 de ellos acompañada de clínica de cefalea y dolor torácico. Cuatro pacientes tenían en el momento del diagnóstico afectación severa de la función renal. Sólo uno de ellos presentaba miocardiopatía hipertensiva. A todos ellos se les practicó Ecografía Doppler abdominal, renograma isotópico y Angio-TAC que confirmó el diagnóstico de estenosis de la arteria renal, en 4 de ellos bilateral y en los otros 3 unilateral (del lado izquierdo). En todos ellos se inició tratamiento médico combinado con tres grupos diferentes de fármacos antihipertensivos sin mejoría de las cifras tensionales. En todos ellos se practicó cirugía consistente en By-pass aorto-renal con vena safena unilateral o bilateral. Sólo en tres de ellos se practicó previamente a la cirugía angioplastia percutánea con balón, sin respuesta clínica. Tras la cirugía todos mantienen cifras de tensión arterial normales para su edad, tres de ellos no precisan ningún fármaco antihipertensivo después del primer año de la cirugía. Los otros cuatro todavía precisan uno o dos fármacos antihipertensivos a dosis bajas. En todos ellos se ha normalizado el filtrado glomerular y ha disminuido la proteinuria, ha desaparecido la clínica y en el caso con miocardiopatía hipertensiva, la ecocardiografía de control es normal.
Conclusiones:La HTA de origen renovascular representa un 5-10 % de los casos de HTA en la infancia. El tratamiento precoz puede evitar las secuelas graves de la HTA y la anulación funcional del riñón afecto. En nuestra revisión el 100 % de los pacientes presenta una buena respuesta al tratamiento quirúrgico, por lo que concluimos que en nuestra experiencia la cirugía mediante by-pass es una opción terapéutica de primera elección en pacientes con HTA renovascular.
P364SEGUIMIENTO MEDIANTE GAMMAGRAFIA RENAL TC99-DMSA EN PACIENTES CON REFLUJO VESICOURETERAL TRATADOS ENDOSCOPICAMENTE MEDIANTE INYECCION INTRAMURAL DE TEFLON
David Peláez Mata, José Antonio Álvarez Zapico, Rafael Pardo de la Vega, Serafín Málaga Guerrero, Julián Rodríguez Suárez, M. Luz Domínguez y Carmen Roiz Gaztelu
Hospital Central de Asturias, Oviedo.
El objetivo del tratamiento endoscópico (TE) del reflujo vésico-ureteral (RVU) es evitar nuevas cicatrices renales y la progresión de las ya existentes. No hemos hallado estudios de seguimiento en pacientes con RVU sometidos a TE e inyección de Teflon®mediante la gammagrafía renal con Tc99-DMSA que, por su mayor sensibilidad y especificidad, constituye el método de elección para el seguimiento de estos pacientes.
Objetivo:Detectar nuevas cicatrices renales y confirmar la progresión de las lesiones renales parenquimatosas existentes en pacientes pediátricos con RVU tratados endoscópicamente mediante la inyección intramural de Teflon®.
Pacientes y métodos:De una serie de 165 enfermos (128 mujeres) con una edad media de 83 meses (rango: 7-183 meses), todos ellos portadores de RVU intervenido mediante inyección subureteral de Teflon® en un período de 10 años (1991 y 2000), se ha estudiado la evolución de 82 pacientes (134 unidades renales) a los que se les había realizado gammagrafía renal Tc-99 DMSA previa al tratamiento. Pre y post-tratamiento, se analizó el número y tamaño de las cicatrices de cada unidad refluyente, así como la variación en la función renal diferencial, mediante una nueva gammagrafía renal Tc-99 DMSA. El intervalo medio entre ambas exploraciones fue de 3,89 años (rango de 1-10 años).
Resultados:Se constató mejoría de las lesiones (disminución o atenuación del número de cicatrices) en 21 unidades renales (15,6 %), empeoramiento (aumento o incremento de las cicatrices) en 8 (5,9 %) y ningún cambio en los 105 restantes (78,3 %). Tres unidades renales desarrollaron nuevas cicatrices. Se constató empeoramiento de la función renal diferencial en 7 casos (25 %), aunque en ninguno de ellos superaba el 6 %.
Conclusión:La incidencia de nuevas cicatrices renales tras el TE del RVU con inyección intramural de Teflon® es baja, lo que confirma que la progresión de la nefropatía por reflujo en pacientes tratados endoscopicamente de su RVU es satisfactoria.
P365HIPERFOSFATASEMIA TRANSITORIA IDIOPATICA DE LA INFANCIA DE LARGA DURACION
Purificación Cárdenas Guerrero, M. Rosario Benavides Román, Carmen Fuentes Gutiérrez, Montserrat de Felipe Jiménez-Casquet, José Antonio Hurtado Suazo y David Barajas de Frutos
Hospital Virgen de las Nieves, Granada.
La Hiperfosfatasemia Transitoria Idiopática de la Infancia es una entidad en la que se observan marcadas elevaciones séricas de fosfatasa alcalina de carácter transitorio tanto en niños sanos como en portadores de clínica inespecífica. En la edad pediátrica existe períodos de aumento de la producción enzimática fisiológicos, durante los primeros meses de vida y en la adolescencia. Presentamos un caso de persistencia de cifras séricas altas de fosfatasa alcalina durante 2 años, que pensamos puede ser interesante ya que series de casos de aumento transitorio de fosfatasa alcalina total sérica, presentan duración de 1 a 6 meses.
Niña de 2 y años y medio que es remitida por su pediatra por presentar en controles analíticos repetidos, cifras altas de fosfatasa alcalina sérica (FA), que oscilan desde 930 hasta 2.296 U/l (normal de 87-462 U/l). A la exploración destaca un perímetro cefálico aumentado (en control de niño sano ha estado siempre en torno al percentil 90), con un cierre tardio de la fontanela (26 meses). Como antecedente familiar se refiere padre macrocefalo en la infancia. Se procede al estudio mediante pruebas complementarias: FA en padres y hermana dentro de la normalidad; Función renal simple, enzimas hepáticas, metabolismo fosfocalcio, TSH e hidroxiprolina en orina dentro de la normalidad; Edad osea a los 3 años corresponde a 2 años y medio; Radiografía de craneo normal; FA sérica total de 983 U/l con 64,8 % isoenzima osea, 35,2 % isoenzima hepática. Se realiza seguimiento clínico y analítico, no apareciendo alteración alguna salvo las cifras de FA sérica elevada. En el último control, después de 2 años la FA sérica es de 332 U/l.
Ante una cifra elevada de FA sérica se plantea un diagnóstico diferencial que comprende: hiperfosfatasemia asintomática no familiar persistente, diagnóstico en el que se pensó en principio, ante un tiempo tan prolongado de FA sérica alta, siendo en otras series publicadas el tiempo máximo descrito de 6 meses; Hiperfosfatasemia familiar benigna, en la que los familiares presentan cifras altas pero no existe clínica; Hiperfosfatasemia con osteoectasia con anomalias esqueléticas; Hiperfosfatasemia hereditaria con aumento de recambio óseo. Nuestro caso es una forma de Hiperfosfatasemia Transitoria Benigna de tiempo más prolongado.
P366 ENFERMEDAD RENAL POLIQUISTICA COMO FORMA DE PRESENTACION DE ESCLEROSIS TUBEROSA EN 2 HERMANOS GEMELOS
José Alberto Macías Pingarrón, Emilia Hidalgo Barquero del Rosal, José M. García Blanco y Juan José Cardesa García
Hospital Universitario Infanta Cristina, Badajoz.
Antecedentes personales:Embarazo gemelar de fecundación In Vitro. Ecografía prenatal detecta nefromegalia bilateral en el 2.º gemelar.
Primer caso clínico:Lactante, con antecedente de hermano gemelar diagnosticado de Enfermedad Poliquística Renal, en estudio familiar nefrológico. Clínicamente se encuentra asintomático. La ecografía abdominal muestra riñones de tamaño y ecogenicidad normales, apreciándose en riñón derecho quiste corticomédular de 3,2 cm y en riñón izquierdo quiste de 0,8 cm en zona interpolar, con microquistes glomerulares y tubulares focales, datos sugerentes de Enfermedad renal Poliquística autonómica dominante (ADPKD). En ecografía cardiaca múltiples nódulos en paredes ventriculares y septo de unos 2-3 cm compatibles con rabdomiomas cardiacos. En TAC craneal: calcificación puntiforme a nivel de la porción retrolenticular de la cápsula interna, datos compatibles con una Esclerosis Tuberosa (ET) con afectación neurológica, cardiaca y renal.
Segundo caso clínico:Lactante que ingresa en UCIP procedente del hospital de origen por insuficiencia respiratoria por precisar cuidados intensivos. Diagnosticado de Enfermedad renal poliquística, probablemente recesiva. Al ingreso destacaba déficit ponderal, abdomen globuloso e insuficiencia respiratoria con TA 164/115. En la Rx de tórax los pulmones impresionan de hipoplásicos con imágenes de condensaciones bilaterales. En ecografía abdominal nefromegalia bilateral (9,7 cm) con quistes bilaterales. Se diagnostica de bronconeumonía bilateral, Enf. Poliquística renal e hipertensión arterial controlada con enalapril. Con motivo del diagnóstico en el hermano gemelo de ET es estudiado mostrando afectación dermatológica y neurológica (TAC craneal con signos radiológicos sugerentes de ET) sin afectación cardiológico (ecocardiograma: hipertrofia del ventrículo izquierdo secundario a la HTA), compatible con ET con afectación renal.
Comentarios:La presentación clínica de nuestro primer caso como una ARPKD y la ausencia de angiomiolipomas en los 2 hermanos, no nos alertó ante la posibilidad de un cuadro de ET. El ADPKD tipo 1 y TSC tipo 2 constituye un síndrome de genes contiguos (16p13,3). La deleción de parte del gen ADPKD1 en casos de TSC2 explica la patología renal quística de estos pacientes.
P367SINDROME NEFROTICO CONGÉNITO
José Alberto Macías Pingarrón, Eva Fernández Calderón, Emilia Hidalgo Barquero del Rosal, José M. García Blanco y Juan José Cardesa García
Hospital Universitario Infanta Cristina, Badajoz.
Introducción:Síndrome nefrótico (SN) de aparición en el nacimiento o en el primer trimestre de vida, siendo el síndrome nefrótico congénito (SNC) más frecuente y mejor conocido. De etiología idiopática y con herencia autosómica recesiva (19q13.1).
Caso clínico:Ingreso en las primeras 24 horas por sospecha de cuadro séptico (distensión abdominal y vómitos hemáticos). En ecografía imágenes compatibles con litiasis en vesícula biliar. En analítica se detecta hipoproteinemia (2,7 g/dl), hipoalbuminemia (0,5 g/dl), proteinuria masiva, elevación de colesterol (417 mg/dl), elevación de triglicéridos y cifras de GGT elevadas. A la exploración edemas generalizados, con palidez de piel, abdomen globuloso, hígado a 2 cm de reborde costal y nefromegalia. Diástasis de suturas craneales; fontanelas amplias. TA 95/55.
Pruebas complementarias:Urea y creatinina plasmáticas normales. Alfa-fetoproteína plasmática elevada. Complemento, ANA y anticoagulante lúpico: negativos. Serología a virus (CMV, rubéola, VEB, hepatitis B y C), toxoplasma y lues: negativos. VIH: negativo. Filtrado glomerular normal. TSH: elevada con disminución de T4. Estudio genético: se descarta mutaciones más frecuentes del SNC Finlandés. Ecografía al nacimiento: vesícula biliar contraída con litiasis en su interior. Riñones grandes hiperecogénicos. Controles: hepatomegalia con hiperecogenicidad difusa (esteatosis). Renomegalia con hiperecogenicidad sin distinción cortico-medular. Biopsia renal compatible con riñón microquístico. Hipertrofia de mesangio con esclerosis del penacho y fibrosis periglomerular
Evolución y comentarios:Se mantuvo inicialmente con perfusiones de albúmina, diuréticos, carnitina, eritropoyetina, magnesio, calcio, vitamina D, antiagregantes plaquetarios e IECA así como dieta hiperproteíca y pobre en lípidos. Eutiroidea por aportes de tiroxina. Procesos intercurrentes: sepsis a E. coli, trombo en vena innominada izquierda y colecistitis. A los 6 meses se inició tratamiento con indometacina para inducir fallo renal. En la actualidad mantiene buen estado general, mínimos edemas y lenta ganancia ponderal Se suele asociar a un pronóstico desfavorable como consecuencia de la resistencia a todo tipo de tratamiento, dificultades en la nutrición y mantenimiento de una homeostasis adecuada, a una predisposición a procesos infecciosos y a la necesidad precoz de tratamiento substitutivo y trasplante. El tratamiento va encaminado a mantener al paciente en condiciones adecuadas para el trasplante renal
P368AFECTACION RENAL EN SINDROME DE PEARSON
Isabel Santos Ruiz, José M. García Blanco, Emilia Hidalgo Barquero del Rosal, J.M. Vagace Valero, Y. Campos y Elena Martín Hernández
Hospital Materno-Infantil, Badajoz, Hospital Universitario Infanta Cristina, Badajoz y Hospital 12 de Octubre, Madrid.
Introducción:El síndrome de Pearson es una citopatía mitocondrial, debida a una delección del DNAmt habitualmente esporádica, que se caracteriza por afectación medular y del páncreas exocrino.
Caso clínico:Paciente sin antecedentes de interés que a los 4 meses de edad comienza a ser estudiado por anemia severa refractaria a tratamiento y pancitopenia. En el estudio medular se observa sideroacrestia y vacuolización de precursores mieloides y eritroides. Con la sospecha de sdre de Pearson se realiza estudio de DNAmt en linfocitos, que pone de manifiesto una delección (6.900 pares de bases) del 75 % de DNAmt, confirmándose el diagnóstico. Durante los meses siguientes permanece asintomático desde el punto de vista hematológico, pero se aprecia retraso ponderoestatural y episodios de vómitos frecuentes siendo diagnosticado de RGE, además de cierta tendencia a hiperlactacidemia. A los 2 años y medio de edad, coincidiendo con cuadro de vómitos presenta descompensación metabólica con acidosis metabólica severa e hiponatremia grave sintomática (convulsiones) que requiere ingreso en UCIP. Se observa además, disfunción tubular compleja en forma de sdre de Fanconi (con ausencia de glucosuria). Este cuadro vuelve a repetirse meses después coincidiendo con un nuevo episodio de vómitos secundarios a proceso catarral. La función renal se recupera tras los episodios agudos quedando como secuela cierta tendencia a la acidosis metabólica por pérdida de bicarbonato e hipermagnesuria que requieren suplementos. Actualmente el paciente no presenta otras manifestaciones salvo leve fibrosis difusa del páncreas sin traducción clínica hasta el momento.
Discusión:Este síndrome suele ser de inicio precoz y cursa con elevada mortalidad en los primeros años de vida. Los pacientes que sobreviven pueden desarrollar otras manifestaciones (endocrinas, digestivas, cardiacas, neurológicas, musculares,...) constituyendo un proceso multiorgánico cuya variabilidad clínica está en relación con la heterogeneidad del DNA mitocondrial en los distintos tejidos. Dentro de estas manifestaciones es frecuente la afectación renal en forma de tubulopatías que dan lugar a trastornos del equilibrio ácido-base o a un sdre de Fanconi.
P369ESQUISTOSOMIASIS VESICAL. UN DIAGNOSTICO A CONSIDERAR EN INMIGRANTES
Susana Fuertes Blas, Esther Trillo Bris, M. Dolores Rodrigo Jiménez y Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca.
Introducción:Debido a viajes intercontinentales y a la inmigración procedente de zonas endémicas, nos encontramos con enfermedades infecciosas hasta ahora poco frecuentes en nuestro medio, como la esquistosomiasis vesical. Es una parasitosis producida por Schistosoma haematobium adquirida al realizar actividades cotidianas en relación con el agua dulce contaminada. La cercaria penetra a través de la piel y realiza su ciclo hasta llegar a su hábitat definitivo (vejiga, próstata y plexo uterino), siendo por último expulsados.
Métodos:Realizamos una revisión de historias clínicas de pacientes con el diagnóstico de esquistosomiasis vesical, en un período de doce años (1990-2002). Los casos diagnosticados los encontramos en los últimos cinco años.
Revisión:Se exponen cuatro casos de pacientes entre 5 y 10 años de edad diagnosticados de esquistosomiasis vesical. Originarios de República de Guinea y Nigeria, de los cuales tres presentan paludismo crónico. Dos consultan por hematuria macroscópica y dolor abdominal de tipo cólico intermitente de un año de evolución, asociando uno de ellos prurito generalizado. Los dos restantes son hallazgos casuales durante su ingreso por paludismo tras objetivarse microhematuria y hacer un screening de parásitos. La ecografía vesical es normal en dos de los pacientes; otro presenta engrosamiento nodular en la pared posteroinferior izquierda de la vejiga de 0,5 por 1,4 cm de grosor; y en el último se detecta aumento en la densidad de la orina con detritus y engrosamiento parietal en la base y pared derecha vesical de 0,5-2 cm de espesor. Hemograma normal, salvo un paciente con eosinofilia (16 % E). En todos se detectan huevos de S. haematobium en orina y se tratan con praziquantel en dosis única de 40 mg/kg.
Conclusiones:1. En los últimos años ha aumentado la incidencia de enfermedades que no son endémicas en nuestro país, debido al aumento de la inmigración procedente de estas zonas.2. La esquistosomiasis ha dejado de ser una enfermedad extraordinaria en nuestro medio. El signo de presentación más frecuente es la hematuria, por lo que debe entrar dentro del diagnóstico diferencial de la misma en pacientes que procedan o hayan viajado a zonas endémicas.3. Nos planteamos la posibilidad de incluir la realización de sedimento urinario, como cribaje, en personas procedentes de dichas zonas, tanto inmigrantes como viajeros expuestos a factores de riesgo para esta enfermedad.
P370SINDROME DE GALLOWAY-MOWAT
Isabel Carvalho, Graça Ferreira, Eduarda Marques y Carla Barbêdo
Centro Hospitalar de Vila Nova de Gaia, Portugal y Centro de Genética Clínica, Porto (Portugal).
Introdução:O Síndrome de Galloway-Mowat (McKusik*251300) corresponde a uma doença rara, autossómica recessiva, caracterizada por anomalias do sistema nervoso central com defeitos da migração neuronal, síndrome nefrótico congénito e, em alguns casos, hérnia de hiato. O defeito genético desta doença é ainda desconhecido. O prognóstico é mau devido à rápida progressão para insuficiência renal terminal.
Este caso clínico trata-se provavelmente da 1.ª descrição deste síndrome na Península Ibérica.
Caso clínico:Os autores descrevem o caso clínico de um lactente do sexo feminino, produto de uma gravidez vigiada e com diagnóstico de microcefalia às 32 S.G., pais não consanguíneos, que aos 3 meses de idade recorreu ao SU do nosso hospital por vómitos alimentares, recusa alimentar parcial e distensão abdominal. Ao exame objectivo apresentava-se hemodinamicamente estável, hemangioma do lábio superior, microcefalia, micrognatia e anasarca.
Analiticamente, apresentava: hemoglobina: 9,8g/dl, leucócitos 24490/mm3, albumina: 1,0g/dl, proteínas totais: 2,6g/dl e triglicerídeos: 673mg/dl, proteinúria de 24h: 749 mg/m2/h e ecografia abdominal com ascite de grande volume e aumento da ecogenicidade renal com diminuição da diferenciação cortico-medular; compatível com S. Nefrótico Congénito.
O cariótipo, estudo metabólico, biopsia muscular e de pele foram normais.
Iniciou tratamento de suporte com albumina (0,8 g/kg/dia), indometacina (1 mg/kg/dia) e captopril (1 mg/kg/dia), vindo, no entanto, a falecer ao 12.º dia de internamento.
O exame anatomopatológico revelou anomalias da migração neuronal e esclerose mesangial difusa.
Conclusão:Os achados clínicos e anatomopatológicos encontrados: dismorfia craniofacial, síndrome nefrótico congénito, alterações da migração neuronal e esclerose mesangial difusa (embora não esteja descrito um padrão histológico renal típico nestes casos) apontam para o Síndrome de Galloway-Mowat.
P371HIPERTENSION ARTERIAL
Sonia Fernández Fernández, José María de Cea Crespo, Carlos Vela Valldecabres y Amparo González Vergaz
Hospital Severo Ochoa, Leganés.
Introducción:Cada vez cobra más importancia el estudio de la hipertensión arterial infantil, por la patología en sí y por la posible repercusión en la vida adulta. Es importante para el diagnóstico una medición correcta, así como una interpretación adecuada de las gráficas de presión para cada edad, sexo y talla.
Objetivos:Descripción de diez casos de HTA (hipertensión arterial) derivados a la consulta de nefrología infantil. Diagnóstico, tratamiento y evolución.
Material y métodos:Revisión retrospectiva de los pacientes enviados a la consulta de nefrología infantil durante los últimos mses, para valoración de HTA.
Resultados:Revisamos retrospectivamente diez pacientes de edades comprendidas entre 6 y 14 años, 6 hombres y 7 mujeres. Todos habían sido derivados por presentar cifras altas de presión arterial en sus revisiones rutinarias. En primer lugar se realiza una cuidadosa anamnesis, donde se recoge la presencia de antecedentes familiares de HTA en 4 de los 10 pacientes; en cuanto a los antecedentes personales (incluido período perinatal), ninguno presentaba otras patologías acompañantes excepto una paciente con síndrome de Turner.
De los 10 pacientes, 6 presentaban un P > 90 de peso y talla; Solo 3 presentaban manifestaciones clínicas (cefalea, mareos...). Como pruebas diagnósticas, a todos los pacientes se les realizó electrocardiograma, radiografía de tórax, estudio de función renal y bioquímica sanguínea, siendo normales en todos los casos.
Al realizar las mediciones en nuestra consulta, no objetivamos HTA en tres pacientes (en varias visitas). Entre los restantes, 2 de ellos mejoraron con adelgazamiento y mejora del estilo de vida; uno fue clasificado como HTA de bata blanca y los cuatro restantes (todos con HTA esencial) recibieron tratamiento médico (en estos últimos el estudio se completó con otras pruebas diagnósticas).
Dos de los cuatro pacientes fueron tratados con Losartan, uno con Enalapril y otro después de ser tratado primero con Losartan y después con Enalapril, mejoró con un antagonista del calcio.
Conclusión:Es fundamental una buena anamnesis así como exploración física, para diferenciar la hipertensión esencial de la secundaria; la correcta medición e interpretación de las gráficas evitará considerar cifras erróneamente altas.
P372ESTUDIO RETROSPECTIVO DEL SEGUIMIENTO DE 63 PACIENTES CON PIELONEFRITIS
Sonia Fernández Fernández, Ana Siles Sánchez-Manjavacas, Cristina Calvo Rey, José María de Cea Crespo, Rafael Díaz Delgado y Enriqueta Román
Hospital Severo Ochoa, Leganés.
Antecedentes:Las pielonefritis en los lactantes pueden producir lesión renal secundaria, por ello un seguimiento correcto después del episodio agudo será importante.
Objetivos:Analizar el seguimiento de los pacientes menores de dos años diagnosticados de pielonefritis. Objetivar la existencia de lesión renal posterior, valorando la posible relación con otros parámetros clínicos y radiológicos.
Material y métodos:Se revisan de manera retrospectiva sesenta y tres casos de pielonefritis en pacientes menores de dos años a los que se les realiza ecografía renal en el período agudo, cistografía seis semanas posteriores al tratamiento y DMSA en los seis o nueve meses posteriores.
Resultados:Revisamos sesenta y tres pacientes con edades comprendidas entre uno y veinte meses diagnosticados de pielonefritis. Se aisló en el 90,5 % de los casos E. coli como germen causal, siendo el 9,5 % restante debida a otros gérmenes. Al ingreso presentaron una temperatura de 38,98 ± 1,6 °C, con una leucocitosis media de 16492,90/mm3 y PCR media de 68,63. El tiempo medio de fiebre al diagnóstico fue de 34,83 horas.
Se realizó ecografía renal que resultó normal en el 65 % de los casos y patológica en el 35 % restante; cistografía renal que resultó normal en el 48,4 %de los casos y patológica en el 51,6 % restante y DMSA que resultó normal en un 82,54 % y patológico en un 17,46 %. Del total de los pacientes con reflujo, el 68,75 % presentó DMSA normal.
Entre los once pacientes con DMSA patológico, cinco presentaron un reflujo de grado IV, cinco grado III o menor y en un único paciente la cistografía fue normal. El tiempo medio de fiebre hasta el tratamiento en los pacientes con DMSA patológico fue de 14,27 horas y de 39,1 horas en el resto.
Comentarios:En nuestra muestra encontramos un bajo número de pacientes con cicatriz renal, sin evidenciarse una clara relación con el grado de reflujo ni con las horas de demora hasta el inicio del tratamiento.
NEONATOLOGIA
P373PARADA CARDIORESPIRATORIA TRAS SCREENING DE RETINOPATIA DEL PREMATURO
Fco. Javier Aguirre Rodríguez, Raúl Sánchez Pérez, M. del Rosario Jiménez Liria, José Luis Gómez Llorente, Pedro Cortés Mora, Moisés Leyva Carmona, M. Ángeles Llamas Guisado, José Arcos Martínez y Juan López Muñoz
Hospital Torrecárdenas del SAS, Almería.
La retinopatía de la prematuridad (ROP) es una enfermedad retiniana vasoproliferativa multifactorial, cuya incidencia aumenta inversamente proporcional a la edad gestacional. Aproximadamente el 65 % de los neonatos < 1.250 g y el 80 % de los menores de 1.000 g desarrollan algún grado de ROP.
Presentamos los casos de 2 prematuros afectos de Displasia Broncopulmonar (DBP) que presentaron pausas de apnea y parada cardiorrespiratoria tras la realización de screnning oftalmológico de la Retinopatía del Prematuro (ROP):
Lactante de 4 meses de vida, con antecedentes de gran inmadurez 25SG y PRN 950 g afecta de DBP en tratamiento con Budesonida,Salbutamol inhalado y oxigenoterapia continua en gafas nasales. Se inicia tratamiento tópico con 1 gota de Fenilefrina 10 % y otra de Cyclopentolato 1 % en cada ojo, se repite la dosis a los 15 minutos y se realiza el examen retiniano. A los 20 minutos de terminado el estudio la paciente comienza con pausas de apnea, desaturación y bradicardia severa que finaliza en parada cardiaca, precisando reanimación cardipopulmonar y ventilación mecánica, a las 24 horas de su estancia en la Unidad de Cuidados Intensivos Pediátricos se encuentra asintomática y sin la aparición de nuevos episodios apneicos.
Lactante de 3 meses de vida, antecedentes de prematuridad EG 29semanas, PRN 1.110 g, afecto de DBP y en tratamiento con Budesonida y Salbutamol inhalado, oxigenoterapia continua en gafas nasales y vitamina D3. Se inicia preparación para screnning ROP según protocolo de nuestro centro. Aproximadamente a la hora de haber finalizado la exploración, aparece episodio de desaturación y bradicardia severa que precisa ventilación con bolsa autohinchable y maniobras de RCP, el paciente es trasladado a la UCIP donde permanece 24 horas sin nuevos episodios.
Esta técnica exploratoria no está exenta de efectos secundarios, predominantemente leves, aunque la posibilidad de aparición de efectos indeseables graves existe, y como mostramos en nuestros casos, hasta una hora después de la realización del estudio; por ello creemos necesario una monitorización estricta tanto durante la exploración como también posterior a la misma, en un servicio con medios y personal entrenado para solventar los efectos adversos que puedan presentarse
P374POTENCIALES EVOCADOS ACUSTICOS TRONCOENCEFALICOS EN NEONATOS CON RIESGO DE HIPOACUSIA
Elisa Cueto Calvo, Leonor Guardia Nieto, Miguel Sarrión Cano, M. Ángeles García Jiménez, Itziar Martínez Badas y Gregorio Garde Saiz
Hospital Virgen de La Luz, Cuenca.
Antecedentes:La detección precoz de los problemas de audición en niños con riesgo de hipoacusia es de vital importancia en la adquisición del lenguaje y en el desarrollo psíquico del niño. Los PEATs y la audiometría mediante PEATs son métodos objetivos de evaluación del umbral auditivo que permiten explorar la vía auditiva hasta el troncoencéfalo.
Objetivo:Determinar la frecuencia de alteraciones de la vía auditiva en neonatos con factores de riesgo de hipoacusia.
Métodos:Se realizó un estudio prospectivo observacional entre el 1 de enero de 2000 y el 31 de octubre de 2002 en 30 recién nacidos de nuestro hospital que presentaban uno o más factores de riesgo establecidos por el Joint Commitee on Infant Hearing (1994). A todos ellos se les realizaron PEATs y audiometría mediante PEATs.
Resultados:Características de la población: de los 30 RN, 19 varones y 11 mujeres, la mayoría, 24, eran RN a término y 6 prematuros, con una edad gestacional media de 38,47 ± 3,26 semanas. La edad media de los pacientes cuando se realizó el estudio fue de 3,97 ± 2,19 meses. Todos los niños seguidos han tenido un desarrollo psicomotor normal. Factores de riesgo: el uso de fármacos ototóxicos es el factor de riesgo más frecuente en la población a estudio, estando presente en el 80 % de los casos. El 20 % de los casos habían presentado asfixia, definida como Apgar menor de 5 al minuto o menor o igual a 6 a los 5 minutos. En el 109 % de los casos, el factor de riesgo principal presente fue una historia familiar de sordera, y sólo uno de nuestros pacientes tenia un peso inferior a 1500 g. 4 niños presentaron más de un factor. Audiometría: la prevalencia de hipoacusia en nuestra población es del 26,7 %, siendo igual de frecuente la afectación uni y bilateral. Todas ellas fueron conductivas y en el 80 % de los casos leve. PEATs: sólo fueron patológicos en 4 casos, con asimetría en uno de ellos.
Conclusiones:La prevalencia de alteraciones auditivas en nuestra población de riesgo es del 26,7 %, porcentaje muy importante que justifica un programa de screening. La audiometría mediante PEATs en los primeros meses de vida es un buen método para esta población.
P375 SECUELAS OFTALMICAS DE RETINOPATIA DE LA PREMATURIDAD
Carmen Medina, F.J. Márquez Báez, Esther Ocete Hita, R. Piñar, F.J. Bermúdez, B. Carreras, M.C. Ramírez, Eduardo Narbona López y Ángela Ruiz Extremera
Hospital Clínico Universitario San Cecilio, Granada.
Antecedentes y objetivos:La retinopatía de la prematuridad (ROP) conlleva el riesgo de padecer numerosas patologías oftalmológicas identificables como secuelas de la ROP. El objetivo fue conocer las secuelas encontradas al año de vida en pacientes que fueron diagnosticados de ROP en la etapa neonatal.
Métodos:Estudio prospectivo de 251 recién nacidos prematuros con edad gestacional (EG) ≤ 32 semanas o prematuros de > 32 semanas de EG sometidos a oxigenoterapia o con curso clínico complicado y que fueron dados de alta del protocolo de ROP en el período neonatal. Al año de vida fueron explorados de: agudeza visual, motilidad extrínseca, estado refractivo, funduscopia, entre otros parámetros.
Resultados:Baja agudeza visual (< 0,5) se encontró en 10/251 niños (4 %), 4 (1,6 %) con amaurosis pero sólo 1 de ellos secundario a ROP. Motilidad ocular extrínseca patológica en 24/238 niños (10 %). Funduscopia patológica en 8/238 (3,36 %). Defectos refractivos en 27/238 niños (11,34 %), con mayor frecuencia de hipermetropía (4,62 %). No encontramos relación entre el padecimiento o no de retinopatía en cualquier estadío y alteraciones de agudeza visual, motilidad ocular, funduscopia o defectos refractivos, pero sí existen diferencias en todos estos parámetros considerando el estadío I frente a los superiores (p < 0,01). Los niños con estadios de ROP en el período neonatal más avanzada, fueron los nacidos con menor peso y edad gestacional tabla 1.

Conclusiones:Las secuelas oftalmológicas son más frecuentes y graves en prematuros con menor edad gestacional y estadíos de ROP más avanzados, necesitando esos niños un seguimiento más exhaustivo especialmente a partir del estadío II. El pronóstico visual con un correcto seguimiento oftalmológico parece esperanzador.
P376FLUTTER AURICULAR DE PRESENTACION NEONATAL
Jorge Rodríguez Ozcoidi, Diana Martínez Cirauqui, María Garatea Rodríguez, Carlos Romero Ibarra, Beatriz Solís Gómez, Fidel Gallinas Victoriano y Carlos Javier Gurbindo Arana
Hospital Virgen del Camino, Pamplona.
Introducción:La monitorización mediante registro electrocardiográfico tipo Holter ha permitido reconocer numerosas arritmias y trastornos de la conducción en el período neonatal. Estos trastornos resultan relativamente frecuentes especialmente en los pretérminos siendo las de mayor incidencia las contracciones auriculares prematuras, los ritmos de la unión, las pausas sinusales y las contracciones ventriculares prematuras. Presentamos a continuación un caso de arritmia tipo flutter auricular de excepcional presentación neonatal.
Caso clínico:Se trata de una gestante primípara de 29 años de edad sana, sin antecedentes personales ni familiares de interés. El embarazo había cursado con normalidad, sin enfermedades intercurrentes y no refería consumo de drogas o tóxicos. Las serologías, ecografías y exploraciones de control fueron normales. En la semana 36 se efectúa un registro cardiotocográfico donde se objetiva una taquicardia fetal por lo que se indica cesárea urgente.
Al nacimiento presenta buen estado general, estabilidad hemodinámica y sin signos de insuficiencia cardíaca. Test de Apgar de 9 al minuto 1 y 5. Peso: 3105 gramos. Se realiza electrocardiograma con tira de ritmo donde se objetiva un flutter auricular con frecuencia auricular de 300 lpm y conducción variable 2:1, 3:1. Dada la estabilidad hemodinámica, se inicia tratamiento con digoxina. Se completó el estudio diagnóstico con radiografía de tórax y ecocardiografía que resultaron normales. La evolución posterior fue buena, con recuperación del ritmo sinusal en las siguientes 48 horas. Se continuó el tratamiento digitálico durante 20 meses, permaneciendo actualmente asintomático y en ritmo sinusal.
Conclusiones:El flutter neonatal es una arritmia de presentación poco frecuente y que se asocia en ocasiones con cardiopatías congénitas (Ebstein, estenosis mitral, atresia tricúspide), miocarditis viral e infecciones sistémicas. La evolución cuando es primaria es en general benigna hacia la recuperación del ritmo sinusal mediante cardioversión si existe compromiso hemodinámico y con digoxina si no lo existe. Si es secundario el pronóstico depende de la enfermedad de base.
El flutter auricular neonatal es una arritmia habitualmente benigna que cede al tratamiento con digital.
P377HIPOPLASIA PULMONAR, HIPERTENSION PULMONAR Y COLESTASIS HEPATICA
José Lorenzo Guerra Díez, Ana Benito Cornejo, Macarena Otero Fernández, Ignacio Banzo Marraco, Elena Pérez Belmonte y José Ricardo Galván Robles
Hospital Universitario Marqués de Valdecilla, Santander.
Introducción:La hipoplasia pulmonar primaria (HPP) es un trastorno congénito de incidencia muy baja y que se acompaña algunas veces de hipertensión pulmonar (HP) en el recién nacido. Presentamos un caso que además se acompaña de síndrome colestásico biliar no descrito en la literatura.
Objetivo:1. Presentar un recién nacido que padece HP con HPP y síndrome colestásico biliar.2. Evaluar cuál puede ser el mecanismo fisiopatológico que explique una asociación entre HPP, HP y colestasis biliar.
Recién nacido de 35 + 5 semanas de gestación, embarazo normal, peso natal 3760g y parto por cesárea urgente por desprendimiento de placenta. En la primera hora de vida inicia distres respiratorio con gasometría normal y radiografía de tórax compatible con poca expansión pulmonar y edema intersticial. Posteriormente aumentan los requerimientos de oxígeno y precisa de ventilación mecánica. Se diagnostica de hipoplasia pulmonar y fallo cardiaco (con persistencia del ductus arteriosus) asociado a hepatomegalia; enzimas hepáticas GOT: 69 U/l; GPT: 241 U/l; d -GT:535 U/l; Bilirrubina total: 3,6 mg/dl; Bilirrubina directa: 2,65 mg/dl Cultivos y serologías de hepatitis B y C, VIH, CMV, Toxoplasma negativas salvo Varicela Zoster IgG(+). Hemograma normal y PCR: 0,3 mg/dl. Alos cinco días de vida requiere sólo el 45 % de oxígeno y ventilado con un volumen tidal bajo alcanza altas presiones el respirador. Se realizó gammagrafía hepato-biliar normal y gammagrafía pulmonar que revelaba una hipoplasia pulmonar bilateral. Posteriormente precisó cerrar el ductus quirúrgicamente y presentó un neumotórax que se resolvió mediante toracocentesis. Tras un mes de evolución y mejora de su situación cardiaca y respiratoria se normalizaron las enzimas hepáticas. El niño fallece posteriormente a los 110 días de vida pendiente de transplante pulmonar.
Conclusión:El desarrollo de un fallo cardiaco congestivo por a la hipertensión pulmonar e hipoplasia pulmonar desencadenó un aumento de las transaminasas, GGT, bilirrubina total y directa; sugiriendo una colestasis biliar que cede tras solucionar el fallo cardiaco.
P378HIGROMA QUISTICO SECUNDARIO A EXTRAVASACION DE NUTRICION PARENTERAL POR UN CATÉTER EPICRANEAL
Gemma Arca Díaz, María José García Boreau, Gemma Ginovart Galiana, Pere Parés Muñoz, Elisenda Moliner Calderón, Israel Anquela Sanz, Jordi Martínez-Baylach, Joan Nadal Amat y Josep Cubells Rieró
Hospital de la Santa Creu y Sant Pau, Barcelona.
Introducción:Los catéteres de silicona de pequeño calibre han contribuido a la mejora asistencial del neonato, pero no están exentos de complicaciones. Presentamos un caso de formación de un higroma quístico secundario a la extravasación de alimentación parenteral a través de un catéter epicraneal.
Caso clínico:Recién nacida de 640 g, de 26 + 1 semanas. Correcto control gestacional. Cesárea urgente por sospecha de corioamnionitis. Apgar 2-7-7. Controles ecográficos cerebrales iniciales normales. A las 48 horas de vida se inicia nutrición parenteral y a los seis dias alimentación enteral trófica que es mal tolerada presentando apneas que requieren intubación orotraqueal y conexión a ventilación mecánica. A las cinco semanas de vida presenta empeoramiento clínico con una marcada hiponatremia, se realiza una ecografía transfontanelar de control y se evidencia un importante higroma quístico en hemisferio cerebral izquierdo que colapsa el ventrículo lateral izquierdo y desplaza la línea media hacia la derecha. Se realiza drenaje quirúrgico por punción a través de la sutura coronal, dando salida a 30 ml de líquido blanquecino espeso. Bioquímica del líquido extraído: glucosa: 10,8 mmol/l, proteínas 2,740 g/l. Tanto el aspecto del líquido como la composición bioquímica del mismo confirman que se trata de una extravasación de nutrición parenteral por progresión hacia espacio subdural del catéter epicraneal en vena temporal izquierda. La ecografía de control postpunción confirma la evacuación del higroma y desaparición del efecto masa con recentralización de la línea media. A las pocas horas se detecta pequeña hemorragia homolateral a nivel de la matriz germinal. En controles ecográficos transfontanelares posteriores presenta ventriculomegaliaex vacuo y microquistes poroencefálicos periventriculares frontales, compatibles con leucomalacia periventricular. La paciente presenta mala evolución clínica con complicaciones digestivas (NEC) e infecciosas que le conducen a la muerte a los tres meses de edad.
Conclusiones:Creemos que la utilización de microcatéteres percutáneos es eficaz si se realiza correctamente, puesto que los riesgos y complicaciones no sobrepasan los de otros métodos empleados. Se han descrito complicaciones respiratorias, mecánicas e infecciosas, pero no se encuentra en la literatura ninguna complicación por extravasación de nutrición parenteral a través de un catéter epicutáneo como la expuesta.
P379INFLUENCIA DE LA GESTACION MULTIPLE EN LA MORBILIDAD NEONATAL
Adelina Pellicer Martínez, Rosario Madero, José Quero Jiménez y Fernando Cabañas González
Hospital Universitario La Paz, Madrid.
Antecedentes:La tasa de partos múltiples se ha incrementado dramáticamente en las últimas décadas debido a la mayor edad de las gestantes y a las técnicas de reproducción asistida. Actualmente, el interés creciente sobre el devenir de estos niños se justifica no sólo por el incremento en el número de pacientes nacidos de parto múltiple, sino porque estudios poblacionales recientes parecen señalar un pronóstico menos favorable cuando se comparan con los fetos únicos.
Hipótesis:La gestación múltiple no afecta a la salud del recién nacido.
Diseño/métodos:Estudio trasversal en un hospital universitario de ámbito terciario.
Pacientes:Productos de todas las gestaciones nacidos entre Mayo '00 y Agosto '02.
Variable principal de evaluación: Morbilidad neonatal, incluyendo acidosis perinatal, bajo peso para la gestación (BPEG), prematuridad, enfermedad de membrana hialina (EMH), ductus persistente (DAP), lesión adquirida del SNC (hemorrágica/isquémica), convulsiones, enterocolitis necrotizante, malformaciones congénitas (cardíaca, cerebral, gastrointestinal, urogenital, oro-facial, hipoplasia pulmonar/hernia diafragmática, arteria umbilical única, músculo-esquelética), anomalías cromosómicas e hidrops.
Resultados:El estudio incluye 21154 pacientes: 20256 productos únicos y 898 múltiples (752 gemelos, 138 trillizos, 8 cuatrillizos). La tasa de ingreso en Serv. Neonatología fue del 7,6 % en únicos y 47,8 % en múltiples (p < 0,001). La presencia de cualquier tipo de malformación congénita, anomalía cromosómica, BPEG o acidosis perinatal fue significativamente superior en los productos únicos. El parto con < 32 sem aconteció con una frecuencia significativamente mayor en las gestaciones múltiples (11,6 % vs 0,9 %, p < 0,001). Sin embargo, la prevalencia de las enfermedades típicas de la prematuridad (hemorragia intracraneal, leucomalacia periventricular, DAP, EMH) fue similar en los dos grupos.
Conclusiones:Nuestro estudio sugiere un buen pronóstico para los niños nacidos de parto múltiple a pesar de la mayor tasa de parto prematuro.Especulamos que la mayor intervención obstétrica, en parte mediada por la indicación del parto pretérmino, unida a los avances en los cuidados neonatales, probablemente estén salvando situaciones comprometidas en las gestaciones de riesgo en nuestro centro.
P380ARTROGRIPOSIS MULTIPLE CONGÉNITA CLASICA
M. Teresa Jiménez Fernández, Itziar Echevarría Matía, Eva M. García Jara, Olga García Bodega, Alicia Sáez de Cabezón, Fco. Javier López Pisón y Víctor Rebaje Moises
Hospital Miguel Servet, Zaragoza.
Introducción:La Artrogriposis Múltiple Congénita Clásica (AMC) es una afección caracterizada por la presencia de contracturas articulares congénitas y no progresivas.
Caso clínico:Recién nacida hembra pretérmino sin antecedentes familiares de interés. Parto por cesárea urgente debido a desprendimiento placentario. Gestación con retraso de crecimiento intrauterino y disminución de movimientos fetales confirmados por Ecografía. Peso: 1830 gr. Talla aproximada: 40 cm. PC: 34 cm.
Exploración:Presenta contracturas articulares múltiples y simétricas de las 4 extremidades, con manos y pies zambas y actitud característica, con mayor afectación distal. Articulaciones de aspecto fusiforme y agrandadas con presencia de hoyuelos o fosetas cutáneas. Desaparición de pliegues y relieves articulares normales. Atrofia muscular generalizada. Facies especial con macrocefalia relativa, micrognatia y platirrinia. Contacto social bueno. Sensibilidad conservada. Reflejos osteotendinosos y arcáicos ausentes, excepto reflejo de succión.
Pruebas complementarias:Todas las exploraciones encaminadas a descartar patología infecciosa o metabólica fueron normales así como el EEG, TAC craneal y fondo de ojo. Cariotipo 46 XX. Ecografía cardiaca, renal y ocular normales. ENG: normal. EMG: denervación activa difusa crónica que parece tener su origen en astas anteriores de médula espinal.
Comentario:Nuestro caso corresponde a una AMC encuadrable dentro de su forma clásica por el cuadro clínico característico, y más exactamente a una secuencia de acinesia-hipocinesia fetal. La inmovilidad fetal intraútero fue comprobada por ecografías, y el nivel de afectación según el estudio electromiográfico se situaría a nivel de las astas anteriores de la médula espinal.
P381¿APENDICITIS NEONATAL COMO ENTIDAD AISLADA?
Núria Tomasa Wörner, Jordi Almar Mendoza, Carles Giné, José Lluis Peiro Ibáñez, Nuria Torán, Élida Vázquez Méndez, Marc Tobeña, Rosa Sementé, Carmen Abellán y Salvador Salcedo Abizanda
Hospital Vall D'Hebrón, Barcelona.
Introducción:La apendicitis es una entidad rara durante el período neonatal que se asocia generalmente a otras patologías: enterocolitis necrotizante (ECN), fibrosis quística de páncreas (FQ), enfermedad de Hirschprung (EH), impactación meconial (IM) y hernia inguinal incarcerada. Presentamos un caso clínico de apendicitis neonatal como entidad aislada basándonos en criterios clínicos y anatomopatológicos.
Caso clínico:Recién nacido a término mediante cesárea debido a sufrimiento fetal. Peso al nacer: 3840 gramos. Apgar: 9/10. Al quinto día de vida presentó fiebre (38 °C) por lo que se instauró tratamiento antibiótico empírico. Los cultivos realizados fueron negativos. Tres días después inició cuadro de vómitos junto con aparición de una masa dolorosa a la palpación en hemiabdomen derecho. La radiografía simple de abdomen mostró un efecto masa en dicha zona, mientras que la ecografía abdominal no fue concluyente. No presentó ningún signo clínico ni radiológico de ECN. La tomografía axial computerizada (TC) demostró la existencia de una masa en fosa ilíaca derecha y un engrosamiento del mesoapéndice. Se realizó apendicectomía a los 15 días de haber iniciado el tratamiento antibiótico. Los hallazgos anatomopatológicos evidenciaron una apendicitis aguda sin signos histológicos de ECN y con presencia de células ganglionares y tejido linfoide hiperplásico. Seis meses después el paciente se encuentra asintomático.
Discusión:Describimos un caso de apendicitis neonatal como entidad aislada diferenciándola de las patologías a las que normalmente se asocia y en especial de la ECN. La histología no mostró signos clásicos de ECN como hemorragia de la mucosa, edema de la submucosa y trombosis de los vasos de la submucosa. En nuestro caso destacó la presencia de un proceso inflamatorio agudo de la mucosa con aumento de celularidad a expensas de polimorfonucleares en glándulas y lámina propia, la existencia de un componente inflamatorio más cronificado en la submucosa y la presencia de celulas ganglionares, por lo que descartamos la posibilidad de ECN y EH. Destacamos la utilidad de la TC como exploración complementaria para el diagnóstico y la importancia del tratamiento antibiótico precoz en el manejo de este proceso.
Conclusiones:Defendemos la existencia de la apendicitis aguda neonatal como entidad aislada y creemos que debería ser considerada en el diagnóstico diferencial de cualquier proceso abdominal en el recién nacido, a pesar de su extraordinaria rareza.
P382SINDROME FEBRIL PROLONGADO ASOCIADO A INFECCION POR VIRUS HERPES HUMANO TIPO 6
Sara Guillén Martín, Pablo Rojo Conejo, Begoña Losada Pinedo, José Tomás Ramos Amador, Carlos Díaz González y M. Isabel González Tomé
Hospital 12 de Octubre, Madrid.
Introducción:El virus herpes humano tipo 6 (VHH-6) es un virus linfocitotropo ubicuo que afecta a niños menores de 3 años y produce habitualmente cuadros leves autolimitados. No obstante se ha asociado a cuadros graves en inmunodeprimidos y, excepcionalmente, en niños inmunocompetentes, incluyendo cuadros de fiebre prolongada, hepatitis, síndrome mononucleósico y síndrome hemofagocítico.
Caso clínico:Paciente varón de 2 años y 8 meses, previamente sano que consulta por fiebre alta (40 °C) y dolor abdominal. La fiebre tiene un curso recurrente de 35 días objetivándose anorexia intensa, afectación del estado general con pérdida de peso, progresiva anemización (Hb 7,9 g/dl), gran hepatomegalia y esplenomegalia. Entre los hallazgos de laboratorio destaca leucocitosis con un 4 % de linfocitos atípicos, VSG de 112 mm/h, PCR de 92 mg/l, e hipergammaglobulinemia. La ecografía abdominal reveló adenopatías en hilio hepático y renal que se confirmaron en TAC. La médula ósea no mostró lesiones histológicas relevantes. Una investigación extensa microbiológica no reveló ningún patógeno habitual. En la serología en paralelo se objetivó un título alto de IgG para VHH-6 y amplificación de DNA mediante PCR plasmática positiva para VHH-6 (tipo A y B). El niño tuvo una remisión espontánea de la sintomatología con normalización de la analítica en los 3 meses siguientes, incluyendo negativización de PCR específica para VHH-6.
Comentarios:Aunque en este caso, no es posible definir si la infección por VHH-6 es una primoinfección o reactivación, queremos resaltar la asociación del VHH-6 con el cuadro grave observado. El VHH-6 debe incluirse entre las posibles etiologías de los síndromes febriles prolongados y recurrentes en la infancia.
P383COMPLICACIONES SECUNDARIAS AL RETRASO EN LA REPARACION QUIRURGICA DE LA HERNIA INGUINAL EN LOS RN PREMATUROS
María González Santacruz, Ana Fernández, Jesús Mira Navarro y Bartolomé Jiménez Cobo
Hospital General Universitario, Alicante.
Objetivo:No existe consenso sobre cuando realizar la reparación quirúrgica de la hernia inguinal (HI) en los prematuros. Por ello, se intento determinar si el retraso en la herniorrafia supone o no un aumento en la tasa de complicaciones, tanto a corto como medio plazo, comparándolo a su vez con lo que ocurre en los neonatos a termino.
Métodos:Se revisaron las historias clínicas de 41 recién nacidos intervenidos de HI en nuestro Centro. Se realizó un seguimiento clínico posterior con el objeto de detectar la presencia de complicaciones postoperatorias tardías. Para el análisis, los niños se distribuyeron en tres gruposA) niños prematuros intervenidos precozmente, < 2 semanas tras el diagnóstico (n = 9);B) niños prematuros intervenidos tardíamente, > 2 semanas tras el diagnóstico (n = 21);C) un grupo control de recién nacidos a término (n = 11). Parámetros tales como la edad gestacional, la duración de la estancia hospitalaria, la morbilidad prequirúrgica y las complicaciones a medio y largo plazo fueron comparados entre los tres grupos.
Resultado:El parámetro que mejor correlacionó con el retraso en la reparación de la HI fue la edad gestacional. En el momento de la intervencion, no hubo diferencias significativas en la edad postconcepcional, en la duración de la herniorrafia, ni en la duración de la estancia postquirúrgica entre los tres grupos. Se identificaron 7 episodios de incarceración antes de la herniorrafia: uno en el grupo A, dos en el grupo B y cuatro en el grupo C. En el seguimiento clínico posterior se detectaron dos atrofias testiculares: una en el grupo A y otra en el grupo C.
Conclusión:El retraso en la reparación quirúrgica de la hernia inguinal en el recién nacido prematuro no parece incrementar el riesgo de episodios de incarceración o la aparición de atrofia testicular secundaria a los mismos. Por tanto, la cirugía en este grupo se podría posponer hasta que el recién nacido esté en una situación estable, se haya recobrado de los problemas derivados de la prematuridad y esté próximo al alta de la unidad neonatal.
P384RECIÉN NACIDOS DE MADRES CONSUMIDORAS DE DROGAS EN NUESTRO HOSPITAL EN LOS AÑOS 2000-2002
Leonor Guardia Nieto, Elisa Cueto Calvo, Miguel Sarrión Cano, M.ª José García Martínez y Ana Usano Carrasco
Hospital Virgen de La Luz, Cuenca.
Antecedentes:El consumo de drogas durante el embarazo es un problema médico y social para la madre y para su hijo, pudiendo producir en el RN un síndrome de abstinencia.
Objetivo:Conocer las características de los RN hijos de madres consumidoras de drogas y del síndrome de abstinencia cuando se presenta.
Métodos:Se realizó un estudio retrospectivo entre el 1 de enero de 2000 y el 31 de diciembre de 2002 de las historias clínicas de los RN hijos de madres consumidoras de drogas. A todos ellos se les aplicó el score de Fennegan. Se analizaron distintas variables.
Resultados:Fueron estudiados 14 niños, 10 mujeres. La edad media de la madre fue de 28 ± 4,79 años. La edad gestacional media fue 37,46 ± 2,37 semanas, siendo 5 RN pretérmino. El 71,4 % de las madres se encontaban en tratamiento sólo con metadona. El resto reconocía consumir otras drogas además de metadona. Todas menos 1 habían tenido controles adecuados de su gestación. La serología más frecuente fue + a VHC, en un 71,4 % de los casos. El 78,6 % de los niños desarrollaron síndrome de abstinencia. La aparición de los síntomas fue a las 46,09 ± 39,32 horas, siendo llamativo que en las tres madres que no consumían exclusivamente metadona, el momento de aparición de la sintomatología se adelantó a las 18 ± 8,49 horas. El tratamiento que se utilizó fue fenobarbital, con una dosis máxima de 7,82 ± 1,46 mg/kg/día, y se mantuvo una media de 15,09 ± 4,99 días. De los RN que desarrollaron un síndrome de abstinencia, todos presentaron hiperexcitabilidad, hipertonía y temblores, siendo también muy frecuentes la fiebre (45,42 % de los casos) y la sintomatología digestiva consistente en vómitos (18,20 %) y diarrea (36,39 %).
Conclusiones:En el momento actual, en nuestro medio la mayoría de las madres adictas a drogas consumen metadona durante el embarazo. Esto hace que cuando aparece un síndrome de abstinencia neonatal, se inicie a partir del segundo día de vida del RN. El síndrome de abstinencia es un diagnóstico clínico, que obliga a una hospitalización prolongada en niños sin otra patología. El fenobarbital es un tratamiento eficaz en el control de los síntomas.
P385RAQUITISMO NEONATAL SECUNDARIO A DÉFICIT MATERNO DE VITAMINA D
Marta Arroyo Martínez, L. Sánchez, E. Ordóñez, C.W. Ruiz, Diego Yeste Fernández, Anna Fina Martí, Salvador Salcedo Abizanda y Antonio Carrascosa Lezcano
Hospital Vall D'Hebrón, Barcelona.
Introducción:El raquitismo congénito es una entidad de presentación excepcional. Su origen,en general, responde a causas maternas.
Caso clínico:Recién nacida a término de 40 s.g. de raza negra, hija de madre de origen subsahariano, remitida a nuestro hospital al 6.º día de vida por presentar tórax displásico, hipotonía muscular y distrés respiratorio progresivo que precisa ventilación mecánica. Ha seguido desde el 1.º día de vida nutrición parenteral exclusiva con aporte de 220 UI/d de vitamina D y de 2,5 mEq/kg/d de Calcio. Examen físico: peso: 2.740 kg, talla: 46,5 cm, PC:32 cm. Fontanela anterior amplia y craneotebes. Tórax estrecho y de aspecto displásico, con escasa excursión bilateral. Rodetes epifisarios en ambas muñecas. Exámenes complementarios: Rx tórax: caja torácica estrecha con costillas onduladas y múltiples fracturas. Scanner torácico: descarta hipoplasia pulmonar. Serie esquelética: osteopenia intensa, fractura de huesos largos, incurvación de ambos fémures y metáfisis cubitales en copa. Metabolismo fosfocálcico del recién nacido: Calcio: 9,2 mg/dl (vn: 8,9-11,5), Fósforo: 3,1 mg/dl (vn: 4-7), Magnesio: 2,1 mg/dl (vn: 1,9-2,5), Fosfatasas alcalinas: 701 U/l (vn: < 500), PTH: 881 pg/dl (vn: 11-62), 25(OH)D: 10 ng/dl (vn: 16-74). Metabolismo fosfocálcico materno: Calcio: 9,1 mg/dl (vn: 9-10,8), Fósforo: 3,2 mg/dl (vn: 2,5-4,8), Fosfatasas alcalinas: 119 UI/l (vn: 50-300), PTH:50 pgdl (vn: 11-62), 25(OH)D:8,1 ng/dl (vn: 16-74), 1,25(OH)2D.16,5 pg/ml (vn: 15-60). Resultados compatibles con estado subclínico de hipovitaminosis D materno y raquitismo neonatal. Aportes maternos estimados: vit. D:260 UI/día de origen nutricional exclusivo por falta de exposición solar; Calcio: 514 mg/día (recomendados en gestación: Vit. D: 1.000 UI/día en 3.º trim, Calcio: 1.500 mg/día). Se inicia tratamiento con vit. D (835 UI/día) y suplemento oral de Calcio (125 mg/día). Al mes de vida los niveles de vit. D y de Calcio son normales y existe remineralización ósea con formación de bandas metafisarias. Se extuba electivamente a los 61 dias de vida.
Conclusión:Los estados carenciales de vit. D son un factor de riesgo de raquitismo neonatal. La reciente corriente inmigratoria de poblaciones subsaharianas, que mantienen hábitos culturales y nutricionales propios (indumentarias que cubren la mayor parte del cuerpo, escasa actividad al aire libre y bajo consumo de derivados lácteos) puede determinar un incremento del raquitismo congénito en nuestro medio.
P386TROMBOCITOPENIA NEONATAL: CASUISTICA DE 2 ANOS NUMA UCI
M. Ana G. del Castillo, Ana Sarmento, Susana Tavares, Ana Margarida Alexandrino, Artur Alegria, José Barbot y Augusta Areias
Maternidade Júlio Dinis, Porto (Portugal) y Hospital de Crianças Maria Pia, Porto (Portugal).
Introdução e objectivos:A trombocitopenia é a alteração hematológica mais frequente no recém-nascido (RN), com incidências de 35 % em Unidades de Cuidados Intensivos Neonatais (UCIN) e de 50 % em RN pré-termo doentes. Fizemos a revisão dos casos de trombocitopenia na nossa UCIN.
Métodos:Estudo dos casos ocorridos em 2001-2002, com análise de factores maternos e neonatais, etiologias, duração e terapeutica transfusional.
Resultados:Entre 964 RN internados, ocorreu trombocitopenia em 99 (10,3 %). Nos RN com peso de nascimento < 1.500 g e < 1.000 g as incidências foram de 30 % e de 49 %, respectivamente. Entre os factores de risco perinatal associados salientaram-se: prematuridade (88), muito baixo peso (65), ACIU (29), asfixia perinatal (21), pré-eclâmpsia (20), fluxo umbilical patológico (16) e oligohidrâmnios (12). Contabilizaram-se 109 episódios. A trombocitopenia ocorreu precocemente (< 72 horas) em 66 deles, sendo etiologias principais a insuficiência placentar, infecções congénitas, processos imunes e asfixia grave. Entre as trombocitopenias precoces, 11 RN receberam plaquetas (14 transfusões). A trombocitopenia foi tardia (> 72 h) em 43 episódios (40 RN), sendo a sépsis a principal etiologia. Treze crianças com trombocitopenia tardia foram transfundidas (65 transfusões). Situações com contagem de plaquetas < 50.000/μl ocorreram em 36 RN, sendo a insuficiência placentar e as doenças imunes os factores etiológicos principais entre os episódios precoces e a sépsis entre os tardios. Vinte e quatro RN foram transfundidos com plaquetas (79 transfusões); 82 % das transfusões foram efectuadas em episódios tardios. A duração dos episódios variou entre 1 e 50 dias, com medianas de 5 dias entre os precoces e de 7 dias entre os tardios.
Conclusões:Em relação ao descrito na literatura, a incidência global de trombocitopenias foi inferior, embora se confirme uma maior incidência em RN de muito baixo peso. As principais etiologias nos episódios precoces foram a insuficiência placentar e as infecções congénitas e nos tardios a sépsis. A trombocitopenia grave ocorreu tanto nos episódios precoces como nos tardios, sendo estes responsáveis pela grande maioria das transfusões plaquetárias, o que decorre da instabilidade clínica inerente à etiologia séptica preponderante neste grupo.
NEUROLOGIA
P387 ACCIDENTE VASCULAR CEREBRAL ISQUÉMICO
Sandra Cristina Alves, Maria Chaves, Paulo Guimarães y M. Rui Carrapato
Hospital São Sebastião, Santa Maria da Feira (Portugal).
Introdução:O Acidente Vascular Cerebral (AVC) é um entidade clínica pouco frequente na criança. Define-se como lesão cerebral focal por obstrução ou ruptura vascular, originando défices neurológicos com duração superior a 24 horas e com evidência de lesão cerebral na Tomografia Axial Cerebral (TAC) ou Ressonância Magnética Nuclear (RMN). O diagnóstico etiológico do AVC na criança é particularmente importante não só para o tratamento, prognóstico, prevenção de recidivas e complicações mas também para a identificação de risco em familiares.
Caso clínico:Os autores apresentam o caso clínico de uma criança de 17 meses, sexo masculino, admitida no Serviço de Urgência por perda da marcha e da força no membro superior direito. Ao exame objectivo apresentava, de relevante, otite média aguda esquerda e hemiparésia direita. Nos antecedentes pessoais de valorizar varicela aos 8 meses. Sem história familiar significativa.
Como hipóteses de diagnóstico consideraram-se: infecção do SNC, traumatismo crânio-encefálico, AVC, tumor cerebral e enxaqueca. Os. exames analíticos excluíram infecção congénita ou adquirida. A função hepática, perfil lipídico, anticorpo antinuclear e anticardiolipina, electrocardiograma e ecocardiograma foram normais. O anticoagulante lúpico foi positivo assim como o nível de lipoproteína (a): 99 mg/dl (N: < 30 mg/dl). A TAC cerebral evidenciou "...lesão hipodensa no território das artérias lenticulo-estriadas..." e a RMN cerebral "...confirma a lesão isquêmica lenticular esquerda...".
A evolução no internamento foi favorável com melhoria progressiva da hemiparésia.
Conclusão:A apresentação deste caso tem como objectivo reforçar o conceito de que a ocorrência de um AVC em idade pediátrica se deve à conjugação de vários factores de risco que determinam a gravidade e o prognóstico. O conhecimento destes factores etiológicos é essencial pois embora a taxa de mortalidade seja baixa, a probabilidade de recidiva é considerável e as sequelas frequentes.
P388TOXICIDAD NEUROLOGICA POR METOTREXATE EN PACIENTE AFECTO DE LEUCEMIA LINFOBLASTICA AGUDA
M. Isabel Llull Ferretjans, Raquel Amo Rodríguez, Nieves Nieto del Rincón, M. de los Ángeles Ruiz Gómez, Mercedes Guibelalde del Castillo y Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca.
Introducción:Está descrito que el tratamiento con metotrexate (MTX) endovenoso (ev) e intratecal (IT) a dosis altas puede producir complicaciones neurológicas de forma aguda, subaguda y crónica. Generalmente aparecen en pacientes con eliminación del MTX disminuída.
Objetivos:Presentar caso de paciente afecto de Leucemia linfoblástica aguda (LLA) con toxicidad neurológica por MTX. Revisión del manejo y evolución posterior.
Caso clínico:Niño de 12 años diagnosticado de LLA de células T en Octubre 2002 en tratamiento según protocolo SHOP-99AR. Antecedente de crisis comicial a los 11 años y otra 3 días tras inicio tratamiento de inducción. En fase de consolidación recibe MTX 3 g/m2 (ev) y 9 mg/m2 (IT) con rescate con folínico según niveles de MTX. A las 60 horas del inicio del tratamiento presentó niveles de MTX de 9,1*107 M por lo que se aumentó dosis de folínico. Al 8.º día tras administración de MTX presenta cuadro de deterioro neurológico progresivo con fases de letargia, delirio, agitación, afasia, crisis convulsiva y coma que precisó ingreso en Cuidados Intensivos. Asociado a este cuadro el paciente presentó cuadro de infección respiratoria con fiebre y aplasia en la semana previa. Debido a Trombopenia (10.000 plaquetas) no se realizó punción lumbar. TAC y RNM normales. El EEG presentaba enlentecimiento marcado difuso. Recibió tratamiento con anticomiciales, dexametasona, antiinfecciosos y folínico. Al aislar en cultivo faríngeo virus influenza B se añadió Amantadina. A las 72 horas inició mejoría progresiva. La recuperación neurológica fue completa tras 20-25 días con EEG normal. Reinició tratamiento con MTX (ev) tras 22 días e IT tras 2 meses, bien tolerados con aumento de dosis de folínico. El LCR obtenido en la 1.ª intratecal fue normal.
Comentarios y conclusiones:1. Sospechar toxicidad por MTX en pacientes con este tratamiento que presentan clínica neurológica.2. Las alteraciones en EEG son difusas y reversibles. 3. Suele presentarse en pacientes con aclaramiento de MTX disminuído.4. La toxicidad por MTX puede presentarse de múltiples formas:convulsiones,paresia,afasia,deterioro cognitivo,radiculopatía o encefalopatía. 5. Destacar la importancia del rescate con folínico en el tratamiento con MTX.
P389 TROMBOSIS VENOSA DEL SENO LONGITUDINAL Y SIGMOIDEO
M. del Carmen Martín Ruiz, Julián Vaquerizo Madrid, Luis Zarallo Cortés, Ana Navarro Dourdil y Nieves Alonso Escobar
Hospital Universitario Infanta Cristina, Badajoz.
Introducción:Presentamos el caso de una trombosis venosa del seno longitudinal superior y sigmoideo derecho, en niño con antecedente de GEA que precisó ingreso y que en el estudio hematológico presentó un déficit de antitrombina III (ATIII).
Caso clínico:Niña de 2 años que ingresa por vómitos biliosos, postración y somnolencia con aparición de estrabismo convergente alternante. El cuadro comienza 24 horas después de ser dada de alta por GEA.
Exploración:BEG. Consciente y algo desorientada, tendencia al sueño aunque responde a estímulos externos. Paresia VI par bilateral. Disminución de fuerza de forma generalizada. ROT presentes y vivos. Discreta rigidez de nuca, signos meníngeos negativos. Glasgow 13-14.
Exámenes complementarios:Hemograma: anemia microcítica e hipocrómica, serie blanca y plaquetas normales. LCR xantocrómico; amonio, ac. Láctico,cloro y ADA normales. EEG: lentificación de la actividad fundamental más acusada en hemisferio derecho (Control normal). IRM craneal y angioIRM: trombosis del seno longitudinal superior y sigmoideo derecho. Estudio de trombofilia: déficit de ATIII. Estudio cardiológico normal.
Evolución:Recuperación del estado de conciencia. Al tercer día presenta episodio de movimientos tónico-clónicos. Tratamiento seguido: aciclovir, ácido valproico, heparina de bajo peso molecular (HBPM).
Conclusiones:1. Los accidentes vásculo cerebrales en la infancia son una entidad infradiagnosticada entre otros factores debido a su baja incidencia. 2. Pueden coexistir varios factores etiológicos, estando indicado el estudio de trombofilia de forma sistemática.3. La forma más frecuente de enfermedad vascular es la hemorragia. La patología oclusiva presenta un predominio de localización arterial sobre venosa.4. En nuestro caso tanto la deshidratación como el déficit de ATIII pueden justificar la trombosis venosa cerebral. No obstante, es necesario confirmar este último aspecto pasada la fase aguda.5. Decidimos tratar con HBPM, aunque el tratamiento anticoagulante es discutido.
P390 COREA DE SYDENHAM: UNA PATOLOGIA DEL PASADO
M. Teresa de Benito Guerra, M. Ester Guerrero Vega, Esther Álvarez García, Myrian Macarena Ley Martos, Rocío Montiel Crespo y Josefina Fornell Forcadas
Hospital Puerta del Mar, Cádiz.
La corea de Sydenham es la corea adquirida más frecuente de la infancia. Constituye uno de los criterios mayores para el diagnóstico de la fiebre reumática y a menudo es el único síntoma de esta enfermedad. Su mayor incidencia se da entre los 5-15 años, con un claro predominio femenino (2:1). Las manifestaciones clínicas principales son movimientos coreicos, hipotonía muscular y labilidad emocional. El diagnóstico es esencialmente clínico por exclusión de otros procesos similares. Se trata de un proceso benigno que puede persistir varios meses o incluso 1-2 años. Aproximadamente un 20 % recurre.
Caso clínico:Niño de 11 años que comienza de forma brusca con movimientos rápidos, incoordinados e involuntarios de predominio en cara y región distal de extremidades. Son movimientos asimétricos, groseros de varias horas de evolución, que le impiden la bipedestación provocándole caídas frecuentes. Se incrementan con el estrés y remiten con el sueño. Discreta labilidad emocional. AP: faringoamigdalitis un mes antes tratada con penicilina im. A destacar en la exploración neurológica: reflejos pendulares en MMII, imposibilidad para la bipedestación y la marcha, tics complejos motóricos y fónicos y hemibalismo.
Dado los antecedentes y la clínica característica, se diagnostica de corea de Sydenham,tras la exclusión de otros procesos mediante la determinación de exámenes complementarios. Se inició tratamiento con penicilina y haloperidol. Este tuvo que ser retirado por la aparición de reacción extrapiramidal.
Conclusiones: 1.Resaltar la atención sobre una patología prácticamente olvidada por su escasa incidencia en la actualidad.2. En todo niño con corea debe descartarse etiología lúpica mediante la determinación de anticuerpos antifosfolípidos, ANA, ANCA,... ya que ésta supone la segunda causa en frecuencia, siendo su pronóstico y tratamiento muy diferentes.3. La corea puede ser la única manifestación de la fiebre reumática y, aunque los exámenes de laboratorio sugestivos de infección estreptocócica contribuyen al diagnóstico, la negatividad de estos no lo excluyen.4. A pesar de la eficacia de los neurolépticos en el tratamiento, la gran sensibilidad de los niños a estos obliga a utilizarlos con precaución.5. Descartar la presencia de corea secundaria a la toma de alguna medicación.
P391SINDROME DE WAGR
Antonia Solas Beltrán, Concepción Sierra Corcoles, Rosa M. Rodríguez García, Ana Arévalo Garrido, M. Dolores Gámez Gómez, Irene Peláez Pleguezuelos, Carmen Vargas, M. del Carmen Fernández Novoa García y Felipe González Rivera
Hospital Universitario Ciudad de Jaén.
Introducción:El síndrome de WAGR forma parte de los llamados síndromes de microdelección o de genes contíguos. En este, la delección afecta a la región p13 del brazo corto del cromosoma 11 lo cual conduce a la aparición de las anomalías que dan nombre al síndrome: Tumor de Wilms (W), aniridia (A), malformaciones génitourinarias (G) y retraso mental (R).
Caso clínico:Niño de 12 años que es derivado a nuestra consulta para estudio por retraso mental.
Antecedentes perinatales:Embarazo normal, parto eutócico a las 38 semanas, peso: 2.190 g (< P10). Al nacimiento presentaba criptorquidia bilateral, hipospadias y aniridia bilateral. Padres sanos, no consanguíneos. 2 hermanos sin patología.
Al año de edad es diagnosticado de tumor de Wilms (TW) y tratado con nefrectomía derecha y posteriormente quimioterapia. Retraso psicomotor: sostén cefálico a los 5 meses, sedestación sin apoyo a los 10 meses, deambulación autónoma a los 19 meses.
Actualmente en la exploración física destaca microcefalia, hipertelorismo, epicantus y aniridia bilateral. Nistagmo horizontal. Macroglosia discreta, orejas de baja implantación. Surco transversal en mano izquierda. Orquidopexia e hipospadias intervenido. Presenta además retraso mental moderado, apreciándose en la Resonancia magnética moderada atrofia córticosubcortical.
El estudio cromosómico aplicando técnicas de bandas GTG muestra una fórmula cromosómica 46 XY con delección de la región p12 p13 del cromosoma 11.
Conclusiones: 1.El Sdr. de WAGR es una entidad clínica muy rara de la que existen pocos casos descritos. 2. La mayor parte de las veces no aparece completo, sin embargo, en nuestro paciente están presentes todas las anomalías que lo definen.3. El Sdr. de WAGR es habitualmente esporádico, por tanto es improbable la recurrencia en hermanos, sin embargo estaría indicado la realización de cariotipo a los padres ya que pueden existir portadores de reordenamientos cromosómicos balanceados que tienen el riesgo de hijos con disbalances cromosómicos.
P392RECONSIDERACION DE PARAMETROS CLINICOS Y DE TRATAMIENTO EN LA MIGRAÑA
M. Isabel Serrano Robles, Mónica Hernández Martínez, Beatriz Beseler Soto, M. Rosario Domingo Jiménez, Alberto Puche Mira, Carlos Casas Fernández y Trinidad Rodríguez Costa
Hospital Universitario Virgen de la Arrixaca, El Palmar y Hospital Infantil "Marina Alta", Denia.
Objetivo:Valoración prospectiva de parámetros clínicos y respuesta al tratamiento en 69 casos de migraña infantil.
Material y métodos:Se recogen 69 casos de migraña infantil, durante 15 meses, según los criterios de la International Headache Society (I H S), en una base de datos de 36 campos referidos a tipo de migraña, sexo, edad, características clínicas, tratamiento del episodio agudo y profiláctico. Se ensaya una pauta de tratamiento analgésico de 24 horas de duración desde el inicio de la cefalea y se recogen los resultados.
Resultados:Correspondían a migraña sin aura 55 casos, con aura 13 y complicada 1. La incidencia de varones y mujeres por encima de 5 años fue similar, y por debajo de los 5 años predominaron los varones. La localización más frecuente ha sido frontal, con incidencia similar de la modalidad pulsátil y opresiva. La sintomatología digestiva se ha presentado en un 50 % de los casos, la fono y fotofobia en más de un 80 %. En la mitad de los niños no se ha identificado un desencadenante específico.
De todos los casos que ya habían sido tratados con analgesia, 70 % no habían recibido nunca una dosificación adecuada. La pauta de tratamiento analgésico de 24 horas de duración, conduce a una mejoría importante en el 76 % de los casos.
Conclusiones:Dificultad para el establecimiento rígido de criterios clínicos en la migraña infantil.
La analgesia inicial incumple la dosificación adecuada en gran número de casos.
La pauta analgésica de 24 horas, puede disminuir la necesidad de profilaxis.
P393ATROFIA MUSCULAR ESPINAL: NUESTRA EXPERIENCIA EN 20 AÑOS (1983-2002)
Rosa M. Merlos Madolell, Leticia Vila Sexto, Fernando Oliver Jiménez, Pedro Barbero Aguirre, Noemí Alentado Morell, Ana Cano, Raquel Escrig Fernández y Rafael Gómez
Hospital Universitario La Fe, Valencia.
Las Atrofias Musculares Espinales (AME), son un grupo de enfermedades familiares caracterizadas por degeneración de las células del asta anterior medular. Se describen tres variantes según edad de comienzo y velocidad de progresión: Tipo I (E. de Werdnig-Hoffman); Tipo II (E. de Dubowitz); Tipo III (E. de Kugelberg-Welander).
Objetivos:Estudiar los casos de AME diagnosticados en nuestro hospital en los últimos 20 años, describiendo antecedentes personales y familiares, edad de inicio, síntomas y signos clínicos, resultado de exploraciones complementarias y evolución de la enfermedad.
Material y métodos:Revisión de historias clínicas de pacientes diagnosticados de AME tipo I (n = 10) y tipo II (n = 11).
Resultados:AME tipo I: 5 pacientes eran varones y 5 mujeres; en 50 % casos había antecedente familiar de enfermedad neuromuscular; edad media de inicio de síntomas 37 ± 42días, predominando hipotonía (90 % de los pacientes) y dificultad respiratoria (60 %); a exploración física destacaban hipotonía (100 %), arreflexia (90 %) y fasciculaciones linguales (70 %); hemograma, bioquímica y estudio metabólico normales; electromiograma (EMG) compatible en 100 %; biopsia muscular realizada en 7 pacientes y estudio genético en 8 (delección de exones 7 y 8, en 40 %); edad media de fallecimiento de 26,8 ± 43,2 meses.
AME tipo II: 5 eran varones y 6 mujeres; 45 % casos había antecedente familiar de enfermedad neuromuscular; edad media de inicio 271 ± 181 días; predominando hipotonía (82 %); a la exploración física, hipotonía (100 %), arreflexia (72 %) y ausencia de sedestación y sostén cefálico (81,8 %); hemograma, bioquímica y estudio metabólico normal; EMG compatible en 100 %; biopsia muscular realizada en 7 pacientes y estudio genético en 6 (delección de exones 7 y 8, en 83 %).
Conclusiones:Ante un niño con hipotonía y arreflexia debemos considerar la posibilidad de AME. Aproximadamente la mitad de los pacientes presentaba antecedentes familiares de enfermedad neuromuscular. La realización de EMG y de biopsia muscular ayudan al diagnóstico, aunque resulta necesario el estudio genético, incluídos familiares de primer grado (riesgo de transmisión).
P394CONVULSIONES NEONATALES. REVISION 1997-2001
Susana Elena Zeballos Sarrato, Ana López, Kay Boris Brandstrup Azuero, Carmen Fernández, M. del Mar Guerrero Soler, Mar Roncero, Patricia Aparicio, Pedro Castro, Dorotea Blanco Bravo y Vicente Pérez Sheriff
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción:Las convulsiones neonatales (CN) constituyen una expresión clínica de disfunción o daño cerebral frecuente en el recien nacido (RN). El objetivo es valorar la incidencia de CN, la evolución de los pacientes y la asociación con posibles factores de riesgo (FR).
Material y métodos:55 RN (1,3 % del total de ingresos en el servicio de Neonatología) presentaron convulsiones clínicas en el período neonatal entre 1997-2001. De estos, se han seguido 40 niños en la consulta de Neuropediatria. Hemos realizado un estudio retrospectivo analizando las siguientes variables:edad gestacional, FR pre y perinatales, edad de presentación de la convulsión, tipo de convulsión, EEG, pruebas de neutoimagen, explación neurológica al alta de neonatología, tratamiento y evolución posterior.
Resultados:Lo más frecuente es presentar distintos tipos de crisis dentro de un mismo episodio convulsivo. La incidencia de crisis clínicas focales y generalizadas es muy parecida. Las CN son más frecuentes en RN a término (estadísticamente significativo). La mayoría de los pacientes tienen FR pre/perinatales. El 90 % tienen alteraciones en el EEG y neuroimagen. Las convulsiones en las primeras horas de vida se asocian a peor pronóstico. La causa más frecuente es la encefalopatía hipóxico isquémica (EHI). Hay una asociación estadísticamente significativa entre la exploración neurológica al alta del servicio de Neonatología y la evolución posterior. El 92,5 % de los neonatos han recibido tratamiento (Fenobarbital). En un 48,5 % se retiró antes del alta. El tiempo medio de tratamiento ha sido de 6 meses. Sólo 3 pacientes continuan con tratamiento en el momento actual.
Conclusiones:1. Encontramos una escasa incidencia de CN en comparación con otras series.2. Ser pretérmino no es un FR por si mismo para presentar crisis convulsivas.3. La causa más importante es la EHI.4. El factor pronóstico más importante es la exploración neurológica al alta del servicio de Neonatología, no la realizada inicialmente tras el episodio convulsivo.5. Tienen peor pronóstico las convulsiones precoces (en las primeras horas de vida).6. Las convulsiones neonatales por si mismas no son un factor de mal pronóstico para la evolución a largo plazo de estos pacientes.
P395HEMIPLEJIA ALTERNANTE DEL LACTANTE
Rafael Maese Heredia, Lourdes Escudero Ruiz de Lacanal, Teodoro José Martínez Arán, Rafael Vera Medialdea, Carmen Vida Fernández, María González López, Jacinto Martínez Antón y Antonio Jurado Ortiz
Hospital Materno Infantil, Málaga.
Introducción:La hemiplejía alternante de la infancia es una entidad rara y de fisiopatología poco clara, al parecer no epiléptica, caracterizada por episodios transitorios y repetitivos de hemiplejia que afectan alternativamente a un hemicuerpo. Suele aparecer en menores de 18 meses. Puede asociarse a una alteración mitocondrial.
Caso clínico:Lactante de 1,16/30 con clínica de movimientos clónicos oculares izquierdos acompañados de contractura en extensión de miembro superior homolateral y en flexión del contralateral, apareciendo de forma episódica (10-20 al día) e intensidad creciente, de 1 minuto de duración y cese espontáneo. No pérdida de reactividad. Exploración con leve hipotonía axial y tendencia a la lateralización derecha de la cabeza. Bioquímica general, perfil metabólico y EEG normales. Discreta atrofia cortical en el TC craneal.
Catalogado de epilepsia parcial simple y compleja secundariamente generalizada, se logra remisión durante 2 meses con VPA. Desde los 4,3/30 a los 8,15/30 sufre episodios similares, afectando de forma cambiante a un hemicuerpo. Aparecen brotes de ondas lentas témporo-parietales derechas. Pruebas de imagen sin cambios. Se asocia VGB y CBZ, lográndose control clínico con CBZ y PRM. Se aprecia discreto retraso psicomotor. A los 11 meses reaparecen las crisis sin respuesta a pautas con CBZ, PRM, LTG y CLB. Estado de mal parcial a los 15 meses. Se evidencia retraso madurativo y hemiparesia derecha adquirida.
A los 18 meses se detecta elevación de cistationina en orina y EEG con lentificación de la actividad centrotemporal derecha. Control con CZP, TPM y LTG. En estudio mitocondrial se evidencia aumento de Cistationina en sangre y orina, elevación de homocisteína sérica, biopsia muscular normal y genotipo HTHFR, heterocigoto 677T, indicadores de déficit en los complejos I y IV de la cadena mitocondrial.
Conclusiones:La hemiplejia alternante del lactante debe considerarse por su evolución y pronóstico en el diagnóstico diferencial de las epilepsias de inicio precoz refractarias al tratamiento, con las que se confunde inicialmente, y de las enfermedades vasculares o mitocondriales. Es fundamental una adecuada anamnesis y exploración, al no existir pruebas complementarias definitivas. Actualmente no existe tratamiento curativo, empleándose la flunaricina en el manejo sintomático.
P396ANISOTROPIA COMO CONSECUENCIA DE UN TRAUMATISMO PERIOCULAR
Jesús Palencia Garrido-Lestache, Belén Díaz Orro, Andrea Sanz y J. Navalón
Hospital San Francisco de Asís, Madrid y Hospital Infantil San Rafael, Madrid.
Los traumatismos craneales en región ocular, orbitaria y periorbitaria, son una causa frecuente de consulta en la practica clínica. Por cada 100.000 miembros de la población pediátrica, sufren traumatismo ocular 15,2 niños. Siendo la mayoría de ellos predecibles y evitables.
Las causas más frecuentes de traumatismos son golpes o caidas en un 37 %, deportes o juegos en el 27 %, quemaduras en el 9 %, accidentes de coche en el 11 % y armas de fuego en el 4 %.
Caso clínico:Lactante de 12 meses de edad sin antecedentes de interés, que presenta un traumatismo de intensidad media en región frontal y periorbitaria del ojo izquierdo, sin repercusión neurológica. Localmente presenta un hematoma periocular que interesa a parpados y tejido celular subcutáneo, sin afectar al resto de estructuras oculares desde el punto de vista anatómico y funcional.
Al mes del traumatismo presenta como secuela, una ptosis palpebral superior del lado izquierdo, etiquetada de paresia postraumática.
A los 8 meses del traumatismo es remitida al oftalmólogo infantil como consecuencia de persistir la ptosis, donde se objetiva una anisotropía, por lo que se realiza un T.A.C. de órbita apreciándose un callo de fractura en el techo de la órbita que comprime el músculo recto superior del ojo izdo, con desplazamiento del globo ocular hacia abajo.
Conclusiones:Llamamos la atención sobre la necesidad de realizar un seguimiento continuado de los traumatismos en región ocular y periocular y ante la más mínima sospecha de alguna secuela, remitir al niño al especialista en oftalmología infantil. Ya que la intervención precoz en dichas secuelas, previenen alteraciones visuales que pueden ser definitivas en el desarrollo de la visión del niño.
P397DOS CASOS GRAVES DE ENCEFALOMIELITIS DISEMINADA AGUDA
José M. Martos Tello, M. Rosario Domingo Jiménez, Encarnación Bastida Sánchez, M. Isabel Serrano Robles, David Gil Ortega, Pedro Torres Tortosa y Trinidad Rodríguez Costa
Hospital Universitario Virgen de la Arrixaca, El Palmar.
Introducción:La encefalomielitis diseminada aguda (EMAD) es una enfermedad inflamatoria desmielinizante del SNC, con variabilidad clínica y características lesiones en la neuroimagen. Presentamos dos casos graves de EMAD y su analisis.
Casos clínicos:
Caso 1:Lactante de 14 meses, con antecedentes de gastroenteritis 8 días antes que presenta debilidad de inicio brusco de un mes de evolución con leve tendencia a la mejoría espontánea. En otro hospital se realizó: TC craneal: normal, y LCR: hiperproteinorraquia y pleocitosis leves. Exploración física: disminución de la actividad espontánea, piramidalismo generalizado. TC craneal: zonas de hipodensidad asimétrica. RM cerebral: múltiples lesiones diseminadas en la sustancia blanca periventricular. Se inicia tratamiento con dexametasona, obteniendo mejoría parcial, persistiendo como secuela grave tetraparesia espástica.
Caso 2:Escolar 9 años, ingresa por vómitos, febrícula y estrabismo. Antecedentes patológicos: hipospadias intervenido, faringitis una semana antes. Exploración física: somnolencia y signos meníngeos. Exámenes complementarios: leucocitosis, LCR: pleocitosis e hiperproteinorraquía leves. TC craneal, normal. Evolución con crisis convulsivas y coma, precisando ventilación mecánica. Posteriormente, arreflexia y oftalmoplejia, se inicia pauta de inmunuglobulinas y se realiza EMG y neurografía, normales. RM cerebral: múltiples lesiones diseminadas en sustancia blanca de hemisferios cerebrales, tronco y tálamos. Se inicia tratamiento con metilprednisolona, con gran mejoría clínica. Sin secuelas.
Discusión:La EMAD es una enfermedad autoinmune que, generalmente, se desencadena tras una infección vírica o vacunación. La presentación clínica es variable aunque predominan los síntomas motores y alteraciones de la conciencia. El diagnostico lo establece los hallazgos en la RM, característicos. El pronóstico vital es bueno, aunque hay formas graves con secuelas posteriores. Se han relacionado las secuelas con la afectación del nervio óptico en el momento de la presentación. El tratamiento se realiza con metilprednisolona a altas dosis con muy buena respuesta. Se preconizan la utilización de inmunoglobulinas iv junto a metilprednisolona en las formas graves de la enfermedad.
P398 ANOMALIAS DEL CROMOSOMA 15: SINDROME DE ANGELMAN Y DE PRADER WILLI
Leonor Bardallo Cruzado, M. Dolores Lluch Fernández, Inmaculada Ramos Sánchez, Carmen Garnacho Montero y Casto Estefanía Gallardo
Hospital Universitario Virgen Macarena, Sevilla.
Antecedentes y objetivos:El síndrome de Angelman (SA) y de Prader Willi (SPW) se asocian genéticamente a anomalías del cromosoma 15 de origen materno o paterno respectivamente. Sin embargo, las manifestaciones clínicas son distintas. El objetivo es conocer las características clínicas y el mecanismo de producción genético en los niños con ambos síndromes en seguimiento en nuestro servicio.
Métodos:Estudio retrospectivo y descriptivo mediante revisión de las historias clínicas de los pacientes con SA y SPW. Analizamos los criterios diagnósticos descritos por Williams y col para el SA y los publicados por Holm para el SPW.
Resultados:Hemos revisado 8 pacientes, 2 diagnosticados de SA y 6 de SPW.
SA: dos niñas en seguimiento en la unidad de maduración una de ellas desde el nacimiento por pretérmino y la otra desde los 14 m por retraso psicomotor. La edad de diagnóstico fue a los 32 m y a los 46m respectivamente. Presentan retraso madurativo (gatean a los 18m, deambulan con apoyo a los 2,5 años, bisílabos), marcha atáxica, risa frecuente, crisis convulsivas generalizadas (una desde las 4 horas de vida y la otra desde los 2 años) controladas con valproato. En los EEGs aparece actividad paroxística y brotes de ondas lentas. En ambas hay hiperactividad, trastornos del sueño, piel clara y estrabismo divergente. La alteración genética fue una delección en el cromosoma 15 (q11-q13) de origen materno.
SPW: tres varones y tres niñas. Tres se diagnosticaron en el período neonatal, 2 alrededor de los 2 años estudiando el retraso psicomotor y en una niña se confirmó el diagnóstico a los 13 años. Presentación fetal anómala y actividad fetal reducida (3 casos). Hipotonía presente en todos y en 4 de ellos succión débil. Retraso psicomotor (sedestación 12,2 m; deambulación 31,2 m). CD medio 75. La mitad desarrollaron hiperfagia, 5 casos obesidad central y la edad media en la que el peso supera el p 90 es 26 m. Ninguno alcanza estatura superior al p 25. Apneas del sueño y estrabismo (3 pacientes). En los 3 varones hay criptorquidia bilateral. 4 en protocolo de tratamiento con GH (sólo lo ha iniciado uno). El mecanismo genético en 5 casos fue delección en el cromosoma 15 (q11-q13) de origen paterno y en 1 traslocación (15-16).
Conclusiones: 1.El mecanismo genético más frecuente es la delección.2. En el SA hay un retraso mental más severo y problemas neurológicos más graves.3. En el SPW predomina la hipotonía neonatal, obesidad, hipogonadismo y el retraso psicomotor.
NEUMOLOGIA
P399
MARCADORES DE LA INFLAMACION EN NIÑOS ASMATICOS TRATADOS CON CORTICOIDES INHALADOS
Bruno Nievas Soriano, Santiago Rueda Esteban, Luis Arruza Gómez, Florencio Balboa de Paz, Begoña Losada Pinedo, Beatriz Agúndez Reigosa y M. Gloria García Hernández
Hospital Clínico San Carlos, Madrid y Hospital 12 de Octubre, Madrid.
Objetivos:Estudiar la evolución clínica, la función pulmonar medida por espirometría y los valores de ONE, proteína catiónica eosinófila (ECP), IgE y eosinófilos en una población de niños con asma leve persistente de nuevo diagnóstico, de edades comprendidas entre los 6 y los 14 años.
Material y métodos:Estudio prospectivo en el que se analizan distintos parámetros de inflamación bronquial en un grupo de 21 pacientes, antes y después de recibir tratamiento con fluticasona inhalada durante 6 semanas. Dichos parámetros son: A) clínica, evaluada mediante encuesta: episodios de sibilancias y síntomas nocturnos a la semana; aparición de dificultad respiratoria con ejercicio; número de semanas que transcurrían entre las exacerbaciones asmáticas. B)función pulmonar, evaluada mediante espirometría. C) cifras de ONE, según las recomendaciones de la American Toracic Society y mediante la técnica en T de exhalación lenta contra resistencia (ELCR), con un analizador por quimioluminiscencia. D)niveles de ECP, mediante técnica FEIA Pharmacia.
Resultados:Se constata: A) mejoría clínicade los pacientes: disminuyen los episodios de sibilancias de 2,68 días a la semana antes del tratamiento a 0,25 tras el tratamiento (p < 0,012); los síntomas nocturnos disminuyen de 2,45 días a la semana antes del tratamiento, a 0,13 días (p < 0,039); de presentar clínica con ejercicio moderado a requerir ejercicio intenso para presentarla (p < 0,011); de sufrir una exacerbación asmática cada 3,64 semanas antes del tratamiento, a una cada 7,94 semanas (p < 0,001). B) en laspruebas de función pulmonar mejoraron todos los parámetros (FVC, FEV1 y MMEF) tras del tratamiento (p < 0,006; p < 0,016; p < 0,011, respectivamente). C) lascifras de ONE pasaron de 22,47 ppb a 7,95 ppb tras el tratamiento (p < 0,0001). D) la medición de ECPpasó de 48 μg/l antes del tratamiento, a 22,47 μg/l (p < 0,025).
Conclusiones:La medición de la concentración de ONE parece que es un medio para detectar y monitorizar la inflamación en el tracto respiratorio inferior, y así poder evaluar la eficacia de tratamientos antiinflamatorios, como la fluticasona. La medición de los niveles sanguíneos de ECP podría ser un método complementario de monitorización y control del paciente asmático, aunque es un método invasivo y diferido en el tiempo, que no da un valor inmediato.
P400TRATAMIENTO ANTIBIOTICO EN EPISODIOS AGUDOS DE ASMA
M. Rosario Benavides Román, Máximo Martínez Gómez, Ana M. Martínez-Cañavate Burgos y Julia C. Martínez Bernal
Hospital Virgen de las Nieves, Granada.
Objetivos:Analizar en un grupo de pacientes con episodio asmático agudo, la indicación o necesidad de asociar antibióticos al tratamiento protocolizado del asma, considerando por la clínica y pruebas complementarias, una posible relación entre la crisis asmática y el proceso infeccioso.
Metodología:Se realiza estudio abierto de 37 pacientes, 23 mujeres y 14 hombres, con edades comprendidas entre 4 y 14 años y que consultan por episodio asmático agudo, presentando también sintomatología compatible con proceso infeccioso. Se consideran síntomas y signos primarios: fiebre, tos, sibilantes, disfonía y disnea; secundarios: estertores húmedos, hiperhemia e hiperplasia faringe amigdalar, laringitis, dolor torácico y mucosidad nasal; se valoran dos síntomas primarios o uno primario y dos secundarios. Se realizan hemograma, velocidad de sedimentación globular (VSG), serología para Mycoplasma Pneumoniae (MP) y Chlamydia Pneumoniae (CP), radiografía de tórax y senos, estudio de atopía en no diagnosticados y Test funcional respiratorio en pacientes colaboradores.
Resultados:Considerando que en algunos pacientes se aisla más de un patógeno, el 91,9 % tenían serología positiva a MP, 19 % a CP y 13,5 % a otros patógenos (E. Barr, rickettsias). Los síntomas predominantes eran tos (97,2 %), sibilantes (72,2 %), disnea (48,6 %) y fiebre (43,3 %); alteraciones radiográficas (64,9 %); alteraciones de hemograma (78,3 %) y VSG (56,5 %). Tenían diagnóstico previo de asma el 62,1 % de los que realizaban tratamiento de mantenimiento el 45,9 %. Se realizó estudio funcional en 21 pacientes, de los que un 67,7 % tenían alteraciones valorables. Se instauró tratamiento antibiótico (macrólidos) en 64,8 % de los pacientes, en períodos que oscilaron entre 2 y 4 semanas (estos últimos con CP positivo).
Conclusiones:En un porcentaje apreciable hay que considerar el proceso infeccioso como desencadenante de un proceso asmático; aunque no es aconsejable utilizar antibioterapia en el tratamiento de fondo del asma, si conviene analizar dicho tratamiento en pacientes con asma y síntomas de infección; hay que considerar que la serología es bastante sensible para el diagnóstica de MP, no así para CP, en la que se precisarían otra técnicas diagnósticas complementarias (cultivo, PCR); al no ser la serología una prueba de diagnóstico inmediato, la sospecha clínica de infección obliga en ocasiones a realizar un tratamiento empírico.
P401PRIMEIRO ESTUDO PROSPECTIVO EM PORTUGAL SOBRE BRONQUIOLITE POR VIRUS SINCICIAL RESPIRATORIO: EPIDEMIOLOGIA, CLINICA E FACTORES DE RISCO
Pedro Flores, Eduarda Neves Sousa, Helena Rebelo de Andrade, Maria João Leiria, Fernando Noronha y José Palminha
Hospital de S. Francisco Xavier, Lisboa (Portugal) y Instituto Nacional de Saúde, Lisboa (Portugal).
Objectivo:Analisar o perfil clínico e epidemiológico das crianças com bronquiolite por vírus sincicial respiratório (VSR), observados num serviço de urgência pediátrica de um Hospital Central de Lisboa; determinar os factores clínicos, epidemiológicos e laboratoriais que se relacionem com uma maior gravidade da doença.
Doentes e métodos:Estudo prospectivo que incluíu todas as crianças internadas no Serviço com idade inferior a 36 meses e o diagnóstico de bronquiolite aguda, durante dois invernos consecutivos (Novembro a Março 2000/01 e 2001/02), bem como uma amostra equivalente, aleatória, de crianças não internadas. A detecção laboratorial do VSR foi efectuada por polymerase chain reaction (PCR).
Resultados:225 doentes foram incluídos no estudo. A mediana das idades foi de 5 meses (min: 0, max: 34) e a razão masculino/feminino de 1,6:1. O VSR foi isolado em 60,9 % dos casos, predominantemente no grupo dos doentes hospitalizados. A razão sub-tipo A:B foi de 7,4:1, semelhante em ambas as épocas e com gravidade clínica sobreponível. Os doentes VSR positivos eram de idade inferior (média: 6,0 ± 5,4 vs 8,4 ± 7,1 meses, p = 0,007), tiveram formas mais graves de bronquiolite e menos alterações do leucograma (20,7 vs 51,4 %, p = 0,001). Entre os doentes com bronquiolite por VSR, os scores de gravidade mais elevados ocorreram no primeiro episódio de broncospasmo (p = 0,015), em doentes residentes em agregados familiares alargados (p = 0,007), que frequentavam o infantário (p = 0,011) e em ex-prematuros com idade gestacional inferior a 36 semanas (p = 0,009).
Conclusões:A bronquiolite aguda por VSR é fundamentalmente uma doença do primeiro semestre de vida, principal responsável pelo primeiro episódio de broncospasmo nos lactentes. Os ex-prematuros, bem como as crianças que frequentam o infantário ou residem em meios familiares alargados têm formas significativamente mais graves de doença. Este primeiro estudo prospectivo realizado em Portugal sobre a epidemiologia do VSR permite estabelecer uma linha de base para programas de vigilância epidemiológica no País. Um estudo multicêntrico futuro será desejável, de forma a delinear orientações quando à profilaxia e à terapêutica da infecção VSR em Portugal.
P402UTILIDAD DE LA FIBROBRONCOSCOPIA EN MENORES DE 1 AÑO
Begoña Pérez-Moneo Agapito, Carmen Martínez Carrasco, M. Isabel Barrio Gómez de Agüero y M. del Carmen Antelo Landeira
Hospital Universitario La Paz, Madrid.
Introducción. La fibrobroncoscopia (FB) es una técnica útil en la exploración de la vía aérea del niño, utilizada cada vez con mayor frecuencia en las unidades de Neumología Infantil.
Objetivos:Recoger la experiencia de nuestra Unidad en niños menores de 1 año.
Pacientes y métodos:Se revisaron las FB realizadas en niños menores de 1 año en los últimos 11 años. De un total de 712, 109 correspondían a este grupo de pacientes. Se emplearon dos tipos de fibrobroncoscopios (Olympus de 3,6 y de 2,2 mm de diámetro externo), realizándose todas las exploraciones bajo sedación y monitorización en una unidad de reanimación o cuidados intensivos. 6 pacientes fueron menores de 1 mes (5,5 %); 65 (59,6 %) niñas y 44 (40,4 %) niños.
Resultados:72 FB (66,1 %) se realizaron durante los meses de invierno y primavera mientras que 37 (33,9 %) en los restantes; el fibrobroncoscopio neonatal se usó en 16 casos (14,7 %). Las indicaciones fueron: exploración para estudio diagnóstico de la vía aérea en 36 casos (33 %), tratamiento de atelectasias en 31 (28,4 %), estridor en 19 (17,4 %), neumopatía intersticial en 9 (8,3 %), dificultad para la extubación en 8 (7,3 %), cuerpo extraño en 3 (2,8 %) e intubación difícil en 3 (2,8 %). Los hallazgos diagnósticos fueron: alguna malformación de vía aérea en 27 (24,8 %), broncorrea severa en 12 (11 %), estenosis de vía aérea inferior en 11 (10,1 %), inflamación en 9 (8,3 %), alteraciones de cuerdas vocales en 4 (3,7 %), compresión extrínseca en 4 (3,7 %), malacia en 4(3,7 %), granuloma traqueal en 3 (2,8 %), en el 30 % restante la anatomía de la vía aérea fue normal. 64 FB (58,7 %) fueron realizadas en pacientes con patología aguda (UCIN y UCIP). Las complicaciones fueron: desaturaciones transitorias 14,7 %, tos 1,8 %, estridor 2,8 % y en un caso espasmo de glotis, con buena tolerancia en un 83,2 %. Se realizó lavado bronco-alveolar en 13,8 % y toma de aspirado bronquial en 34,9 % delos casos.
Conclusiones:1. Un 30 % de las FB tenían indicación terapéutica, por lo que al hablar de FB no debe considerarse solo su faceta exploratorio-diagnóstica.2. En general es una técnica bien tolerada, que podremos considerar para la evaluación de nuestros pacientes menores de un año. 3. Es una técnica adecuada para la valoración del niño agudo y grave.4. Su uso se está extendiendo en los últimos años, tanto para la exploración de vía aérea, obtención de muestras y tratamiento.
P403NEUMOTORAX IZQUIERDO MASIVO Y COLONIZACION POR BURKHOLDERIA CEPACIA EN FIBROSIS QUISTICA
Florencio Jiménez Fernández, Marta Ruiz Jiménez, M.ª Elisa Corrales del Río, Enrique Salcedo Lobato, Enrique Alberto Criado Vega y Ernesto Antonio Sáez Pérez
Hospital Universitario de Getafe.
Antecedentes y objetivos:La Fibrosis Quística es una de las enfermedades genéticas más frecuentes en la población caucásica con una importante morbimortalidad que está relacionada con la afectación pulmonar y sus complicaciones, responsables del 95 % de los fallecimientos de los pacientes que la padecen, la presencia de neumotórax masivo en el período infantil es rara, aunque aumenta con la edad, llegando a padecer algún episodio el 20 % de los pacientes en la edad adulta, así mismo se considera un hecho emergente la colonización por Burkholderia cepacia.
Material y métodos:Paciente de 6 años y 8 meses, con fibrosis quística, colonizado por Burkholderia cepacia, en tratamiento con Tobramicina inhalada y alfa-Dornasa, con FVC: 1,02 (77,8 %), FEV1: 0,64 (55,7 %), PEF: 1,42 L/S (77,8 %), con un escore de Brasfield inicial de 13, y de Schawchman: 58, con un escore de Crispin y Norman:13, con colonización por Burkholderia cepacia (+ + + +) actualmente, que recibió terapia i.v. con Meropenem más Ceftazidima durante un mes con disminución transitoria de la colonización. En el 2001 presentó neumotórax masivo izquierdo resuelto tras colocación de drenaje precisando estancia en la unidad de cuidados intensivos pediátricos, recibiendo tratamiento con ceftazidima más vancomicina durante dicho ingreso.
Conclusiones:A pesar del avance en el manejo y tratamiento de la fibrosis quística, y de las mejoras conseguidas en el tratamiento antibiótico de la Pseudomona Aeruginosa, la aparición de nuevos gérmenes emergentes, como Burkholderia cepacia, Stenotrophomona Maltophilia, y Achromobacter spp. Constituyen un nuevo reto terapéutico en la prevención del deterioro de los pacientes con fibrosis quística.
P404RENTABILIDAD DE LAS TÉCNICAS ENDOSCOPICAS EN LA FIBROSIS QUISTICA
Carmen Serrano Recio, Leticia Olivares Sánchez, M. Esther Rojas Gracia, Isabel M. Luque Gómez, Estela M. Pérez Ruiz, Fco. Javier Pérez Frías y Antonio Jurado Ortiz
Hospital General Carlos Haya, Málaga.
Antecedentes y objetivo:La Fibrosis Quística (FQ) es la enfermedad letal hereditaria más frecuente en la raza blanca (1/2500 recién nacidos). Su morbimortalidad depende de la afectación pulmonar. Para el estudio de los problemas pulmonares se utiliza el fibrobroncoscopio (Fb) flexible bajo sedación. El objetivo es analizar la rentabilidad de la fibrobroncoscopia (FB) en la FQ.
Métodos:Pacientes: de 1990 al 2002 se realizaron en nuestro servicio 650 FB, 19 de ellas se realizaron a 12 niños con FQ, de un total de 120 niños con FQ (60 % < 16 años). Instrumentación: Fb de 3,5 mm de diámetro externo y 1,2 mm de canal de trabajo; en los pacientes > 10 años Fb de 4,9 mm/2,2 mm.
Resultados:Edades: 3-13 años. Sexos: varón/mujer 2/1. Sedación: Diacepam + Ketamina en 15/19, Midazolam y Fentanilo 4/19. Acceso: en 10 pacientes fue nasotraqueal, y en 2 a través de tubo endotraqueal. FB terapéuticas: las 19 FB tuvieron fines terapéuticos, en 9 ocasiones para resolver atelectasias, en 1 ocasión para tratamiento de hemoptisis. Se instilaron fármacos en 15 ocasiones (11 mucolíticos, 4 DNAasa). FB diagnósticas: 10/19. Se obtuvo nueva información en 6/10 lavados broncoalveolares (LBA) (60 %): nuevos patógenos y hallazgos en el análisis citológico. Valoración de la mejoría: radiológica: 5/19, clínica: 3/19, exploratoria: 2/19, espirométrica: 2/19, saturación de O2: 2/19.
Discusión:Nuestros hallazgos coinciden con otros estudios que sugieren que la FB ofrece beneficios a corto plazo como procedimiento terapéutico, pero es útil para obtener información en circunstancias en las que existen dudas acerca del patógeno responsable de los síntomas respiratorios. Otra posibilidad diagnóstica del LBA es la presencia de lípidos en los macrófagos alveolares, valorado como indicador de aspiración.
Conclusiones:En manos experimentadas tiene bajos riesgos de complicaciones, y puede ser realizada bajo sedación como un procedimiento de uso habitual. La información obtenida del LBA puede significar un avance en el cuidado de los pacientes, por identificar los microorganismos responsables y tomarse medidas específicas en cada caso; como ha ocurrido en 6 de nuestros casos. Todos los niños con FQ deberían tener acceso a una Unidad de FQ donde estos procedimientos sean posibles.
P405EXPERIENCIA CON VENTILACION MECANICA NO INVASIVA EN UNA UNIDAD DE NEUMOLOGIA INFANTIL
Carmen Martínez Carrasco, M. Isabel Barrio Gómez de Agüero, Begoña Pérez-Moneo Agapito y M. del Carmen Antelo Landeira
Hospital Universitario La Paz, Madrid.
Antecedentes y objetivo:El empleo de la ventilación mecánica no invasiva (VMNI) para tratamiento del niño con insuficiencia respiratoria se ha extendido enormemente en la última década, tanto a nivel de UCI como a nivel domiciliario. Presentamos nuestra experiencia en esta técnica.
Métodos:En los últimos 11 años hemos empleado VMNI en 41 pacientes con edades comprendidas entre 3 meses y 19 años. Se han utilizado sistemas de BIPAP así como ventiladores volumétricos. La interfase ha consistido en mascarillas nasales o faciales.
Resultados:Han recibido VMNI pacientes con patología obstructiva (Fibrosis Quística): 14, apneas obstructivas del sueño (SAOS): 2, síndromes de hipoventilación central (S. de Arnold-Chiari, disgenesia cerebral, acondroplasia, S. de hipoventilación-obesidad): 5, enfermedades neuromusculares:18, alteraciones de la caja torácica: 2. La duración del tratamiento va de 1 mes a 7 años. No ha habido complicaciones importantes debidas a la VMNI.
Conclusiones:1. La VMNI es una modalidad terapéutica muy útil en el manejo de niños con insuficiencia respiratoria crónica secundaria a distintas patologías.2. Los neumólogos pediatras deben estar familiarizados con esta técnica para sentar su indicación y realizar el seguimiento de la misma.
P406BRONQUIOLITIS OBLITERANTE CON NEUMONIA ORGANIZATIVA DE ETIOLOGIA IDIOPATICA, DIAGNOSTICO ANATOMOPATOLOGICO DE INFILTRADO PULMONAR PERSISTENTE
Alejandro López Escobar, Ana Siles Sánchez-Manjavacas, Luis Echeverría Zudaire, Teresa Bracamonte Bermejo, M. Luz García García y José María de Cea Crespo
Hospital Severo Ochoa, Leganés.
La Bronquiolitis Obliterante con Neumonía Organizativa (BONO) es un hallazgo patológico infrecuente a la vez que inespecífico, relacionado con el daño sufrido por el pulmón por una variedad de agentes conocidos y desconocidos.
Caso clínico:Varón de 10 años remitido a la consulta de Neumología Infantil en Julio de 1999 por infiltrado pulmonar persistente en lóbulo superior izquierdo (LSI) a raíz de un control radiográfico por neumonía diagnosticada en Marzo de 1999 y con buena evolución clínica con tratamiento antibiótico ambulatorio con eritromicina oral.
La exploración al inicio del estudio era completamente normal. Se realizó analítica completa (bioquímica y hemograma), serología (virus respiratorios, hidatidosis y tuberculosis), parámetros de autoinmunidad, Mantoux y un Ionotest, siendo todos los resultados normales. La espirometría y la phmetría fueron también normales. En las pruebas cutáneas se detectó alergia a pólenes. En Agosto de 1999 se le realizó un TAC sin contraste en el que se objetivó condensación en LSI con adenopatías hiliares y pequeñas adenopatías mediastínicas. En la misma fecha se le practicó una broncoscopia en la que se apreció una estenosis bronquial secundaria a una adenopatía. El lavado broncoalveolar y las pruebas microbiológicas fueron normales. Finalmente se obtuvo una biopsia pulmonar en Septiembre de 1999 mediante toracoscopia. El informe anatomopatológico fue diagnóstico de BONO. Al haber permanecido asintomático durante toda la evolución del cuadro se decidió no iniciar tratamiento corticoideo y hasta la actualidad ha continuado asintomático, con controles espirométricos normales y controles radiológicos, incluido un TAC con contraste, en los que se objetiva disminución progresiva del infiltrado.
Comentarios:La entidad BONO es un hallazgo patológico infrecuente en la edad pediátrica y debe incluirse dentro del diagnóstico diferencial del infiltrado pulmonar persistente. Tras su diagnóstico debe investigarse la etiología de esta entidad. En cuanto a su manejo, clásicamente se ha descrito la mejoría y resolución de los infiltrados con terapia corticoidea, aunque puede haber casos como el que presentamos, con tendencia a la resolución espontánea.
P407LACTANTE CON INFECCION ACTIVA POR CITOMEGALOVIRUS Y FIEBRE PERSISTENTE
Matilde Somoza Martín, Sonia Marcos Alonso, Carmen Curros Novo, J. Ángel Porto Arceo, Elena V. Rodrigo Sáez y Manuel Castro Gago
Hospital Clínico Universitario. Complejo Hospitalario Universitario, Santiago de Compostela.
Objetivo:Se presenta la observación clínica de un lactante de 4 meses que de forma secuencial y en el plazo de dos meses padece una infección activa por CMV y una tuberculosis pulmonar.
Caso clínico:Lactante que como antecedente consta al mes de vida un ingreso por Neumonía y Bronquiolitis. Nuevo ingreso a los 4 meses por fiebre demostrándose una infección activa por CMV cursando en este momento con proceso neumónico, PPD negativo, cultivos negativos, BAAR en aspirado gástrico (negativos), reactantes de fase aguda (PCR y Procalcitonina) positivas, VSG 35 mm. Buena respuesta al tratamiento. Al alta radigrafía de tórax: normal. Tres ingresos posteriores en un intervalo de 2 meses por fiebre elevada, sin foco evidente. Estudios de inmunidad, PPD, cultivos, radiografía de tórax no revelaron patología salvo una zona hipocaptante en polo superior del riñón derecho evidenciada en el DMSA, no encontrándose en ningún ingreso urocultivos positivos. Como dato exploratorio persistía una discreta taquipnea con sibilancias aisladas que se interpretaron como una hiperreactividad leve. Por este motivo y la persistencia de la fiebre, se le realizaron repetidos aspirados gástricos y bronquiales para identificación de BAAR y PCR. En un aspirado bronquial creció Mycobacteria tuberculosa. La TAC torácica demostró adenopatía paratraqueal derecha. Ante el diagnóstico de Tuberculosis pulmonar se inicio tratamiento con triple terapia.
Conclusiones:Ante un lactante con fiebre persistente que puede ser atribuible a su primer diagnóstico se debe de buscar otra etiología, entre ellas la Tuberculosis, si se suman como datos clínicos taquipnea y sibilancias aisladas a pesar de ser las pruebas básicas convencionales (PPD, radiografía de tórax, VSG y reactantes de fase aguda) negativas. La TAC torácica es clave en estos procesos en los que, como en nuestro caso, puso en evidencia la enfermedad.
P408SOMATOSTATINA COMO TRATAMIENTO DE QUILOTORAX PERSISTENTE EN PACIENTE PEDIATRICO AFECTO DE LEUCEMIA LINFOBLASTICA AGUDA DE CÉLULAS T
Margarita Cañellas Fuster, Sonia Yeste, Borja Osona Rodríguez de Torre, Mercedes Guibelalde del Castillo, Nieves Nieto del Rincón y Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca.
Introducción:El quilotórax (QT) se define como líquido pleural > 1.000 células, > 70 % linfocitos, triglicéridos > 100 mg/dl, proteínas > 20 g/dl, y cultivos negativos. Puede ser debido a traumatismos directos (65 %), trombosis o/y aumento de la presión venosa en el territorio de la vena cava superior (27 %) o congénito (8 %). El tratamiento convencional consiste en nutrición enteral especial, nutrición parenteral total,y si no remite pleurodesis u otros procedimientos quirúrgicos. Recientemente se ha descrito la somatostatina como una nueva alternativa terapéutica. Aunque su mecanismo final de acción es desconocido, se presume que actúa mediante vasoconstricción de los vasos linfáticos. Por ello es útil en el manejo de pacientes pediátricos que presentan quilotórax y no son susceptibles de otros tratamientos.
Caso clínico:Niño de 11 años diagnosticado de Leucemia Linfoblástic Aguda de Células T, cuya forma de presentación fue masa mediástinica, síndrome de vena cava superior y derrame pleural masivo. Desde su ingreso precisó colocación de drenaje torácico, con estudio del líquido pleural (LP) positivo para malignidad por estudio de citometría de flujo de las poblaciones linfocitarias (CD3:70 %, CD5:70 %, DR + CD3 +: 14 %, Gamma/Delta + CD3 +: 20 %, Tdt), con volúmenes máximos de 2 l/día. Se inició tratamiento quimioterápico según protocolo LLA-SHOP 99 para alto riesgo. A partir del día + 27 cambiaron las características del LP, adquiriendo un aspecto cremoso-blanquecino, aumentando niveles de triglicéridos hasta 225 mg/dl, con predominio de celularidad linfocitaria sin signos de malignidad. Se pautó tratamiento conservador con dieta exenta de grasa y nutrición parenteral sin evidenciar buena respuesta, mostrando el día + 38 en TAC torácico aumento importante de la efusión pleural. El día + 36 entró en remisión completa de su LLA. El día + 40 se decidió iniciar perfusión de somatostatina (dosis 3,5 μg/kg/h en incremento hasta 10 μg/kg/h) siendo la respuesta favorable a los 5 días de tratamiento, manteniéndola durante 10 días con resolución del QT. El tratamiento fue bien tolerado sin aparición de efectos secundarios
Conclusiones:El tratamiento con perfusión continua de somatostatina demostró ser efectivo en el tratamiento del QT persistente en pacientes en edad pediátrica, no tributarios de otros tratamientos, sin presentar efectos secundarios.
P409AGENESIA AISLADA DE ARTERIA PULMONAR DERECHA: HALLAZGO CASUAL EN LA ADOLESCENCIA
Juana Barja Tur, Mónica Rodríguez Fernández, J. Ignacio Herraiz Sarachaga, Ricardo Díez García, M. Luisa Herreros Fernández, Enrique Fanjul y Alfonso González Laguillo
Clínica Moncloa, Madrid.
Caso clínico:Escolar varón de 12 años que acude a Urgencias por fiebre y síntomas respiratorios de una semana de evolución, sin mejoria con tratamiento antibiótico.Antecedentes personales: 3 infecciones respiratorias con broncoespasmo, tratadas con broncodilatadores. No disnea ni hemoptisis. Exploración física:Por aparatos normal, salvo mínima hipoventilación basal derecha y dudosa asimetría torácica con leve disminución de hemitórax derecho.Exploraciones complementarias: Rx tórax: disminución de campo pulmonar derecho con desplazamiento mediastínico a la dcha, que no varía en espiración, e hipertrofia de arteria pulmonar izquierda. Sangre: hemograma, fórmula, recuento, PCR y gasometría normales. Mantoux.Evolución: Se ingresa con tratamiento antibiótico y broncodilatadores para estudio. Rápida mejoría clínica manteniéndose asintomático al 3.º día del ingreso.Exploraciones complementarias posteriores:TAC: disminución de volúmen de hemitórax dcho, y marcada hipoplasia de arteria pulmonar dcha. Aumentos de densidad periféricos focales en lóbulos superior e inferior dchos, compatibles con áreas de consolidación.AngioRMN: hipoplasia pulmonar dcha 2.ª a agenesia de arteria pulmonar derecha, con hipertrofia compensadora pulmonar y arterial izquierdas.Electrocardiograma: BIRD.Ecocardiograma: Dentro de la normalidad.Gammagrafia pulmonar: Ausencia total de perfusión en pulmón derecho.Cateterismo cardiaco: Agenesia de arteria pulmonar derecha con circulación colateral de vasos cervicales. PSP y en API basal al 40 % de la sistémica con presión diastólica baja.
Conclusiones:A pesar de la baja frecuencia de esta patología, sobre todo aislada sin cardiopatía asociada, se debe de tener presente en el dco diferencial de la disminución del volúmen pulmonar unilateral, aún con escasez o ausencia de síntomas en la infancia.
Señalar la importancia de la realización de una Rx tórax en todo niño con infecciones respiratorias y/o broncoespasmos recurrentes para descartar patologías de base.
P410TUBERCULOSIS CAVITADA
Vanessa Alonso Morales, Yolanda María Chica Fuentes, Leticia Olivares Sánchez, Olga M. Escobosa Sánchez, M. Pilar Ranchal Pérez, José Manuel Jiménez Hinojosa, Antonio Madrid Madrid, Estela M. Pérez Ruiz, Fco. Javier Pérez Frías y Antonio Jurado Ortiz
Hospital Materno Infantil, Málaga y Hospital General Carlos Haya, Málaga.
Introducción:La tuberculosis (TBC) pulmonar es una enfermedad infecciosa producida principalmente por M. tuberculosis. Después de unos decenios de menor incidencia,el número de casos de TBC ha aumentado en la última década produciéndose 1,3 millones de casos/año en el mundo.
Caso clínico:Motivo de ingreso: Niña de 13 años con 6-7 meses de evolución de astenia, anorexia y sensación febril no termometrada. En los últimos dos meses pérdidas de peso (47 kg al inicio) y tos escasa. Exploración:Regular estado general. Palidez de piel y mucosas. Escaso panículo adiposo. Peso: 27,4 kg. Cabeza y cuello: Normoconformado. Adenopatías laterocervicales rodaderas.Tórax: Normoconformado. ACR: hipoventilación bilateral en 2/3 inferiores, más marcada en hemotórax derecho. No distrés. Resto de exploración: Normal.Pruebas complementarias: Hemograma: 19200 leuc. (87 %N); Hb: 10; Plq: 534.000. VSG: 36 mm/h. Hemocultivo, urocultivo y cultivo LCR: (). Mantoux: 5 mm. Eco abdomen: normal. Rx tórax: condensación y cavidades en pulmón izquierdo y LSD. TAC tórax: infiltrados alveolares en LSD y LM y en todo el pulmón izquierdo con imágenes cavitadas bilaterales. Tinción Zhiel-Nielsen de LABA: 100 BAAR/campo.Diagnóstico: Tuberculosis cavitada.Evolución: Recibe tratamiento con isoniacida, rifampicina y piracinamida durante 6 meses. Se repite Mantoux: 15 mm. En TACAR se observan lesiones residuales: neumatoceles múltiples y de gran tamaño en pulmón izquierdo y de menor tamaño, junto con bronquiectasias cilíndricas en LSD y LM.
Conclusiones:La TBC cavitada en un adolescente puede deberse a una diseminación de un foco primario o a la reactivación de un foco TBC previo. Con tratamiento, la recuperación suele ser favorable. En nuestro caso, dada la gran afectación parenquimatosa mantenemos una actitud expectante acerca de la evolución.
P411ANALISIS DE LAS NEUMONIAS ADQUIRIDAS EN LA COMUNIDAD INGRESADAS EN UN HOSPITAL PEDIATRICO
M. Isabel Llull Ferretjans, José Antonio Gil Sánchez, Borja Osona Rodríguez de Torre, Juan Figuerola Mulet y Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca.
Objetivos:Evaluación de las Neumonías adquiridas en la comunidad (NAC) que precisaron ingreso en el Servicio de Pediatría de un Hospital terciario. Se analiza la epidemiología, motivo de ingreso, manifestaciones radiológicas y de laboratorio, etiología, duración del ingreso y tratamiento.
Método:Estudio retrospectivo de los pacientes inferiores a 14 años ingresados por NAC en un período de 13 meses desde Octubre 2001 a Noviembre 2002.
Resultados:Un total de 113 casos de NAC precisaron ingreso hospitalario. La edad media fue de 3 años y 4 meses. 64 niños y 49 niñas. El período de mayor incidencia fue de Octubre a Mayo. Entre los motivos de ingreso destacan en más del 50 % de los casos la afectación del estado general y la necesidad de oxigenoterapia, siendo otros motivos de menor importancia la mala tolerancia oral al antibiótico, la edad o patología subyacente. 6 casos precisaron ingreso en Cuidados Intensivos precisando 4 de ellos ventilación mecánica. Un 55 % no habían recibido ningún tratamiento antibiótico previo. En 81 casos la alteración radiológica fue lobar de los cuales un 30 % presentaban afectación multilobar. Se apreció leucocitois en 62 % con valores de PCR alto (> 4) en el 50 %. En el estudio etiológio destaca el escaso rendimiento obtenido en nuestro medio con los Hemocultivos (un solo caso positivo a streptococo pneumoniae) y cultivos de líquido pleural (ninguno positivo). Presentaron serologías compatibles con neumonía atípica 7 casos y un probable origen viral (cultivos y serologías) en 28 casos. La estancia media fue de 7,3 días. Precisaron antibiótico endovenoso el 66 % de los casos durante una media de 6 días; 85 casos recibieron beta-lactámicos y/o macrólidos precisando asociación con otros fármacos 28 casos. El 46 % precisó oxigenoterapia una media de 3,7 días.
Comentarios y conclusiones: 1.Destaca el elevado número de casos (63) que no recibieron tratamiento previo y precisaron ingreso por su gravedad de presentación.2. El diagnóstico etiológico es escaso a pesar de que las manifestaciones radiológicas y de laboratorio sugieran una causa bacteriana. 3. A pesar de la buena respuesta al tratamiento antibiótico, la estancia media es elevada (probablemente debido a la presencia de complicaciones en algunos casos como por ejemplo derrame pleural).
P412PRESENTACION ATIPICA DE UNA MALFORMACION ADENOMATOIDEA QUISTICA CONGÉNITA
Raquel Amo Rodríguez, Ana Jiménez, Manel Herrera, Sonia Yeste, Borja Osona Rodríguez de Torre y Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca.
Introducción:La MAQ de pulmón es una anomalía congénita poco frecuente. Se caracteriza por una masa multiquística de tejido pulmonar inmaduro. Suele afectar a un lóbulo provocando hipoplasia del resto del pulmón.
Objetivo:Presentar caso clínico de MAQ con manifestaciones clínicas y de imagen peculiares.
Caso clínico:Recién nacido a término varón que ingresa a los 2 días de vida por distres respiratorio (Silverman 2-3) con resto de exploración norma; sin precisar oxigenoterapia y con gasometrías en límites normales. En ecografías intrautero se objetiva masa en pulmón izquierdo sospechosa de MAQ, sin hidrops, que desaparece a partir de la 34 semana de gestación; sin otros antecedentes de interés. Los estudios de imagen (Rx y TAC) no muestran claramente lesiones bullosas, visualizándose un pulmón izquierdo hiperinsuflado que se hernia hacia el hemotórax derecho, desplaza mediastino y un pulmón derecho hipoplásico. Mediante ecografía-doppler se descartan vasos anómalos que perfundan áreas del pulmón y en la gammagrafía se objetiva hipoperfusión del lóbulo superior izquierdo. Los hallazgos de la angioRMN sugieren un enfisema lobar congénito del lóbulo superior izquierdo y vasos pulmonares normales. El paciente permanece estable hasta el mes de vida en que aumenta dificultad respiratoria y desaturación con las tomas, por lo que se decide intervención quirúrgica (lobectomía superior izquierda). Posteriormente evolución favorable con reexpansión pulmonar, persistiendo desplazamiento mediastínico. La anatomía patológica del pulmón extirpado revela hallazgos sugerentes de MAQ tipo II y cambios de displasia bronquial segmentaria.
Conclusiones: 1.La desaparición de imágenes ecográficas prenatales no descarta la existencia de malformación. 2. La presentación clínica precoz puede aparecer incluso en aquellos casos en que se resolvieron las imágenes intrautero.3. La dificultad para diferenciar las distintas malformaciones pulmonares congénitas y su coexistencia en ocasiones, sugieren un posible origen embriológico común (MAC, enfisema lobar congénito, hipoplasia pulmonar). 4. El manejo terapéutico debe ser individualizado según la evolución clínica.
NUTRICION
P413ACEITE DE OLIVA Y METABOLISMO LIPIDICO EN NIÑOS OBESOS. RESULTADOS PRELIMINARES
Jesús Fleta Zaragozano, José Luis Olivares López, Luis Moreno, Leonor Roda, Armando Giner, Antonio Sarria y Gloria Bueno Lozano
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción:Se conocen los efectos beneficiosos por la ingesta de aceite de oliva virgen extra (AOVE), en adultos, sin embargo, se desconocen cuando es ingerido por niños e inmediatamente tras su ingesta. Comunicamos las modificaciones observadas tras la ingesta de AOVE en niños obesos.
Material y método. Se han estudiado 8 niños obesos de 12,6 ± 1,7 años de edad (rango: 11,7-14,9) con IMC superior al percentil 95. La dieta de los niños era completa y equilibrada, de 2.200 a 2.500 kcal/día y su actividad física normal. Se les ha administrado, en la cena, AOVE (DO: Bajo Aragón), en una cantidad que representa el 10 % de la ingesta energética diaria que necesitan (RDA): de 20 a 30 ml, según edad y sexo. Estudio bioquímico: Tiempo 1: en ayunas (08,00 h), Tiempo 2: a las 4 h de la ingesta de la cena más el aceite (24,00 h), Tiempo 3: en ayunas del día siguiente (08,00 h). El colesterol (C) se ha determinado por método colorimétrico enzimático, el HDL-C, mediante método enzimático y el LDL-C según la fórmula de Friedewald et al: LDL-C = C-(Trigl/5 + HDL-C). Índice aterogénico = C/HDL-C. La comparación entre grupos se realizó mediante el test de ANOVA.
Resultados:Se observa descenso de las tasas de colesterol y de LDL-C, e incremento de las tasas de HDL-C, aunque sin diferencias significativas. Sin embargo, el índice aterogénico desciende significativamente (p = 0,040):

Conclusiones:Se comprueba que el AOVE modifica el metabolismo lipídico en niños obesos, a las 12 horas de su ingesta. Existe un efecto beneficioso, que se manifiesta con el descenso de niveles de colesterol, de LDL-C y del índice aterogénico, así como incremento de las tasas de HDL-C. Son necesarios estudios más amplios, realizados en niños, tanto obesos, como normales, para confirmar los hallazgos y obtener conclusiones definitivas.
P414MÃES DE CRIANÇAS DESNUTRIDAS E OBESAS, ATENDIDAS NO PROGRAMA EINSTEIN DE NUTRIÇÃO NA COMUNIDADE (PENC) DE PARAISOPOLIS
Kazue Sato, Ana L.G. Demarchi, Christianne F.L. Nascimento, Custódia V. de N. Mäder, Daniella M. Mazzaferro, Lilian C.Q. Mattoso, María Arlete Escrivão, Patricia V. Spada, Vanda M. Falcone y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil), Programa Einstein de Nutrição na Comunidade de Paraisópolis, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Trabalho realizado na Favela de Paraisópolis (50 mil habitantes) São Paulo Brasil pelo PENC.
Objetivos:Comparar algumas características entre mães de desnutridos e de obesos. Atendidos 86 desnutridos e 112 obesos. As mães, sempre acompanhando as crianças, prestaram informações sócio-econômicas sobre a família e gestação do filho e receberam atendimento específico em nutrição, com ênfase em alimentação saudável (incluindo preparo de refeições pelas mães, em nossa cozinha), psicológico e pedagógico, para estimular e possibilitar desenvolvimento psicomotor mais adequado ao filho. Em relação à idade, 73,3 % das mães de desnutridos tinham menos de 30 anos, e 65,2 % das mães de obesos, mais. A condição nutricional, expressa em índice de massa corporal, mostrou que das mães de desnutridos, 67,1 % eram eutróficas, contra 29,0 % das de obesos e 28,9 % estavam com excesso de peso e 71,0 % das mães de obesos (p = 0,0000). As famílias, em sua maioria tinham um ou dois filhos. Poucas famílias viviam com outros parentes: 10 (11,6 %), entre os desnutridos e 8 (7,3 %), entre os obesos. As mães de obesos eram mais anêmicas (34,4 %) do que as dos desnutridos (27,9 %). Não houve diferenças, em relação à escolaridade. Quanto ao nível intelectual, ouve diferenças nos níveis inferiores: p = 0,0191, no nível baixo e p = 0,0019, no nível de deficiência mental. Houve, também, diferenças significantes no desempenho psicopedagógico, entre as mães de desnutridos: o ruim caiu de 76,0 % para 20,0 % (p = 0,0000), após, no mínimo 3 meses, e entre as mães de obesos, o desempenho psicopedagógico ruim caiu de 59,3 % para 10,3 % (p = 0,0000). Entre os desnutridos, as atitudes maternas ruins caíram de 74,0 % para 34,0 % (p = 0,0000) e com as mães dos obesos, caíram de 77,0 % para 25,3 % (p = 0,0000). Na cozinha, as mães aprenderam a preparar alimentação mais saudável, com menos gordura e mais hortaliças, com mais higiene e menor risco de contaminação cruzada. As mães de desnutridos aprenderam a preparar os alimentos de modo que ficassem mais apetitosos e saborosos. Ressalte-se que as mães de obesos eram mais disponíveis no aprendizado na cozinha e se mostraram mais interessada nas trocas de receitas culinárias.
P415 RELAÇÃO DA CONDIÇÃO NUTRICIONAL DAS CRIANÇAS E SUAS MÃES NO AMBULATORIO DE PEDIATRIA DO PROGRAMA EINSTEIN NA COMUNIDADE DE PARAISOPOLIS
Kazue Sato, Lilian C.Q. Mattoso, Denise T. Schirch, María Arlete Escrivão, Suzana de Souza Queiroz y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil), Programa Einstein de Nutrição na Comunidade de Paraisópolis, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Nas últimas décadas, vem se observando no mundo e no Brasil, o que se convencionou chamar de transição nutricional, isto é, a coexistência, em crianças, da desnutrição e da obesidade, com evidente aumento desta última.
Objetivos:Avaliar a condição nutricional das crianças e de suas mães; relacionar a condição nutricional das crianças com o número de irmãos, peso de nascimento, idade gestacional, condição nutricional materna e relacionar a idade materna, o número de gestações, a condição nutricional dos filhos com a condição nutricional materna.. Foram estudadas 296 crianças e suas mães que freqüentaram o Ambulatório de Pediatria do Complexo Telma Sobolh do Programa Einstein na Comunidade, na favela de Paraisópolis São Paulo/Brasil. As crianças pesadas e medidas e classificadas segundo idade: para crianças menores de 2 anos, usou-se o indicador peso/idade e a classificação de Gómez. Para crianças maiores de 2 anos, os indicadores foram peso/estatura e estatura/idade, adotando-se a classificação de Waterlow, modificada por Batista. Para as mães, o indicador foi o índice de massa corporal (IMC) e a classificação preconizada pela OMS. Os resultados foram, entre as crianças, 56,8 % eram eutróficas, 22,3 %, obesas e 20,9 %, desnutridas. Entre as mães: 50 % eram eutróficas; 45,3 %, obesas, 4,7 %, desnutridas. A relação da condição nutricional entre mãe e filho mostrou que das crianças eutróficas, 49 % tinham mães eutróficas. Das crianças com sobrepeso e obesas, 54 % tinham mães obesas. Das crianças desnutridas, 57 % tinham mães eutróficas e 40 %, obesas e apenas 3 %, desnutridas. A maioria das mães (68,6 %) tiveram de uma a três gestações. A idade materna parece influir na condição nutricional das mães: entre aquelas com idade inferior a 20 anos, 60,6 % eram eutróficas e das que tinham mais de 30 anos, 60,9 %, obesas. O baixo peso ao nascer foi de 10,8 %, peso insuficiente, 25,3 % e peso adequado, 59,1 %. Das crianças que nasceram com baixo peso, 43,8 % eram desnutridas e 46,9 %, eutróficas; das que nasceram com peso insuficiente, 25,3 % eram desnutridas e 58,7 %, eutróficas e das que nasceram com peso adequado, 15,4 % eram desnutridas e 57,1 %. Com a obesidade ocorreu caminho inverso. a obesidade aumenta com o aumento do peso ao nascer (9,4 %, 16,0 % e 27,4 %, respectivamente). Verificou-se, ainda, que mães obesas tendem a ter filhos obesos, parecendo ser erro alimentar na família.
P416CRIANÇAS DESNUTRIDAS E OBESAS, ATENDIDAS NO PROGRAMA EINSTEIN DE NUTRIÇÃO NA COMUNIDADE (PENC) DE PARAISOPOLIS
Kazue Sato, Ana L.G. Demarchi, Christianne F.L. Nascimento, Custódia V. de N. Mäder, Daniella M. Mazzaferro, Lilian C.Q. Mattoso, Patricia V. Spada, María Arlete Escrivão, Vanda M. Falcone y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil), Programa Einstein de Nutrição na Comunidade de Paraisópolis, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Trabalho realizado na Favela de Paraisópolis (50 mil habitantes) São Paulo Brasil pelo PENC.
Objetivos:

