

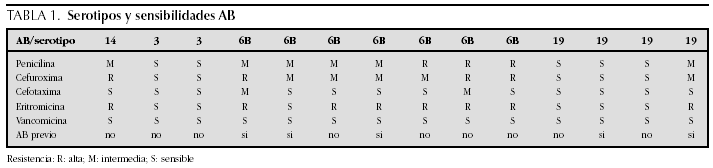
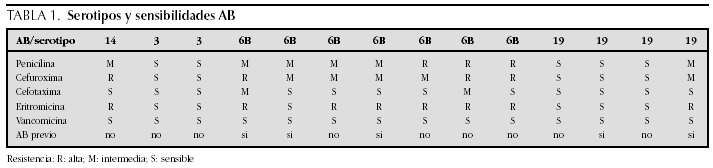
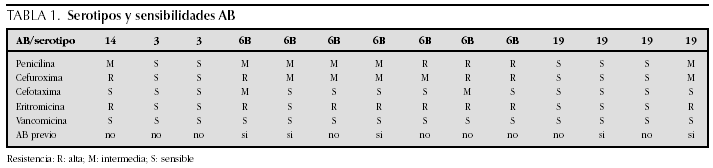
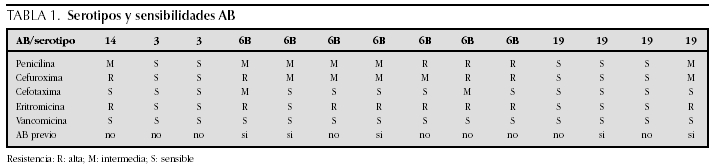
ATENCION PRIMARIA Y PEDIATRIA EXTRAHOSPITALARIA
P53PATRON DE CONSUMO DE TABACO ENTRE LOS JOVENES 1.º Y 2.º DE LA E.S.O. DE LA PROVINCIA DE SALAMANCA
Raquel Ruano García, M. Dolores Plaza Martín, Miguel Torrecilla García, Miguel Ángel Hernández Mezquita, Alejandro Alonso Díaz, Manuel Martín Temprano, Roberto Pérez Santos, Mar González Bustos, Esperanza Corrales Arganda y Almudena Morales Sánchez
Centro de Salud San Juan Salamanca y Unidad Docente de Medicina Familiar de Salamanca.
Antecedentes y objetivo:Conocer el patrón de consumo de tabaco entre los jóvenes de 1.º y 2.º de la E.S.O., periodo en el que se suele iniciar el consumo de tabaco (13,2 años según el observatorio español sobre drogas).
Metodología:Estudio descriptivo sobre un total de 7.284 jóvenes mediante una encuesta anónima. El estudio estadístico se realizó con las pruebas de chi cuadrado y t de Student. Valor p < 0,05.
Resultados:Contestaron el 67,8 % (4.907) de los jóvenes sobre los que se mandaron la encuesta, reconociéndose fumador el 20,5 % de los que el 56,4 % eran varones (53,3-59,5 %) (p < 0,0001). El 42,6 % (41,2-44 %) reconocían haber probado el tabaco. Eran fumadores habituales, de forma diaria, el 7,6 % de los chicos y el 4,8 % de las chicas (37,1 % y 23,2 % de los que se consideraban fumadores) (p < 0,0001). La edad de inicio de los que fumaban fue de 11,2 ± 3,9 años (11,04 ± 3,6 años de los que en general habían probado el tabaco en alguna ocasión) siendo la curiosidad el motivo por el que se fumaron su primer cigarrillo en la mayoría de las situaciones, en un 73,5 % (70,6-76,2 %) y el no atreverse a decir no en un 6,2 % (4,8-7,9 %) (p < 0,0001); generalmente en compañía de los amigos, concretamente el 73,3 % (70,4-76 %), aunque un 6,2 % (4,8-7,9 %) lo hizo en compañía de un hermano y un 2,5 % (1,6-3,7 %) en presencia de sus padres (p < 0,0001). Les gustó el primer cigarrillo al 34,7 % de los que probaron en alguna ocasión (p < 0,0001). El consumo de cigarrillos era ocasional, es decir, alguno a la semana pero no de forma diaria en el 21,6 % de los que se declaraban fumadores (4,1 % del total) y menor de 5 cigarrillos al día en el 93,1 %, 71,6 % si se excluye el grupo anterior (13,2 % del total) (p < 0,0001).
Conclusiones:La edad media de consumo de tabaco entre los jóvenes de 12 y 13 años de Salamanca fue de 11,2 años. La experimentación y el grupo de iguales son determinantes a la hora del inicio del consumo de tabaco, así como la permisividad existente. Dada la temprana edad en la que se tiene contacto con el tabaco y que el establecimiento del hábito tabáquico es propio de edades más avanzadas, la intervención en el medio escolar es fundamental en la prevención del tabaquismo.
P54MEJORIA DEL ASMA INFANTIL CON EL USO DE LA COMBINACION INHALADA DE SALMETEROL Y FLUTICASONA
Eduardo Antonio Lara-Pérez
Universidad Veracruzana, Veracruz (México) y Academia Mexicana de Pediatría, Veracruz (México).
Introducción:Para el buen control del asma debe ofrecerse un anti-inflamatorio y broncodilatador.
Objetivo:Evaluar mejoría de escolares asmáticos con el uso de Salmeterol y Fluticasona (Seretide®).
Material y métodos:Se ofrece 3 meses Salmeterol 50 microgramos y Fluticasona 100 microgramos cada 12 h, inhalados con Aerochamber en niños asmáticos de 5 a 10 años, midiéndose al inicio y cada mes de tratamiento el flujo espiratorio máximo (FEM) en porcentaje comparado con el esperado.
Resultados:41 escolares, 15 femeninos y 26 masculinos, edad media de 7,5 años. Femeninos valores medios de talla 120 cm y FEM al inicio 72 %, primer mes de 89,7 % p < 0,005, segundo mes 109,6 % p < 0,012 y tercer mes con 115,1 % p < 0,056. Masculinos valores medios de talla 119,5 cm y FEM inicial de 88,5 %, primer mes 89 % p < 0,0001, segundo mes 104,1 % p < 0,0001, tercer mes 111,3 % p < 0,003.
Discusión:Es mejor la combinación de Salmeterol y Fluticasona sobre todo si es inhalado en el control del asma que utilizarlos por separado.
Conclusiones:El Salmeterol y Fluticasona mejora el FEM en los asmáticos progresivamente cada mes de tratamiento.
P55COMPORTAMIENTO DEL FLUJO ESPIRATORIO MAXIMO SEGUN EL INDICE DE MASA CORPORAL EN ESTUDIANTES UNIVERSITARIOS
Carolina Palmeros-Exsome, Eduardo Antonio Lara-Pérez y Enrique González-Deschamps
Universidad Veracruzana, Veracruz (México) y Academia Mexicana de Pediatría, Veracruz (México).
Introducción:El flujo espiratorio máximo (FEM) varia de acuerdo a talla y sexo, se desconoce si existe relación con el índice de masa corporal (IMC).
Objetivo:Determinar relación del FEM respecto al IMC.
Material y métodos:Se encuestaron universitarios, tomándose; peso, talla, y FEM, se utilizaron pruebas estadísticas de tendencia central, tablas de contingencia y correlación de; Shapiro-Wilks, Spearman, y Wilcoxon.
Resultados:Hay diferencias de IMC y FEM por genero (masculino-femenino) p < 0,005, el IMC y FEM no están relacionadas en las mujeres p 0,12345, los valores medios no mostraron diferencias estadísticas. En hombres el IMC y FEM si están relacionados p 0,00385, los valores medios estadísticamente demostraron que a mayor IMC mayor FEM.
Discusión:El FEM y el IMC son unas excelentes herramientas clínicas, el FEM evaluando función pulmonar y el IMC la relación de talla/peso. Actualmente subutilizadas a pesar de sus ventajas sin que se hayan realizado estudios con FEM y su relación con el IMC.
Conclusiones:El IMC y el FEM son diferentes según el genero (masculino-femenino), no existe relación entre el FEM y el IMC en mujeres (femeninos) pero si en hombres (masculinos); a mayor IMC mayor FEM.
P56COMPARACION DE FLUJO ESPIRATORIO MAXIMO SENTADO Y DE PIE EN ESTUDIANTES UNIVERSITARIOS
Eduardo Antonio Lara-Pérez, Enrique González-Deschamps y Karina Lupercio-Mora
Universidad Veracruzana, Veracruz (México) y Academia Mexicana de Pediatría, Veracruz (México).
Introducción:Existen pocos estudios de flujo espiratorio máximo (FEM) desconociéndose su comportamiento de acuerdo a la posición, con la finalidad de comparar los valores del FEM de pie y sentado.
Material y métodos:Se encuestaron estudiantes universitarios que se le tomo; edad, talla y FEM sentado y de pie con el flujómetro de Tru-Zone®.
Resultados:228 universitarios; 111 femeninos y 117 masculinos con edad promedio de 21,5 años, desviación estándar de 2,1213, en los femeninos el promedio de FEM sentado fue de 384,7 lts x min con desviación estándar de 0,7071, el FEM de pie fue de 394,7 lts xmin con desviación estándar de 1,4142. Para los masculinos el FEM sentado fue de 418,3 lts xmin con desviación estándar de 2,8284, el FEM de pie fue de 448,7 lts x min con desviación estándar de 10.6066.
Discusión:El FEM es una excelente herramienta objetiva para evaluar función pulmonar, tanto en salud como en enfermedad.
Conclusiones:El FEM es mayor en posición de pie que en posición sentado, los valores de FEM son mayores para masculinos en ambas posiciones, se recomienda al evaluar el FEM especificar la posición en que se tomo.
P57ÍNDICE DE MASA CORPORAL Y FLUJO ESPIRATORIO MAXIMO EN ESCOLARES SANOS DE VERACRUZ
Eduardo Antonio Lara-Pérez y Enrique González-Deschamps
Universidad Veracruzana, Veracruz (México) y Academia Mexicana de Pediatría, Veracruz (México).
Introducción:No se utilizan: índice de masa corporal (IMC) y flujo espiratorio máximo (FEM) para evaluar salud.
Objetivo:Clasificar IMC y FEM en escolares sanos.
Material y métodos:En escolares se tomó: peso, talla, y FEM (flujómetro TruZone®), IMC bajo; 14, IMC normal 14 a 25 y IMC sobrepeso + 25, diferencia del FEM obtenido con el esperado.
Resultados:3,423 escolares; 48,61 % masculinos, edad promedio 11,5 años y 51,38 % femeninos, edad promedio 9 años. Masculinos: IMC bajo: 75 (4,5 %) promedio 13,97, promedio FEM 72,78 %. IMC normal: 1476 (88,7 %) promedio 16,8, promedio FEM 324,95 %. IMC Sobrepeso: 113 (6,79 %) promedio 32,18, promedio FEM 90,54 %. Femeninos: IMC bajo: 81 (4,6 %) promedio 12,46, promedio FEM 85,90 %. IMC normal: 1568 (89,14 %) promedio 18,03, promedio FEM 144,52 %. IMC sobrepeso: 110 (6,25 %) promedio 29,55, promedio FEM 80,27 %.
Discusión:El IMC varia según raza, etnia, edad, género y condiciones sociales, también es diferente para escolares, IMC y FEM pueden alterar la calidad de vida, tanto sobrepeso y desnutrición, se necesita; orientación nutricional y fomento del ejercicio.
Conclusiones:El IMC influye sobre FEM, con IMC normal se tiene FEM por encima del esperado.
P58PREVENCION DEL TABAQUISMO. UN OBJETIVO DE TODOS
M. Dolores Plaza Martín, Raquel Ruano García, Miguel Torrecilla García, Miguel Ángel Hernández Mezquita, María José Marcos García, Generoso Gómez Cruz, Alejandro Alonso Díaz, Rosario Sánchez González, Mariví Salvador García y Aurora Ramos Monge
Asociación para la Prevención y Control del Tabaquismo de Castilla y León.
Antecedentes y objetivo:La edad media del consumo de tabaco entre los jóvenes españoles de 14 a 18 años es 13,2 años. Por ello, el objetivo de la intervención es ofrecer una información seria pero a la vez atractiva, adaptada a las características de la población infantil para concienciarles del problema real derivado del consumo de tabaco y evitarlo en lo posible; o al menos retrasar la edad de inicio.
Metodología:Estudio descriptivo sobre los jóvenes de 12 y 13 años (1.º y 2.º de la E.S.O.) de la provincia de Salamanca (n = 7.284). Se confeccionó un cómic en el que se abordaban los distintos aspectos derivados del consumo de tabaco (perjuicios para la salud, perjuicios estéticos, falta de independencia, engaño de la publicidad, ventajas de no fumar...). Al final del mismo había una encuesta para valorar su aceptación e impacto. El cómic fue financiado por la Obra Social de Caja Duero y en su distribución colaboró la Delegación de Educación.
Resultados:La práctica totalidad de los 2.056 jóvenes que contestaron a la encuesta valoró positivamente el programa de intervención (99,9 %). El mensaje que más se valoró fue la influencia negativa del tabaco en la relación chico/chica y lo que más impactó fue el gran número de enfermedades y aspectos negativos derivados del consumo de tabaco (al 28 %). La forma de presentar la información, a través del cómic fue lo que más gustó al 41 % de los chavales y las estrategias para decir "no" al tabaco llamó la atención al 9,7 % de los chavales. Tras la lectura del cómic el 99,1 % consideraban al tabaco como una droga y un 97,8 % identifican a la nicotina como responsable de la adicción. El 99 % considera que fumar es perjudicial.
Conclusiones:Los pediatras tenemos que implicarnos en la prevención del tabaquismo pues afecta a una población de nuestra competencia. El diseño de programas de información adecuados a la población sobre la que se vaya a actuar es fundamental para conseguir los objetivos marcados.
P59SEGUIMIENTO DE RECIÉN NACIDOS DE MUY BAJO PESO (RNMBP) EN UN CENTRO UNIVERSITARIO EN CHILE
Jorge Fabres, M. Rosario Moore Valdés, Paula Bedregal, Ana Verdugo, Raúl Escobar y Tomás Mesa
Pontificia Universidad Católica de Chile, Santiago de Chile (Chile).
Introducción:La sobrevida de los RNMBP ha mejorado. Se necesita conocer el pronóstico a largo plazo para estudios de costo-efectividad.
Objetivos:Comparar el estado nutricional (EN), desarrollo psicomotor (DPM) y severas discapacidades (DIS) a los 24 meses de edad corregida (EC), entre niños < 1.000 g y 1.000-1.500 g, e identificar factores de riesgo asociados con DIS y retraso DPM (RDPM).
Método:Se evaluó a 24 meses EC a niños ingresados al Seguimiento entre Enero 1995 y Octubre 1999. El EN se evaluó con curvas del National Center for Health Statistics (NCHS) y el DPM con tests de Griffiths (1995-1998) o Bayley (1998-2001). Se comparó a los niños < 1.000 g con ≥ 1.000 g. Se usó análisis de regresión logística, para determinar factores de riesgo asociados con un mal pronóstico.
Resultados:142 niños (88 % de los egresados) ingresaron al Seguimiento. El Peso de Nacimiento (promedio ± DS) fue 1.070g (± 245), edad gestacional 29 semanas (± 2), 56 (39 %) eran < 1.000 g, 48 (34 %) abandonaron el seguimiento. No se encontró diferencias significativas en variables neonatales entre perdidos y esos que completaron el seguimiento. Se identificó la Hemorragia Intra-Ventricular (HIV) como factor de riesgo independiente para DIS (OR = 3,1; IC: 1,6-5,9) y RDPM (OR = 1,8; IC: 1,1-3,2). La Tabla compara niños < y ≥ 1.000 g.
Conclusiones:Niños < 1.000 g presentan más compromiso del EN, RDPM y DIS; se observan problemas en niños ≥ 1.000 g HIV, el principal factor de riesgo para daño neurológico. Se requieren programas de seguimiento y se requieren estrategias para mantener adherencia a estos programas.

P60DISURIA: NO ES INFECCION URINARIA TODO LO QUE MOLESTA
Carmen Martínez González y Juana Cacho Calvo
Centro de Salud San Blas, Parla y Hospital Universitario de Getafe.
Introducción:La infección urinaria (ITU) es el proceso infeccioso más frecuente en la infancia, tras la infección respiratoria de vías altas. Aunque el diagnóstico se confirma mediante urocultivo, la clínica de disuria con o sin síntomas acompañantes, es fundamental para el diagnóstico.
Caso clínico:Varón de 2 años y 6 meses sin antecedentes familiares ni personales de interés, que presenta en tres ocasiones, cuadro de disuria sin síntomas asociados. El primer episodio a los 18 meses de edad, se inicia con la clínica descrita, exploración local normal, sistemático de orina y urocultivo negativo. Los síntomas remitieron espontáneamente.
En el 2.º episodio a los 2 años de edad, presentaba dos pequeñas pápulas puntiformes en glande, con punto central erosionado, y leve hiperemia generalizada de glande y surco balano prepucial. Se inicia tratamiento con nistatina tópica por sospecha de Candidiasis, tras la toma exudado para hongos, con resultado posterior negativo.
El tercer episodio 6 meses después, debuta desde el inicio con tres lesiones puntiformes, y una pequeña úlcera en surco balanoprepucial (foto 1). Se toma muestra para cultivo de virus herpes simplex (VHS) tipo 1 y 2, mediante raspado de lesión y procesamienteo por técnica de inmunofluorescencia directa (MicroTrak HSV1/HSV2, Ireland) confirmándose la etiología por VHS 1.
Conclusiones:El VHS 1 de localización genital es infrecuente en niños. El síntoma aislado de disuria y la edad del niño, hicieron sospechar inicialmente las causas más frecuentes, pero no debemos olvidar que además de la anamnesis y los estudios complementarios, la exploración minuciosa sigue siendo imprescindible para la orientación diagnóstica.
P61HISTORIA NATURAL DE LAS REGURGITACIONES EN EL LACTANTE: ESTUDIO PROSPECTIVO DE 12 MESES EN LACTANTES MADRILEÑOS
María García-Onieva Artazcoz, María Aparicio Rodrigo y M. Julia Bustos Fonseca
Centro de Salud Entrevías, Madrid.
Antecedentes:A pesar de la elevada prevalencia de las regurgitaciones en el lactante, su historia natural sigue estando escasamente documentada.
Objetivo:Evaluar la historia natural de las regurgitaciones en el primer año de vida.
Métodos:Se realizó un estudio prospectivo longitudinal en un Centro de Atención Primaria de Madrid. Todos los lactantes nacidos entre el 1-04-99 y el 31-03-00 fueron incluidos. Se utilizó un cuestionario estandarizado en las visitas de 1, 2, 4, 6, 9 y 12 meses para identificar lactantes con sospecha de reflujo gastroesofágico (RGE)/regurgitaciones. La información fue obtenida interrogando a los padres. Los lactantes se dividieron en cuatro grupos:a) no regurgitadores,b) regurgitan 1-3 veces/día,c) regurgitan 3-5 veces/día yd) regurgitan > 5 veces/día. La frecuencia de hipo, llanto excesivo, rechazo de alimento y problemas respiratorios de vías aéreas superiortes e inferiores fue recogida interrogando a los padres. La edad gestacional, peso al nacimiento, tipo de lactancia (materna vs fórmula) y exposición al humo de tabaco fueron también registrados.
Resultados:Se incluyeron 260 lactantes (100 % de los nacidos en el año de estudio). 191 (73,4 %) completaron las 6 visitas. Al mes de vida el 53 % de los lactantes regurgitaba. Este porcentaje descendió al 46 % a los 2 meses, 40 % a los 4 meses, 16,5 % a los 6 meses, 3,5 % a los 9 meses para llegar al 1 % a los 12 meses de edad. La mayoría regurgitaba entre 1 y 3 veces al día y en cantidades pequeñas. El sexo, lactancia materna frente a fórmula y la exposición ambiental al tabaco no se relacionaron cola frecuencia de regurgitaciones en ninguna de las visitas. Los nacidos con menos de 2,5 kg regurgitaban con más frecuencia que los que pesaron > de 2,5 kg durante los primeros 4 meses (p < 0,05 a los 4 m). El hipo y llanto excesivo fueron más frecuentes (p < 0,05) en los primeros 4 meses. Ningún lactante tuvo signos de enfermedad por RGE (p. ej. fallo de crecimiento, esofagitis o enfermedad respiratoria crónica).
Conclusiones:Nuestro estudio confirma que las regurgitaciones en niños españoles, como en otros países, son un problema frecuente que alcanza su máximo a los 1-2 meses. Confirmamos que la mayoría tiene un reflujo no complicado que se resuelve espontáneamente en el primer año sin precisar estudios o tratamientos agresivos.
P62FIEBRE PERIODICA AFTOSA FARINGOAMIGDALAR
Paloma Martínez Mateo, M. Julia Bustos Fonseca y Francisca Menchero Pinos
Área I de Atención Primaria.
Objetivo:Demostrar la existencia de procesos febriles, sin evidencia infecciosa en la infancia.
Material y método:Seguimiento, estudio y evolución clínica de un niño con fiebre recurrente desde una consulta de Atención Primaria.
Paciente con episodios de fiebre de presentación brusca y elevada, de 3-6 días de duración, con una periocidad de 3-8 semanas; ocasionalmente se acompaña de estomatitis aftosa, faringitis y adenitis cervical. Las pruebas complementarias, tanto en período febril como en fases intercríticas no han evidenciado otra patología. Los antitérmicos habituales y los antibióticos resultan ineficaces como tratamiento. Cumple criterios diagnósticos de síndrome de Marshall, descrito en 1.989 denominando a este cuadro como síndrome FPAFA. Los episodios han sido controlados con un corto curso de prednisona.
Resultados:Respuesta espectacular de un caso de fiebre periódica aftosa faringoamigdalar al tratamiento con corticoides.
Disminución del absentismo escolar y de la ansiedad familiar.
Conclusiones:Aunque la causa más frecuente de procesos febriles en la infancia es la infección, no es la única.
El conocimiento de este síndrome evita la necesidad de realizar pruebas complementarias adicionales.
P63TUMORACION LATEROCERVICAL EN EL NIÑO. EXPERIENCIA DE 4 AÑOS
Fidel Gallinas Victoriano, Marta González, Beatriz Solís Gómez, Joaquín Duarte, María Sagaseta de Ilurdoz Uranga y Francisco Javier Molina Garicano
Servicio de Pediatría. Hospital Virgen del Camino, Pamplona.
Objetivos:Presentamos la experiencia de tumoraciones laterocervicales en edad pediatrica durante el periodo comprendido entre 1/1999 a 12/2002. Describimos las manifestaciones clínicas, diagnósticos etiológicos y planteamos una actitud práctica para su tratamiento.
Material y métodos:Hemos revisado las historias de 32 niños en las que hemos estudiado: edad, sexo, duracion de la sintomatologia, tratamiento previos, manifestacines clínicas, exámenes complementarios, distribucion por años y evolucion.
Resultados:La edad estaba comprendida entre los 7 meses y 14 años, predominando en menores de 6 años, con un discreto predominio en varones. La mayor parte de los pacientes se remitió en las primeras dos semanas de iniciado el proceso y se evidencia un discreto mayor numero de casos en el año 2000. La tumoración fue mayor de 2 cm en 70 % de los casos (21/32). Presentaban manifestaciones inflamatorias locales el 81 % de los pacientes. Se realizó radiología simple de torax en el 66 % y estudio histológico en el 53 %. Se obtiene estudio serológico en el 65 % sin significado clinico dado la escasez de positividades obtenidas. Fiebre se presento en un 38 % de los casos. La mayor parte de los niños recibieron antinflamatorios no esterorideos, antibióticos y en ocasiones hielo local. De los 32 pacientes, 5 (16 %) fueron tumores malignos, una un quiste branquial infectado, adenopatía inflamatoria más flemón 17 casos (53 %) y abceso en 9 (28 %).
La evolución fue favorable en 25/32 casos (78 %), 2 niños precisaron más de una intervención quirúrgica y los 5 procesos malignos están actualmente en remisión completa con o sin tratamiento.
Conclusiones: 1.Se trata de una patologia lo suficinetmente frecuente como para conocer su manejo. 2. Procuremos diferenciar una etilogia infecciosa de patologías neoplásicas.3. Proponemos tratar a aquellas tumoraciones sugerentes por la clínica de infección, con antibioterapia y antinflamatorio de 1 a 2 semanas antes del estudio complementario.4. Derivar el resto de tumoraciones, y las que su evolución lo requieran, para su correcto diagnóstico y tratamiento.
CARDIOLOGIA
P64TRASPLANTE CARDIACO EN LACTANTE CON FIBROMA GIGANTE
Teresa Álvarez Martín, Manuela Camino López, José Luis Zunzunegui Martínez, Carlos Maroto Monedero, Enrique Maroto Álvaro, Rubén Greco Martínez, Fernando Ballesteros Tejerizo, Águeda Herrera Chamorro, M. del Rosario Jiménez Liria y M. Ángeles Carrasco Azcona
Hospital General Universitario Gregorio Marañón, Madrid, Clínica Virgen de la Vega, S. A., Murcia y Hospital Torrecárdenas del SAS, Almería.
Introducción:Los tumores cardíacos en edad pediátrica son habitualmente benignos. Cuando causan problemas hemodinámicos o arritmogénicos son tributarios de tratamiento quirúrgico. En algunas ocasiones por tamaño o inabordaje quirúrgico convencional pueden ser tributarios de trasplante cardíaco.
Descripción del caso:Lactante de 5 meses de edad que debutó con un episodio de parada cardiorrespiratoria, comprobándose fibrilación ventricular y resucitada mediante maniobras de RCP avanzada. La ecocardiografía 2D-doppler y TAC torácico demostró masa gigante tumoral sin bordes definidos de clivaje con respecto a miocardio en la pared inferior, lateral y posterior cardíaca sin producir alteraciones hemodinámicas. la biopsia a cielo abierto demostró fibroma sin signos de malignidad. Se consideró inextirpable quirurgicamente.
Por episodios repetidos de taquicardia ventricular, a pesar del tratamiento antiarritmico, se indicó trasplante cardíaco ortotópico que se realizó sin complicaciones. En la actualidad 4 meses después permanece asintomática desde el punto de vista cardíaco y en tratamiento con triple terapia inmunosupresora (ciclosporina, azatioprina y corticoides)
Conclusión:El trasplante cardíaco es una alternativa válida en el tratamiento de los tumores cardíacos que no son abordables mediante cirugía convencional por su situación o tamaño.
P65TRATAMIENTO CON COLCHICINA DE LA PERICARDITIS RECURRENTE EN NIÑOS
M. Elvira Garrido-Lestache Rodríguez-Monte, M. Soledad Jiménez Casso, M. Teresa Penela Vélez de Guevara, M. Cristina Puente Sánchez, Celia Reig del Moral, M. Dolores Romero Escos y Pedro Cuadrado Bello
Hospital General de Segovia, Segovia.
Antecedentes y objetivos:La colchicina se utiliza como tratamiento de la Fiebre Mediterránea Familiar. Basándose en este hecho, se comenzó a usar en el tratamiento de la pericarditis recurrente en adultos con muy buenos resultados. En niños, el porcentaje de pericarditis recurrente oscila entre un 15-30 %, existiendo muy pocos estudios de la utilización de colchicina. Presentamos un caso de una niña de estas características que respondió a la misma.
Caso clínico:Niña de 11 años, sin antecedentes de interés, que acude por dolor precordial, disnea de decúbito y fiebre. Exploración física: Peso: 36,200 kg. Taquipneíca, taquicárdica. Auscultación cardiaca: rítmica. No soplos. No hepatomegalia. ECG: ritmo sinusal con aplanamiento generalizado del ST y ondas T. Ecocardiograma: derrame pericárdico leve. Hemograma y bioquímica normales, ASLO 305 UI/ml, ANA negativos, serología positiva a Influenza A 1/128. Ante el diagnóstico de pericarditis aguda se pauta tratamiento con antinflamatorios a pesar de lo cual presenta empeoramiento clínico precisando añadir corticoides. Presenta mejoría parcial por lo que al mes se retiran los antinflamatorios y se inicia pauta descendente de los corticoides. Al mes y medio del inicio presenta nueva reagudización precisando aumento de la dosis de corticoides y reintrodución de antinflamatorios. Ante la persistencia de la sintomatología, se decide iniciar tratamiento con colchicina a 1 mg/día mejorando espectacularmente a los 3 días, quedando asintomática a los 12 días permitiendo la retirada de los corticoides. Asintomática durante tres meses se suspende la misma. Tres meses más tarde presenta un nuevo episodio reiniciándose el tratamiento.
Conclusión:La colchicina parece ser un tratamiento efectivo como alternativa a los corticoides en los casos de pericarditis recurrente idiopática en la población pediátrica. Todavia desconocemos el tiempo que se debería mantener (entre seis meses y un año) cuales son las dosis adecuadas para cada edad. Asi mismo, si su uso podría extenderse a otras pericarditis rebeldes al tratamiento convencional como los síndromes pos-pericardiectomía relativamente frecuentes en Pediatría.
P66MIOCARDIOPATIAS EN LA INFANCIA
Antonio Vázquez Florido, M. Ángeles Murillo Pozo, Juan Antonio Soult Rubio, Alberto Varona García, Cristina Montero Valladares, Víctor Manuel Navas López, Juan Antonio García Hernández, Miguel Muñoz Sáez, José Domingo López Castilla y Antonio Romero Parreño
Hospital Universitario Virgen del Rocío, Sevilla.
Antecedentes y objetivo:Las miocardiopatías son enfermedades graves que afectan a la funcionalidad y/o estructura del miocardio. Su etiología es muy diversa.
Presentamos las miocardiopatias diagnosticadas en nuestra Unidad de Cuidados Intensivos Pediátricos (UCIP) en los últimos años.
Métodos:Estudio retrospectivo de las miocardiopatías que ingresaron en UCIP desde 1993 hasta 2001. Se analizan los parámetros epidemiológicos y clínicos.
Resultados:Se diagnosticaron 11 casos. El 64 % fueron niños y el 36 % niñas. La edad estab comprendida entre los 16 días y los 10 años. El motivo de ingreso fue la insuficiencia cardiaca congestiva, en 9 casos (82 %) y la parada cardíaca en los otros 2 (18 %). La ecocardiografía aportó datos compatibles con miocardiopatía en todos ellos. El 91 % de los casos correspondieron a miocardiopatía dilatada y un caso a hipertrófica. Todos los niños precisaron ventilación mecánica, así como tratamiento inotrópico y/o vasodilatador. Se realizó diagnóstico etiológico en el 82 % de los casos: 2 miocardiopatías tóxicas por Adriamicina, 1 por virus Cosackie, 1 por hemocromatosis, 1 secundaria a taquicardia ventricular incesante, 1 por déficit de carnitina, 1 secundaria a coronaria anómala, 1 en un caso de enfermedad de Duchenne y 1 hipertrófica en niño con síndrome de Noonan. Dos casos se encuadraron en el grupo de miocardiopatias dilatadas idiopáticas, al no llegarse a un diagnóstico etiológico. Estos 2 niños entraron en programa de trasplante cardíaco, falleciendo antes de realizarlo. Sobrevivieron 2 niños (18 %): el caso de miocarditis vírica y el de taquicardia ventricular incesante, que precisó tratamiento quirúrgico del foco ventricular ectópico mediante crioablación.
Conclusiones:La miocardiopatías son anomalías poco frecuentes en la edad pediátrica, que ponen en grave peligro la vida del niño.
La etiología es muy diversa y el diagnóstico etiológico suele ser dificultoso.
En la mayoría de los casos el único tratamiento es la terapia de mantenimiento y el trasplante cardíaco.
En casos de miocardiopatía dilatada idiopática debe plantearse precozmente el trasplante cardíaco, ya que suele ser refractaria al tratamiento médico.
P67REVISION DE TUMORES CARDIACOS PRIMARIOS
Yolanda María Chica Fuentes, Javier Blasco Alonso, Olga M. Escobosa Sánchez, José Manuel Jiménez Hinojosa, Vanessa Alonso Morales, M. Pilar Ranchal Pérez, Antonio Madrid Madrid, Victorio Cuenca Peiró y Antonio Jurado Ortiz
Hospital Materno Infantil, Málaga y Hospital General Carlos Haya, Málaga.
Introducción:Los tumores cardiacos primarios en niños son extremadamente raros. El rabdomioma (RC) es el más frecuente de todos. Es un tumor benigno de células musculares que se localiza principalmente en ventrículos y que se asocia en la mayoría de los casos a esclerosis tuberosa (ET). Puede tener diversas manifestaciones clínicas como arritmias, insuficiencia cardiaca congestiva o muerte súbita.
Material y métodos:Estudio retrospectivo de los 5 casos de tumores cardiacos diagnosticados en nuestro centro desde Enero de 1996 a Febrero de 2003. Se analizan principalmente el momento del diagnóstico, presentación clínica, características del tumor, evolución, necesidad de tratamiento y existencia de manifestaciones de ET.
Resultados:El diagnóstico de tumoración intracardiaca se realizó prenatalmente en 3 casos (semana 33-35 de gestación). Al nacimiento no presentaron manifestaciones clínicas. La ecocardiografía postnatal confirma la existencia de tumoración intracardiaca que se califica como RC. Se realiza despistaje de ET con ecografía, TAC y RMN de cráneo, ecografía renal y fondo de ojo. Uno de ellos presenta angiofibromas faciales a los 3 años sin otras manifestaciones de la enfermedad.
En los otros 2 casos el diagnóstico se realiza a los 5 y 7 meses de edad, debutando con clínica neurológica (convulsiones). Se aprecian angiofibromas faciales en uno de ellos y manchas hipomelánicas en los dos. Las pruebas de imagen craneales detectan tubers periventriculares. Con el diagnóstico de ET se realiza ecocardiografía objetivándose tumoraciones intraventriculares. Todos los tumores se localizaban a nivel ventricular: tres únicos en ventrículo izquierdo y el resto múltiples en ambos ventrículos. Ninguno de ellos ha provocado alteración funcional ni ha precisado tratamiento.
Conclusiones:Una masa intracardiaca en niños debe hacernos pensar en el RC. La clínica depende del tamaño tumoral, si es único o múltiple y de su expansión intracavitaria. Suele regresar espontáneamente por lo que no suelen tratarse con cirugía salvo si produce manifestaciones clínicas. No se realiza biopsia por el alto cociente riesgo/beneficio. La incidencia de ET en niños que tienen un RC es difícil de conocer, sobre todo por la aparición frecuente de la clínica en etapas posteriores.
P68ENFERMEDAD DE KAWASAKI. AFECTACION CORONARIA PRECOZ
Rosa M. Rodríguez García, Juan Francisco Expósito Montes, Ana Arévalo Garrido, Antonia Solas Beltrán, Irene Peláez Pleguezuelos, M. Dolores Gámez Gómez, África Ávila Casas, Francisca Serra Llorente, Jesús de la Cruz Moreno y Felipe González Rivera
Hospital Universitario Ciudad de Jaén.
Antecedentes y objetivo:La enfermedad de Kawasaki (EK) es una enfermedad febril multisistémica aguda, principal causa de enfermedad cardiaca adquirida en niños del mundo desarrollado ya que produce aneurismas coronarios hasta en el 25 % de los casos que no reciben tratamiento. Existen formas típicas, y formas atípicas que no cumplen todos los criterios diagnósticos pero que deben ser tenidas en cuenta dada la importancia del diagnóstico y el tratamiento precoz. En la EK atípica, pueden ser útiles los examenes de laboratorio, ya que más de 2 de los siguientes criterios: albúmina baja, hematocrito bajo, GPT elevada y VSG elevada, parecen mostrar una sensibilidad diagnóstica del 93 % en los pacientes que desarrollaban aneurismas coronarios.
Métodos:Presentación de un caso clínico con desarrollo de dilatación coronaria en la primera semana de evolución.
Resultados: Caso clínico: Niña de 2 años que ingresa en nuestro servicio por fiebre eleveda de 5 días de evolución, irritabilidad y quebrantamiento generalizado.Exploración física: Febril, sensación de enfermedad, hiperemia conjuntival bilateral no secretora, labios secos rojos y agrietados, mucosa orofaringea enrojecida, sequedad de piel en cara. Adenopatias laterocervicales menores de 1,5 cm. Eritema perianal. Resto de exploración normal. Exámenes complementarios: VSG 101 mm, Ht.º 30 %, leucocitosis (18430) N82 % L8 %, proteinas totales bajas. Plaquetas, PCR, transaminasas, iones, función renal, y estudio de coagulación normales. Ecocardiodoppler: dilatación de coronaria izquierda sin aneurisma, coronaria derecha de tamaño y morfología normales. Se inicia tratamiento con IGIV a 2 g/kg y AAS a dosis antiinflamatorias hasta 48 h después de ceder la fiebre, continuando a dosis antiagregantes. Tras 2 meses de evolución persiste dilatación coronaria sin aparición de aneurismas.
Conclusiones: 1.Debemos pensar en la EK en pacientes con fiebre inexplicada teniendo en cuenta la EK atípica en la que no se cumplen todos los criterios clásicos de la enfermedad. 2.Las alteraciones cardiacas pueden aparecer muy precozmente en la EK, por lo que debe realizarse estudio ecocardiográfico ante la sospecha de la enfermedad.
P69ENFERMEDAD DE KAWASAKI CON ANEURISMAS CORONARIOS
Manuel Haro Gómez, María José Villalobos Linares, M. Teresa Rojo Jurado, Fco. Javier Moya-Angeler Plazas, Joaquín Romero Cachaza, Ángel Alejo García-Mauricio y José González Hachero
Hospital Universitario Virgen Macarena, Sevilla.
Objetivo:Presentación de un caso de Enfermedad de Kawasaki (EK) diagnosticado en fase subaguda, con aneurismas coronarios en los tres troncos principales. Nos parece de interés por la escasa incidencia de dichas complicaciones actualmente.
Material y método: Anamnesis:Varón de 8 meses sin antecedentes de interés que ingresa por presentar fiebre de 20 días de evolución acompañada de síntomas catarrales, irritabilidad, conjuntivitis bilateral, fisuras labiales y exantema micropapular.Exploración: Regular estado general. Febril. Fontanela normotensa. Adenopatías laterocervicales bilaterales y rodaderas. No exantemas. Tonos cardíacos rítmicos, apagados, con soplo sistólico II/VI en mesocardio. Pulsos periféricos normales. Orofaringe y tímpanos hiperémicos. A las pocas horas del ingreso presenta fenómeno de Raynaud en mano y pie derechos. Exámenes complementarios: Hemograma: leucocitosis (23.400/mm3), trombocitosis (733.000 mm3) y anemia microcítica (Hb 8,2 g/dl, Hto. 24,8 %, VCM 75,4 fl). VSG: elevada (126 mm, 1.ª hora). PCR elevada (154 mg/l). Bioquímica sanguínea: GOT elevada (93 U/l). Serología a VEB, CMV, Toxoplasma, Borrelia, Brucella, Salmonella, Virus respiratorios y exantemáticos: negativa. Rx tórax: cardiomegalia. ECG: normal. SPECT: defecto de perfusión en caras anterior y septo. Ecografía y angiografía: demuestran la presencia de aneurismas en las tres ramas coronarias, dos de ellos gigantes.Tratamiento y evolución: Se inició a las 24 horas del ingreso (22 días de evolución) tratamiento con Gammaglobulina en dosis única a 2 g/kg en perfusión de 12 horas tras lo cual la fiebre desaparece. ASS a 80 mg/kg durante 5 días, continuando posteriormente a dosis de 5mg/dl de manera indefinida. Se estableció estadío 4 de afectación vascular, con un nivel de riesgo coronario 4/5, lo cual supone tratamiento con heparina subcutánea de bajo peso molecular y una valoración de las unidades de Enfermedades infecciosas, Cardiología y Hematología.
Comentario:La EK es una vasculitis aguda idiopática que afecta sobre todo a niños < 5 años, y cuya importancia radica en la posible afectación de arterias coronarias. Ésta complicación se puede prevenir con altas dosis de gammaglobulina intravenosa en la fase aguda de la enfermedad. El retraso en el inicio del tratamiento por un diagnóstico tardío, como ocurrió en nuestro caso podría justificar la aparición de los aneurismas. En resumen, destacamos el diagnóstico y tratamiento precoz.
P70RESULTADOS DE LA CIRUGIA DE NORWOOD
Teresa Álvarez Martín, Manuela Camino López, José Luis Zunzunegui Martínez, Carlos Maroto Monedero, Enrique Maroto Álvaro, Rubén Greco Martínez, Juan Carlos de Agustín Asensio, Belén Bernardo Atienza, M. del Rosario Jiménez Liria y Ana Olivar Gallardo
Hospital General Universitario Gregorio Marañón, Madrid, Hospital Torrecárdenas del SAS, Almería y Hospital Universitario Virgen del Rocío, Sevilla.
Introducción:Presentamos nuestra experiencia en la cirugía de Norwood en el Síndrome de Corazón Izquierdo Hipoplásico (SCIH) o equivalentes.
Material y métodos:Desde 1997 a 2003 se ha practicado intervención de Norwood, estadío I a 49 pacientes con SCIH (33) o equivalentes (16). Todos fueron diagnosticados ecocardiograficamente (5 intraútero) y a 3 se les practicó estudio hemodinámico. 3 SCIH presentaban persistencia de vena atriocardinal izquierda. La cirugía se practicó sin parada cardiocirculatoria, evitando la utilización de material protésico en la reconstrucción de la neoaorta. La fístula sistémico-pulmonar fue, salvo excepciones puntuales, de 3,5 mm. En los últimos 11 casos la conexión a pulmonar se realizó desde ventrículo derecho con injerto de gore-tex de 5-6 mm. Desde 1998 se han practicado 17 Norwood estadío II (Glenn bidireccional) bajo circulación extracorpórea (CEC).
Resultados:La edad media en el estadío I fue de 14,7 días (5-45 días) y el peso medio de 6,5 kg (5,1-7,4 kg). Necesitaron de ECMO 21 pacientes (disfunción ventricular, hipoxia o ambas). Pudieron ser dados de alta hospitalaria 29, de los cuales fallecieron súbitamente 5. De los 24 restantes 1 fue sometido a trasplante cardíaco por insuficiencia severa de la válvula neoaórtica, 6 esperan estadío II y a 17 se les ha efectuado Glenn bidireccional, de los cuales fallecieron 2 tardiamente por trombosis venosas. Quedan en seguimiento 22 niños (44,8 %).
Conclusiones:La intervención de Norwood es en la actualidad el tratamiento de elección en el SCIH o equivalentes. La mejoría en el tratamiento pre y post operatorio así como en la técnica quirúrgica y sus modificaciones hacen aumentar la supervivencia.
P71MIOCARDIO VENTRICULAR NO COMPACTO. ESTUDIO DE TRES CASOS
Ana Pérez Aragón, M. del Mar Rodríguez Vázquez del Rey, Montserrat de Felipe Jiménez-Casquet, Pilar Azcón González de Aguilar, Gabriela Lobo Tamer, Caridad Llopis Baño, José Miguel Pérez de la Cruz, Encarnación Montes Pecete, Mercedes Casado Rodríguez y Silvia García Huete
Hospital Virgen de las Nieves, Granada.
El miocardio ventricular no compacto (MVNC) es una miocardiopatía congénita muy rara, descrita recientemente, que se caracteriza por una alteración en la morfogénesis endomiocárdica, probablemente por una detención en su normal desarrollo que originan numerosas trabeculaciones miocárdicas.
Los pacientes pueden estar asintomáticos o presentar insuficiencia cardiaca, por disfunción sistólica, embolismos sistémicos y trastornos del ritmo. El diagnóstico se realiza por Ecocardiografía, donde se ven trabeculaciones localizadas en la región inferoapical del VI. La función ventricular puede estar conservada o deprimida al diagnóstico, y evolucionar hacia una disminución progresiva de la contractilidad. La función diastólica puede estar alterada provocando patrones restrictivos.
Presentamos tres casos, atendidos en nuestro Servicio, en los últimos 3 años.
Caso 1:Niño de 2 años diagnosticado al nacer de CIV, CoAo y ductus corregidos quirúrgicamente. En los sucesivos controles ECO-DOPPLER presentaba VI hipertrófico con aspecto desestructurado y función sistólica normal. Actualmente asintomático.
Caso 2:Niña de 4 años con S. dismórfico craneofacial y estudio citogenético normal, diagnosticada de CIV y CoAo severa con un postoperatorio tormentoso con insuficiencia cardiaca y signos de miocardiopatía dilatada crónica con MCNC que requiere tratamiento anticongestivo mantenido.
Caso 3:Niño 1,5 años, que debuta con un cuadro de ICC. El ECO demuestra mala contractilidad de ventrículo izquierdo con MVNC. Tras breve mejoría de 20 días vuelve a presentar insuficiencia cardiaca refractaria al tratamiento que termina en exitus.
Comentario:El MVNC entidad de reciente diagnóstico descrita inicialmente en niños, pueden afectar a ambos ventrículos y causar evolutivamente una severa disfunción sistodiastólica del VI. Se presenta de forma aislada sin patología añadida e infrecuente y asociada a arritmias y dismorfia facial. La base genética no es bien conocida, aunque puede presentarse una recurrencia familiar de hasta el 45 %. El tratamiento es sintomático, con posibilidad de transplante como última solución.
P72ASISTENCIA VENTRICULAR Y RESCATE CARDIOPULMONAR CON OXIGENACION CON MEMBRANA EXTRACORPOREA EN PACIENTES PEDIATRICOS
José Luis Zunzunegui Martínez, Manuela Camino López, Teresa Álvarez Martín, Carlos Maroto Monedero, Enrique Maroto Álvaro, M. Luisa Franco Martínez, Rubén Greco Martínez, Fernando Ballesteros Tejerizo, M. del Rosario Jiménez Liria y Raquel Gómez Casares
Hospital General Universitario Gregorio Marañón, Madrid, Hospital Torrecárdenas del SAS, Almería y Hospital Clínico San Carlos, Madrid.
Introducción:Las técnicas de soporte vital ECMO y los dispositivos de asistencia ventricular (DAV) suponen la posibilidad de recuperar algunos pacientes con cardiopatías congénitas que de otra forma fallecerían. Por"Rescate cardiopulmonar" entendemos aquellos pacientes que presentan un fracaso cardiopulmonar refractario a las medidas terapéuticas y que precisan estas técnicas de soporte para sobrevivir bien antes de la entrada en Circulación Extracorpórea (CEC) o una vez que han salido de CEC en la Unidad de Cuidados Intensivos. Excluímos aquellos pacientes que han precisado soporte cardiopulmonar por imposibilidad de salir de bomba.
Material y métodos:Desde 1998 a Enero de 2003 se han realizado 12 rescates cardiopulmonares. La edad fue de 9 días a 7 años y el peso de 2.500 gramos a 22 kg.
En 3 pacientes el rescate se efectuó antes de la cirugía, 2 miocardiopatías dilatadas con fallo 9 ventricular refractario y 1 atresia pulmonar con hipoplasia extrema de ramas pulmonares e hipoxia severa. En 9 pacientes se realizó en el postoperatorio (3 D-TGA con corrección anatómica, 3 Norwood por Hipoplasia de ventrículo izquierdo y en 2 ventrículos únicos con hipoplasia aórtica, 2 ventrículos únicos uno con fístula sistémico-pulmonar,y uno con banding de la arteria pulmonar y estenosis traqueal congénita, 1 corrección completa de ventrículo derecho de doble salida). Los motivos de precisar el soporte cardiopulmonar fueron: una hemorragia pulmonar, una taquicardia automática de la unión con acidosis láctica, dos hipoxemias refractarias, dos paradas cardíacas, una rotura bronquial, un fallo ventricular izquierdo y una crisis de hipertensión pulmonar.
Resultados:ECMO Venoarterial: 5 pacientes, duración 7 días, decanulación en 4 (80 %), alta hospitalaria en 3 (60 %). ECMO Venovenoso: % pacientes, duración 5,4 días (1-19), decanulación en 3 (60 %), alta hospitalaria en 2 (40 %). DAV (Asistencia ventricular izquierda): 2 pacientes, duración 8,5 días (5-12). Ninguno fue decanulado y no se consiguió alta. Las causas de mortalidad fueron en todos los casos por fallo multiorgánico y/o mal pronóstico neurológico con interrupción de la asistencia.
Conclusiones:El 58 % de los pacientes son rescatados (decanulación) con éxito y el 42 % recibieron el alta hospitalaria. ECMO y DAV son recursos eficaces que consiguen rescatar algunos pacientes críticos.
P73TRASPLANTE CARDIACO EN MIOCARDIOPATIA HIPERTROFICA
Teresa Álvarez Martín, Manuela Camino López, José Luis Zunzunegui Martínez, Carlos Maroto Monedero, Enrique Maroto Álvaro, Rubén Greco Martínez, Fernando Ballesteros Tejerizo, Águeda Herrera Chamorro, Raquel Gómez Casares y Ana Olivar Gallardo
Hospital General Universitario Gregorio Marañón, Madrid, Hospital Clínico San Carlos, Madrid y Hospital Universitario Virgen del Rocío, Sevilla.
Introducción:El tratamiento de la miocardiopatía hipertrófica es controvertido. Desde médico a quirúrgico, eléctrico o mediante cardiología intervencionista, el trasplante cardíaco es una opción, no preventivo de la muerte súbita pero indicado en casos seleccionados.
Descripción del caso:Paciente de 13 años de edad diagnosticado desde los 3 meses de vida de miocardiopatía hipertrófica. La causa del diagnóstico fue un estudio realizado por antecedentes de 3 casos de muerte súbita en familiares de primer grado con miocardiopatía hipertrófica, comprobada en un caso. El niño seguía tratamiento con beta-bloqueantes y estaba asintomático hastsa los 12 años de edad. Presentaba progresiva disminución de capacidad funcional hasta disnea de mínimos esfuerzos. Examen físico:Normal, salvo soplo sistólico III/VI en borde esternal izquierdo.Examenes complementarios: ECG: ritmo sinusal. Hemiblolqueo anterior izquierdo. Hipertrofia biventricular y auricualr izquierda. Rx Tórax: cardiomegalia moderada. Ecocardiografía 2D-Doppler: hipertrofia septal asimétrica con gradiente medio apical izquierdo ventricular significativo. SAM mitral. Insuficiencia mitral ligera. Gradiente infundibular ventricular derecho significativo por hipertrofia septal.
El estudio Holter reveló que caminando pocos metros aparecíaan episodios frecuentes de taquicardia monomórfica ventricular derecha.
Por grado funcional, episodios de taquicardia ventricular, no respuesta a tratamiento médico y probable no respuesta a tratamiento convencional (marcapasos bicameral, cirugía u oclusión de aterias coronarias septales) se indicó trasplante cardíaco ortotópico que se realizó sin complicaciones. Dos años después el paciente se encuentra en clase funcional I con Tacrolimus y azatioprina y sin corticoides.
Conclusiones:El trasplante cardíaco es una alternativa en caso de una miocardiopatía hipertrófica.
No debe emplearse como preventivo de muerte súbita y debe indicarse por sintomatología funcional y cuando las perspectivas de tratamiento convencional o no hallan dado resultados o sea de dudosa eficacia.
P74RESCATE CON ECMO DE PACIENTE CON TAQUICARDIA NO PAROXISTICA DE LA UNION AV EN EL POSTOPERATORIO DE TETRALOGIA DE FALLOT
Teresa Álvarez Martín, Manuela Camino López, José Luis Zunzunegui Martínez, Carlos Maroto Monedero, Enrique Maroto Álvaro, Rubén Greco Martínez, Fernando Ballesteros Tejerizo, Águeda Herrera Chamorro, Raquel Gómez Casares y Ana Olivar Gallardo
Hospital General Universitario Gregorio Marañón, Madrid, Hospital Clínico San Carlos, Madrid y Hospital Universitario Virgen del Rocío, Sevilla.
Introducción:La taquicardia no paroxística de la unión AV (TPNU) en el postoperatorio de cirugía cardíaca pediátrica es una complicación grave con mortalidad no despreciable. Se han intentado diversas modalidades de tratamiento.
Descripción del caso:Lactante de 3 meses de edad y 5 kg de peso diagnosticado clínica, eléctrica, radiológica y ecocardiográficamente de Tetralogía de Fallot con hipoplasia de anillo y tronco pulmonar con anomalías coronarias.
Fue intervenido bajo circulación extracorpórea con cierre de comunicación interventricular y homoinjerto pulmonar valvulado.
En el postoperatorio inmediato desarrolló TPNU rebelde a las medidas habituales con disminución de inotrópicos, ATP, digital, amiodarona, e hipotermia con marcapasos bicamenral.
En estado de shock cardiogénico se conectó a ECMO Venoarterial (Canulación carótida-yugular) que se mantuvo durante 5 días con ritmo sinusal, recuperado 24 horas antes. Estabilidad hemodinámica y respiratoria posterior con alta hospitalaria a los 16 días del porstoperatorio.
Conclusión:La oxigenación con membrana extracorpórea es una terapeutica de rescate útil e imprescindible en la TPNU en la que han fracasado medidas terapéuticas convencionales.
P75PERICARDITIS BACTERIANA EN UN LACTANTE
M. del Carmen Torres Torres, M.ª José González García, Gabriel Cebriá Tornos, Amparo López Lafuente, M.ª José López Rodríguez y Valentín Carretero Díaz
Hospital San Pedro de Alcántara, Cáceres.
Introducción:El Estafilococo aureus es el germen causal más frecuente el las pericarditis bacterianas (PB). Los síntomas habituales son: dolor precordial, respiración entrecortada, tos, fiebre y taquicardia. El diagnóstico de presunción se realiza mediante el ecocardiograma. El tratamiento se basa en la evacuación del derrame mediante una ventana pericárdica, antibióticos durante 3 semanas.
Caso clínico:Lactante de 19 meses con fiebre elevada de 6 días de evolución y decaimiento del estado general. Aspecto catarral con rinorrea. AF: sin interés. AP: invaginación intestinal a los 12 meses de edad resuelta con enema de bario.Exploración física: T.ª 39,5 °C. TA: 100/60 mmHg. Regular estado general. No exantema ni petequias. Signos meníngeos negativos. No rigidez de nuca. Orofaringe eritematosa. Auscultación Pulmonar normal. Auscultación cardíaca: rítmica, ruidos discretamente apagados, sin soplos. FC: 150 lpm. Pulsos periféricos normales. Abdomen: blando, depresible, hígado a 1 cm de reborde costal.Pruebas complementarias: Hemograma: leucocitosis con desviación a la izquierda. Coagulación: Fibrinógeno: 986 mg %. PDF: positivo. Dímero D: 1146. PCR: 194,6 mg/l. Bioquímica basal, perfil hepático, LCR y Sistemático de orina: normal. Rx Tórax: cardiomegalia (ICT: 0,71), vascularización pulmonar normal. ECG: ritmo sinusal, elevación de ST en DII, aVF y precordiales izquierdas. Ecocardiograma: derrame pericárdico dividido por tabiques fibrinoides en tres compartimentos (lateral izquierdo, lateral derecho y apical).Evolución: El derrame pericárdico fue evacuado mediante realización de una ventana pericárdica (esternotomía). Bacteriología: gérmenes aerobios y anaerobios (contaminación). Tratamiento antibiótico Vancomicina,Clindamicina, Penicilina G) durante 3 semanas.
Comentarios:1. Cardiomegalia sin Cardiopatía congénita conocida debe hacer pensar en Pericarditis como primera sospecha diagnóstica.2. La sospecha diagnóstica de PB exige la realización de una ventana pericárdica con drenaje continuo del derrame pericárdico, no siendo suficiente la práctica de Pericardiocentesis seriadas.3. La PB exige un seguimiento ecográfico por su posible evolución a Pericarditis Constrictiva.
CUIDADOS INTENSIVOS
P76 HEMOCROMATOSIS NEONATAL. PRESENTACION DE TRES CASOS
Ester Barrios Miras, Miguel Ángel Delgado Domínguez y Francisco Javier Ruza Tarrío
Hospital Universitario La Paz, Madrid.
Forma de hepatopatía caracterizada por un depósito progresivo de hierro en células parenquimatosas. Causa frecuente de fallo hepático fulminante neonatal con pronóstico fatal en la mayoría de casos.
Caso 1:Paciente varon de 30 dias que ingresa para estudio de hipotonía, ictericia y coagulopatía severa. A su llegada presenta hemorragia pulmonar masiva ingresando en Cuidados Intensivos Pediátricos (CIP). En analítica se objetiva fallo hepático severo (tabla 1) por lo que se inicia estudio, durante su estancia signos de encefalopatía grado III y patron de "Burst supresión" en EEG. Fallece a los 9 dias por PCR. Resultado de autopsia:Hemocromatosis Neonatal (H.N)
Caso 2:Paciente mujer de 30 dias con vómitos e ictericia desde nacimiento. Bradicardia severa que requiere intubación y traslado a CIP al llegar al servicio de Urgencias. En analítica de ingreso (tabla 1) fallo hepático y en punción de médula ósea proliferación anómala de linfocitos B. Fallece a los 18 dias por sangrado por foco ORL y PCR. Resultado de la autopsia: H.N.
Caso 3:Paciente varon de 30 dias con ictericia prolongada, en analítica del servicio de Urgencias (tabla 1) se diagnostica de fallo hepático severo ingresando en CIP. Se inicia estudio y se solicita metabolismo del hierro que sugiere hemocromatosis neonatal. Se inicia tratamiento con quelantes y se pone en urgencia 0 para trasplante hepático. Fallece a los 10 dias por hemorragia pulmonar con PCR. Resultado de la autopsia: H.N.

P77USO DE SOLUCION SALINA HIPERTONICA EN COMPARACION A MANITOL EN PACIENTES PEDIATRICOS CON TRAUMA CRANEOENCEFALICO GRAVE
José Rigoberto Canché Fernández, Emma Carbajal Reyes, J. Félix Urbina Hernández y Patricia Beltrán Ramírez
Hospital para el Niño Poblano, Puebla (México).
Antecedentes y objetivo:El trauma craneoencefálico grave es una importante causa de muerte infantil. Uno de los objetivos en el tratamiento es disminuir la presión intracraneana debido a edema cerebral. Para esto se ha usado distintos tratamientos. El uso de solución hipertónica es un tratamiento económico y accesible que puede ser usado en terapias intensivas de países en vías de desarrollo. El objetivo de este ensayo es estudiar su seguridad y eficacia en comparación con el Manitol.
Métodos:Se incluyeron 9 pacientes pediátricos con TCE grave con hipertensión intracraneana a pesar del manejo convencional, todos monitorizados con catéter de presión intracraneana, saturación de bulbo de la yugular, línea arterial y venosa central dividiéndose a su ingreso en dos grupos: grupo A al que se administró solución hipertónica de Sodio al 3 % a 3 ml/kg/dosis y el grupo B con manitol al 8 % 3 ml/kg/dosis, con medición posterior de distintas variables hemodinámicas y cerebrales así como su sobrevida y morbilidad.
Resultados:La edad promedio fue de 6 años, con 4 mujeres y 5 hombres. La presión intracraneana en el Grupo A disminuyó más rápido que en los del grupo B pero sin diferencia significativa (p = 0,09); La presión de perfusión cerebral en el Grupo A aumentó con diferencia significativa con respecto al grupo B (p = 0,0008) incrementado la presión arterial media. La mortalidad en el Grupo A fue menor de manera porcentual (33 % vs 67 % en el grupo B). En el grupo B la disminución de la PIC fue semejante a los pacientes del Grupo A aunque se registró en los pacientes del grupo B disminución de la presión arterial media.
Conclusiones:De acuerdo a nuestros resultados, la solución hipertónica de Sodio al 3 % es eficaz en el tratamiento para disminuir la presión intracraneana con efectos benéficos sobre la presión arterial y la presión de perfusión cerebral, influyendo en la morbimortalidad de los pacientes con pediátricos con trauma craneoencefálico grave.
P78SINDROME CENTROMEDULAR CERVICAL DE ORIGEN TRAUMATICO
Sonia Martínez González, Cristina Calvo Monge, Pablo Diego Gayte, Yolanda López Fernández, Julio López Bayón, Fco. Javier Pilar Orive, Julián Latorre García y M. Teresa Hermana Tezanos
Hospital de Cruces, Baracaldo.
Introducción:El síndrome centromedular se produce por una hiperextensión traumática de la columna cervical. La afectación medular sin alteración radiológica, SCIWORA (spinal cord injury without radiologiocal anormality), es más frecuente en niños, probablemente por una mayor elasticidad y laxitud ligamentosa, permitiendo un movimiento excesivo durante el traumatismo, que causa lesión medular en ausencia de fractura o luxación vertebral.
Caso clínico:Niña de 12 años de edad, con antecedente 3 meses antes de esguince cervical, tras caída del caballo, con normalidad clinica posterior. Ingresa por presentar de forma brusca, dolor cervical y debilidad muscular generalizada, a las 24 horas de experimentar un mecanismo de hiperextensión cervical repetido mientras practicaba equitación. A su exploración inicial presenta una tetraparesia de predominio braquial, con reflejos osteotendinosos disminuidos, sin afectación sensitiva, y con retención urinaria. La radiografía de columna cervical, la TAC craneo-cervical, la analítica sanguínea y el LCR fueron normales. Presenta una progresión de la debilidad muscular, asociando dificultad respiratoria, en el habla, y en la deglución, ingresando en UCIP. El tratamiento fue conservador con oxigeno en reservorio (hasta 3 L/min), y dexametasona iv. En la RNM cervical se objetiva una imagen de hiperseñal lineal centromedular en los niveles C2 hasta C4. La AngioRNM, ecodoppler de troncos-supraórticos, PES, EMG fueron normales. La evolución fue favorable, y al alta la tetraparesia era menor, de predominio braquial y proximal, asimétrica, con sensibilidad conservada, y control de esfinteres. En el control evolutivo (a los 40 días) presentaba atrofia muscular de la cintura escapular izquierda, con una afectación mayor de dicha extremidad, y recuperación casi completa de la derecha y las inferiores.
Comentarios:La lesión medular por "latigazo cervical", sin luxación o fractura, no es frecuente fuera del periodo de lactante. La lesión centromedular se asocia a un pronóstico más favorable, como ocurrió en este caso, sin poder descartarse futuras lesiones de siringomielia.
P79BRONQUIOLITIS EN UCIP (2002-2003). ESTUDIO DESCRIPTIVO Y COMPARATIVO RESPECTO A LA TEMPORADA PREVIA
Pablo Diego Gayte, Sonia Martínez González, María González Balenciaga, Cristina Calvo Monge, Fco. Javier Pilar Orive, Julián Latorre García, Julio López Bayón, Yolanda López Fernández y M. Teresa Hermana Tezanos
Hospital de Cruces, Baracaldo.
Antecedentes y objetivo:Análisis y comparación de los datos obtenidos de los pacientes hospitalizados en nuestra UCIP durante la presente época epidémica de bronquiolitis (2002-2003) respecto a los que fueron hospitalizados en UCIP durante la temporada 2001-2002.
Métodos:Para ello hemos recopilado todas las historias clínicas de los niños hospitalizados en UCIP con el diagnóstico de bronquilitis entre octubre del 2001 y marzo del 2003 para realizar un estudio retrospectivo y comparativo.
Resultados:Este último año no ha habido un incremento significativo de los niños hospitalizados por bronquiolitis; no obstante, un 20 % de los niños hospitalizados por bronquiolitis han precisado ingresar en UCIP frente a un 10,52 % del año anterior. El mayor pico en cuanto al número de ingresos en UCIP se ha producido en diciembre, manteniéndose elevado en enero. Hemos comprobado que el aumento de ingresos ha sido a costa de niños menores de un mes de edad, sin haber dentro de éstos una tasa mayor de niños exprematuros que el año anterior. Un 66 % de los niños ingresan en UCIP con menos de 72 horas de evolución presentando en el momento del ingreso signos clínicos y gasométricos de gravedad, llamando la atención que un 26 % de ellos tiene una hiponatremia marcada. Dentro del tratamiento empleado en UCIP, en un 80 % de los casos se utiliza adrenalina y salbutamol nebulizados y corticoide IV y en un 40 % de ocasiones teofilina IV; solo un caso ha precisado VMC y ventilación en prono. El tiempo de estancia media ha sido de 9,8 días y las complicaciones descritas han sido: apneas (2 casos), SDRA, neumotórax y broncoaspiración.
Conclusiones:Este año la epidemia de bronquiolitis se ha adelantado un mes repecto a la temporada anterior y hemos asistido en UCIP a un mayor número de niños < 1 mes afectos de bronquiolitis sin haber habido un incremento similar en otras edades. Este aumento no ha sido a costa de un mayor porcentaje de exprematuros. A pesar de ello, ha habido una tasa baja de complicaciones.
P80ANALGESIA CONTROLADA POR EL NIÑO VERSUS POR LA ENFERMERA EN CUIDADOS INTENSIVOS PEDIATRICOS
Sonia Marcos Alonso, Antonio Rodríguez Núñez, Manuel Fernández Sanmartín, Montserrat López Franco, Federico Martinón Torres y José M.ª Martinón Sánchez
Hospital Clínico Universitario. Complejo Hospitalario Universitario, Santiago de Compostela.
Antecedentes personales y objetivos: La prevención y el tratamiento del dolor infantil debe ser una prioridad para el pediatra. El objetivo del presente trabajo es comparar la aplicación clínica de la analgesia controlada por el niño (ACN) o por la enfermera (ACE) en una Unidad de Cuidados Intensivos Pediátricos (UCIP).
Métodos:De forma prospectiva se les administró fentanilo a un grupo de 91 niños con dolor moderado y severo ingresados en nuestra UCIP durante un período ininterrumpido de dos años. Los niños mayores de 6 años con capacidad intelectual y sin limitación física recibieron ACN y en los demás la enfermera administraba los bolus adicionales de analgésico. El protocolo inicial incluyó una infusión basal de 1 μg/kg/h, con bolus de 0,5 μg/kg, tiempo de bloqueo de 5 minutos y un máximo de 10 bolus/hora. Cada 24 horas recogíamos las cifras de consumo, bolus y efectos adversos observados, modificando los parámetros según la situación clínica. Monitorizamos a todos nuestros pacientes de forma continua (ECG, frecuencia respiratoria y pulsioximetría).
Resultados:66 pacientes recibieron ACN frente a 25 que recibieron ACE. En los dos grupos el tiempo de mantenimiento coincidió (mediana: 4). El consumo medio de analgesia el 1.º día fue similar en ambos grupos, descendiendo progresivamente siendo dicho descenso más lento en el grupo ACE. Los bolus administrados fueron significativamente mayor en el grupo ACN reflejándose este dato en la observación de mayor número de efectos adversos menores (náuseas, vómitos, prurito y retención urinaria) y un efecto adverso mayor (depresión respiratoria, que revertida con oxígeno y estimulación física).
Conclusiones:Los sistemas de analgesia controlada son aplicables y eficaces en los niños de cualquier edad en el tratamiento del dolor moderado y severo. Según nuestros resultados es posible que la enfermera minusvalore el dolor del niño, o bien tema provocarle efectos adversos; aunque la incidencia de estos es aceptable los niños deben estar monitorizados de forma adecuada.
P81INTOXICACION POR EXPOSICION PASIVA A CARBAMATOS EN UN LACTANTE
Raquel Perera Soler, Ángel Antonio Hernández Borges, José Manuel Rial Rodríguez, José Luis Aparicio Sánchez y Raúl Cabrera Rodríguez
Hospital Universitario de Canarias, La Laguna y Hospital Universitario Ntra. Sra. de Candelaria, Santa Cruz de Tenerife.
Paciente de 12 meses de edad que ingresó en nuestra UCIP por irritabilidad, dificultad respiratoria, decaimiento generalizado y broncorrea intensa de aparición brusca tras estar en contacto con un familiar que llegaba de trabajar en el campo. Exploración física al ingreso: FC 180 lpm; FR 28 rpm; TA 110/70; T.ª 36 °C; dificultad respiratoria con retracción esternal, auscultación cardiorrespiratoria con murmullo vesicular conservado y ruidos transmitidos de vías respiratorias altas. SN: pupilas mióticas (puntiformes) e importante hipotonía, con nivel de conciencia conservado. Cardiaco y abdomen, normal.
Pruebas complementarias:1. Analíticas y perfil bioquímico, y EAB venoso: normal. 2. Tóxicos (cocaína, opiáceos, benzodiazepinas y cannabis): negativo.3. Colinesterasa sérica: 2 UI/l (N: 4,2-14,3 UI/l). 4. Muestra de orina enviada al Instituto Nacional de Toxicología (Delegación de Canarias): presencia de carbofurán fenol y cetocarbofurán-fenol, metabolitos del carbofurano.
Comentario:Debido a la sospecha de intoxicación por anticolinesterásicos se inicia tratamiento con atropina (hasta 5 dosis) y pralidoxima en perfusión continua, lográndose la extubación y la normalización clínica y de los niveles de colinesterasa a las 12 horas del ingreso. Aunque nuestro caso apuntaba a una intoxicación leve por organosfosforados quedaba la duda de otros tóxicos que pudieran producir un síndrome colinérgico ya que la evolución fue benigna y corta, se manifestaron tanto síntomas muscarínicos como nicotínicos y no presentaba afectación del SNC (ni convulsiones, ni coma). En nuestro caso se detectó carbofurán fenol en orina, un plaguicida del grupo de los carbamatos.
Ante un niño con síndrome colinérgico agudo se debe tener presente como posible causa la intoxicación por organofosforados o carbamatos, siendo los últimos más probables si la clínica es leve, sin afectación del SNC, y con evolución benigna. Aunque la forma de intoxicación suele ser la ingestión de pesticidas, no hay que descartar el contacto cutáneo sobre todo en niños que no deambulan.
P82INTOXICACION SEVERA POR CAUSTICO
Cristina Montero Valladares, José Carlos Flores González, Lidia M. Santalo González, Alberto Varona García, José Domingo López Castilla, Antonio Vázquez Florido, Miguel Muñoz Sáez, Juan Antonio Soult Rubio, Elia Sánchez Valderrábanos y Mercedes Carranza Conde
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción:Las intoxicaciones son una causa frecuente de consulta en los servicios de Urgencias pediátricos. Sin embargo su incidencia ha disminuido en los últimos 10 años, tanto las debidas a medicamentos como las producidas por productos del hogar. Dentro de éstas últimas los álcalis-cáusticos constituyen la causa más frecuente de intoxicación. En la edad pediátrica la ingesta del tóxico suele ser accidental y en su mayoría ocurre en el domicilio.
Caso clínico:Lactante de 16 meses de edad que consulta por ingesta accidental de hidróxido sódico (desatascador) en su domicilio y vómitos repetidos. A la exploración presenta quemaduras cáusticas severas en tracto digestivo alto y vía aérea superior, procediéndose a su intubación e ingreso en UCIP. Precisa conexión a ventilación mecánica, sueroterapia intravenosa, dieta absoluta, protección gástrica con ranitidina, sedoanalgesia y corticoterapia a dosis de 25 mg/kg/día (7 días). Se inicia nutrición parenteral total a las 24 horas del ingreso manteniéndose 19 días y tolera alimentación oral al 16.º día. Durante la terapia con corticoides aparecen hiperglucemias que se normalizan al disminuir las dosis de éstos. Presenta hemorragia digestiva alta que requiere tratamiento con sucralfato y omeprazol. Se realiza laringoscopia directa al 6.º día de ingreso donde se observan escaras y lesiones sangrantes con edema en base de lengua, vallécula y epiglotis con retracción de la misma, aconsejándose el mantenimiento de la ventilación mecánica. Se instaura tratamiento antibiótico por la presencia de imagen radiográfica compatible con neumonitis química. Extubación al 13.º día tras apreciarse mejoría de las lesiones en la segunda laringofibrobroncoscopia. Al alta no presenta dificultad respiratoria, recibe alimentación oral exclusiva y el estudio esofagogástrico baritado es completamente normal.
Comentarios:Los corticoides a altas dosis deben mantenerse al inicio del cuadro para controlar el proceso inflamatorio y evitar estenosis cicatriciales posteriores. Las laringofibrobroncoscopias sucesivas indicaron la necesidad de intubación electiva para proteger la vía aérea y el momento adecuado de la extubación. El tratamiento agresivo y precoz evitó, en este caso, la aparición de secuelas severas que alterasen su calidad de vida.
P83SINDROME DE SHOCK TOXICO POR ESTREPTOTOCO PYOGENES
José Carlos Flores González, Cristina Montero Valladares, Alberto Varona García, José Domingo López Castilla, Miguel Muñoz Sáez, Elia Sánchez Valderrábanos, Antonio Vázquez Florido, Lidia M. Santalo González, Juan Antonio Soult Rubio y Mercedes Carranza Conde
Hospital Universitario Virgen del Rocío, Sevilla.
Introducción:El síndrome del shock tóxico (SST) es una entidad poco frecuente en la infancia, descrita por primera vez en 1978. El Estreptococo del grupo A no se relacionó como agente causal del SST hasta 10 años después. La reacción hiperinmune producida por las toxinas del S. pyogenes pueden llegar a provocar un fracaso multiorgánico con una elevada mortalidad.
Caso clínico:Niña de 5 años que acude a Urgencias por fiebre alta y coxalgia de 3 días de evolución. A su ingreso en UCIP presenta mal estado general, letárgica, taquicárdica, taquipneica, eritrodermia macular difusa, hiperemia bucofaríngea, relleno capilar enlentecido con mala perfusión generalizada, pulsos periféricos no palpables y micropetequias en miembros. Ante la sospecha de sepsis se inicia tratamiento con expansores de volumen, inotrópicos y cobertura antibiótica. A las 3-4 horas aparece una agitación progresiva coincidiendo con mayor trabajo respiratorio sin hipoxemia ni hipercapnia y cuadro de coagulopatía de consumo. En los miembros superiores se observan lesiones equimótico-ampollosas que desarrollan un síndrome compartimental. A las 18 horas del ingreso presenta dos parada cardiorrespiratoria sucesivas que remontan con maniobras de RCP avanzada. Posteriormente se establece un cuadro de shock refractario con hipotensión severa y oligoanuria que requiere perfusiones de inotrópicos a altas dosis (dopamina, dobutamina y noradrenalina), hemodiafiltración y perfusión de proteína C coagulativa. Ante la persistencia de la hipotensión marcada iniciamos tratamiento con bolos de terlipresina, consiguiendo normalización de sus tensiones. En el hemocultivo se aisla Estreptococo pyogenes añadiendo al tratamiento clindamicina y penicilina. Tras confirmación microbiológica del SST se administra gammaglobulina hiperinmune. A las 72 horas presenta hipoxemia severa por cuadro de SDRA que precisa parámetros agresivos de ventilación mecánica y óxido nítrico, complicándose con la aparición de neumotórax bilateral que requirió drenaje. A pesar del tratamiento intensivo es éxitus a los 6 días del ingreso.
Comentarios:El síndrome del shock tóxico debe incluirse en el diagnóstico diferencial de una sepsis. La terlipresina es un recurso útil en el cuadro de hipotensión refractaria. No obtuvimos respuesta con la gammaglobulina hiperinmune. El concentrado de proteina C mejoró el cuadro de CID.
P84SINDROME DE WIC
Bruno Nievas Soriano, Esther Aleo Luján, Luis Arruza Gómez, Olga Pérez Rodríguez y José Luis Ruibal Francisco
Hospital Clínico San Carlos, Madrid.
Introducción:Se presenta el caso de una lactante de dos meses de edad que sufrió un episodio de estatus convulsivo por hiponatremia dilucional secundaria a la alimentación exclusiva con biberones de agua.
Caso clínico:Niña de 2 meses que es traída a nuestro servicio de urgencias por cuadro de agitación, tras haber recibido una toma de agua con canela. A su ingreso presenta buen estado general pero con marcada irritabilidad. Bien hidratada, la exploración general fue normal. Durante su estancia en urgencias inició un cuadro de movimientos tónico-clónicos generalizados y mioclonias de predominio en hemicuerpo derecho, que no respondieron a la terapia anticonvulsivante. Se realizó bioquímica urgente que mostró una hiponatremia de 115 mEq/l. Se corrigió el déficit hasta 120 mEq/l en 30 minutos, cediendo entonces el cuadro convulsivo. En la anamnesis dirigida, la madre relató que la niña llevaba siete días en alimentación exclusiva con agua endulzada con canela. De entre las exploraciones complementarias realizadas no se evidenciaron otras alteraciones salvo la severa hiponatremia antes referida con una natriuria acompañante de 30 mEq/l. La evolución clínica fue satisfactoria llegando a la corrección completa del sodio en sangre a las 24 horas de su ingreso. La madre era inmigrante, procedente de Ecuador, y carecía de recursos económicos con los que atender adecuadamente a su hija. Durante su ingreso se contactó con los servicios de asistencia social para resolver su situación socioeconómica.
Conclusiones:En Estados Unidos la causa más frecuente de hiponatremia por aporte de sodio insuficiente es el síndrome de WIC, así llamado por el programa norteamericano de ayuda a mujeres, lactantes y niños (Women, Infants, Children). Estos lactantes,sin embargo, son alimentados con fórmulas diluídas o agua exclusivamente, debido a que los recursos administrados resultan insuficientes. Dado el claro aumento de la población inmigrante en nuestro país, este síndrome debe ser tenido en cuenta a la hora del diagnóstico diferencial de la hiponatremia en lactantes de poblaciones con escasos recursos económicos.
ENDOCRINOLOGIA
P85ACIDOSIS TUBULAR RENAL DISTAL CON GRAVE AFECTACION OSEA Y FUNCIONAL
Bruno Nievas Soriano, Vitoria San Antonio Arce, Olga Pérez Rodríguez, José Luis Ruibal Francisco, Alejandro Pazos García y Ramón Sánchez Jacob
Hospital Clínico San Carlos, Madrid.
Introducción:Se presenta un caso de acidosis tubular renal (ATR) distal en una paciente de 16 años de edad, inmigrante, no diagnosticado previamente y que le condicionaba severas secuelas funcionales.
Caso clínico:Paciente mujer de 16 años de edad, procedente de Ecuador, con un cuadro de talla baja muy patológica (113 cm), y grave incapacidad funcional por deformidades óseas importantes.
Entre sus antecedentes personales destaca la presencia, desde su infancia precoz, de dolores óseos generalizados y múltiples fracturas patológicas. En el momento actual sus deformidades le impiden la bipedestación y la deambulación.
A la exploración destacan las deformidades óseas, genu valgo bilateral y cifoescoliosis. No sordera, ni ceguera, ni presencia de escleróticas azules.
Radiológicamentepresenta osteoporosis generalizada, diversas fracturas en diferentes estadíos de consolidación y signos de raquitismo generalizado, así como nefrocalcinosis bilateral.
Bioquímicamentellaman la atención la presencia de una acidosis metabólica con anión GAP normal, e hipopotasemia (pH = 7,30; Bic:18; Cl:106; K:2,2). En orina, pH = 7,50; aumento de fosfaturia, calciuria, kaliuria y bicarbonaturia. En sangre presenta urea y creatinina normales, con Ca:8,5; P:2,2 (normal de 2,5 a 4,5). El aclaramiento de creatinina corregido es de 60 ml/min.
En losestudios de procesos que secundariamente producen este síndrome se han descartado enfermedades autoinmunes, alteraciones primarias del riñón y vías urinarias y enfermedades primarias del metabolismo fosfo-cálcico, infiriendo por ello que se trata de una ATR tipo I (distal) congénita, pendiente de confirmar mediante estudio genético y molecular.
Discusión:La ATR distal congénita es un proceso que se suele presentar a edades precoces, a diferencia de la secundaria, que suele aparecer más tardíamente. Su sospecha diagnóstica debe ser precoz, ya que un temprano tratamiento de la acidosis evita, en su mayor parte, las graves afecciones óseas y metabólicas de estos pacientes.
P86HIPERTIROIDISMO NEONATAL TRAS TIROIDECTOMIA MATERNA
Azucena Díez Suárez, Mirella Gaboli, Cristina Azcona San Julián y Valentín Alzina de Aguilar
Clínica Universitaria de Navarra, Pamplona.
La enfermedad de Graves neonatal es una afección poco frecuente, que ocurre tan sólo en el 1 % de los recién nacidos de madres afectas de enfermedad de Graves, y se debe al paso transplacentario de inmunoglobulinas estimulantes del tiroides (TSI). En ocasiones, la producción de estas inmunoglobulinas persiste tras la realización de una tiroidectomía. El hipertiroidismo neonatal se caracteriza por la presencia de prematuridad, bajo peso, escasa ganancia ponderal, oftalmopatía, bocio, irritabilidad, taquicardia, e incluso insuficiencia cardiaca.
Presentamos el caso de un neonato pretérmino de 35 semanas de edad gestacional con enfermedad de Graves neonatal, cuya madre había sido intervenida mediante tiroidectomía total 6 años antes del embarazo. La madre estaba asintomática y eutiroidea durante la gestación. Se induce el parto por taquicardia fetal mantenida; el recién nacido, de sexo femenino, tiene peso, talla y perímetro craneal adecuados para la edad gestacional. Desde los primeros días de vida presenta exoftalmos, bocio, irritabilidad, hiperexcitabilidad, aumento en el número de deposiciones y escasa ganancia ponderal, con avidez por las tomas. Recibe tratamiento con propiltiouracilo y solución de lugol, con normalización d los niveles hormonales. Al mes de vida desarrolla un hipotiroidismo por disfunción hipofisaria, por lo que precisa tratamiento sustitutivo con levotiroxina.
A pesar de que una mujer gestante con antecedente de enfermedad de Graves haya sido tiroidectomizada, es importante tener en cuenta que es posible que se desarrolle la enfermedad en el neonato, siendo el título de TSI durante el embarazo es el principal factor de riesgo.
Es importante conocer la historia de esta enfermedad para realizar un seguimiento adecuado durante el embarazo, hacer un diagnóstico fetal precoz y aplicar el tratamiento necesario, con el fin de evitar las posibles secuelas del hipertiroidismo neonatal. Por tanto, es necesario realizar un enfoque multidisciplinar de la enfermedad, que implique a especialistas en ginecología, endocrinología y neonatología.
P87CARCINOMA PAPILAR EN NODULO TIROIDEO HIPERFUNCIONANTE
José Antonio López Medina, Montserrat de Felipe Jiménez-Casquet, José Luis Barrionuevo Porras, Rosa Espigares Martín, Rocío López-Jurado Romero, Catalina González Hervás, Antonio Vicente Pintor, Javier Pacheco Sánchez-Lafuente y Luis Ortega Martos
Hospital Virgen de las Nieves, Granada.
Antecedentes:Los nódulos tiroideos hipofuncionantes o "fríos" mediante gammagrafía con Tc-99 o I-123 presentan mayor riesgo de ser malignos que los nódulos eutiroideos o hiperfuncionantes. El carcinoma papilar de tiroides dentro de un nódulo hiperfuncionante es un hallazgo raro, y los casos descritos en pediatría son escasos.
Caso clínico:Niña de 8 años remitida por presentar tumoración en línea media del cuello de 2 meses de evolución. Los antecedentes familiares y personales carecían de interés. En la exploración inicial se observa tumoración de aproximadamente 1 x 1 cm algo desplazada a la derecha y que parece depender de tiroides, móvil a la deglución, dura, sin signos inflamatorios ni adenopatías. Las pruebas complementarias presentaron TSH: 3,4 μu/ml (rango normal [rn] 0,26-4 μu/ml), FT4: 1,2 ng/dl (rn 0,65-1,90 ng/dl). Ecografía tiroidea: tumoración sólida en lóbulo derecho e itsmo del tiroides de 14 x12 mm, bien delimitada. No se aprecian adenopatías cervicales. Gammagrafía tiroidea: imagen de hipercaptación en lóbulo derecho. Punción aspiración con aguja fina (PAAF): carcinoma papilar de tiroides. Tras las pruebas complementarias se decidió tiroidectomía subtotal y el informe definitivo de anatomía patológica informaba de un carcinoma papilar bien diferenciado de 1,2 cm de diámetro mayor que afectaba a lóbulo derecho, sin evidencia de multifocalidad, con áreas sugestivas de invasión capsular y con los márgenes quirúrgicos libres de tumor. Se remitió posteriormente a la paciente a medicina nuclear para tratamiento ablativo con I-131.
Comentarios:Aunque el carcinoma de tiroides dentro de un nódulo hiperfuncionante es una excepción, siempre debe tenerse en cuenta esta posibilidad. La gammagrafía tiroidea dentro del protocolo de estudio del nódulo tiroideo es sólo orientativa y debemos subrayar a la PAAF como técnica diagnóstica de elección.
P88 HIPOTIROIDISMO CONGÉNITO EN EL SIGLO XXI
José Antonio López Medina, Carmen Fuentes Gutiérrez, Montserrat de Felipe Jiménez-Casquet, José Luis Barrionuevo Porras, Rosa Espigares Martín, Rocío López-Jurado Romero, M. del Mar Rodríguez del Águla, Antonio Vicente Pintor, Catalina González Hervás y Luis Ortega Martos
Hospital Virgen de las Nieves, Granada.
Antecedentes y objetivo:Estudiar la evolución en la detección y tratamiento precoz que el hipotiroidismo congénito tiene en nuestro hospital en los últimos 15 años.
Métodos:Se han incluido todos los niños que han sido diagnosticados en nuestro hospital desde el 1 de enero de 1986 hasta el año 2003 y que cumplen los criterios diagnósticos de hipotiroidismo congénito. Se realizó un protocolo de recogida de datos que incluía: edad, sexo, procedencia, fecha de nacimiento, antecedentes personales y familiares, clínica inicial, diagnóstico final y fecha de inicio de tratamiento. Los datos fueron recogidos en Access-2000 y tratados mediante el programa SPSS.
Resultados:Se diagnosticaron 70 nuevos casos de hipotiroidismo congénito. Por sexos el 74,2 % (52) eran niñas (f) y el 25,8 % (18) niños (m). La etiología del hipotiroidismo congénito fue: tiroides ectópico en el 48,6 % (34), proporcion f/m de 27/7, con TSH en el momento de diagnóstico de 176 ± 98 μu/ml (X ± DS); atireosis en el 25,7 % (18), proporción f/m 13/5, TSH de 229 ± 63 μU/ml; y, por último, dishormonogénesis en el 25,7 % (18), proporción f/m 13/5 y TSH de 162 ± 100. La tabla 1 expone la evolución en el retraso de diagnóstico desde el año 1986 hasta la actualidad:

Conclusiones:La principal causa del hipotiroidismo congénito es la disgenesia tiroidea, y dentro de ella la ectopia tiroidea. El predominio femenino en todos los tipos de disgenesia tiroidea es manifiesto. Parece observarse una mejora en el procedimiento de despistaje, aunque todavía son casi 2 semanas la demora media que existe hasta iniciar el tratamiento.
P89RITMOS CIRCADIANOS DE SUEÑO Y MELATONINA EN NIÑOS CON LACTANCIA MATERNA
Javier Cubero Juánez, Vicente Valero Cumplido, Ana Beatriz Rodríguez Moratinos, Juan Durán Retamar, Montserrat Ribero Urgell, Rubén V. Rial Planas y Carmen Barriga Ibars
Universidad de Extremadura, Badajoz, Hospital Materno-Infantil, Badajoz y Laboratorios Ordesa, S. L., Barcelona.
La producción rítmica de melatonina por la glándula pineal ocurre en los pinealocitos. Su síntesis es inhibida por la luz siendo liberada a la circulación en el periodo de oscuridad. Esta hormona constituye la señal endógena de la noche, y es responsable de la regulación de diferentes procesos fisiológicos, desempeñando además un papel antioxidante endógeno. A partir del tercer o cuarto mes de vida extrauterina el niño comienza a cambiar el ritmo biológico de tres a cuatro horas (ultradianos) al de 24 horas (circadianos) alargándose así su periodo de sueño nocturno, que de lo contrario podría ocasionar graves trastornos tanto al bebé como a sus padres.
Teniendo en cuenta que esta hormona es responsable directa de ritmo sueño-vigilia, el presente trabajo ha sido llevado a cabo para valorar si niños con lactancia materna presentan ritmos circadianos de melatonina y de sueño nocturno, y si existe una correlación entre los mismos y el ritmo materno de secreción de melatonina. Este estudio ha sido llevado a cabo en diez niños a partir de tres meses y con lactancia materna a los cuales se les realizaron los registros de sueño mediante Actiwach colocado en el tobillo derecho durante cinco días. Las muestras de orina tanto a las madres (n = 10) como a los niños se tomaron durante 24 horas, analizándose en la misma, mediante ELISA, los niveles de 6-sulfatoximelatonina, metabolito de excreción de la hormona en orina.
Los parámetros cronobiológicos muestran que los niños presentan un ritmo circadiano de melatonina, siendo la acrofase (momento del ritmo en el que el valor es máximo) de excreción de 6-sulfatoximelatonina igual en las madres que en los niños, y en ambos a las 10:00 h. El mesor (media del valor en un ritmo biológico) de dicho metabolito en bebes fue de 9,26 ng/ml y en las madres 15,02 ng/ml. A su vez la duración media del sueño nocturno resultó ser de 9 horas y 37 minutos ± 44 minutos, valores superiores a la media de los niños de dicha edad.
Podemos concluir que los bebés a partir de los tres meses y con lactancia materna poseen un ritmo endógeno de la hormona melatonina con máximos niveles nocturnos, y que el mismo se encuentra influenciado, a través de la lactancia, por el ritmo circadiano de melatonina que presentan las madres, apareciendo en estos niños un ciclo circadiano de sueño/vigilia consolidado.
P90RELACION ENTRE PESO AL NACIMIENTO, INICIO Y PROGRESION DE LA PUBERTAD Y TALLA FINAL
Ana Isabel Curcoy Barcenilla, Victoria Trenchs Sáinzde la Maza, Lourdes Ibáñez Toda y Francisco Rodríguez Hierro
Hospital San Joan de Deu, Barcelona.
Antecedentes y objetivos:La pubarquia precoz idiopática (PP) no parece tener efectos adversos sobre el desarrollo puberal y/o sobre la talla final. Se ha objetivado que las niñas con PP tienden a presentar un bajo peso al nacer (BPN). Por otro lado, las niñas con BPN tienen mayor riesgo de presentar un inicio y progresión más rápido de la pubertad, que resulta en una talla final más baja. La finalidad de este estudio es valorar la relación entre el peso al nacer (PN), la evolución de la pubertad y la talla final en las niñas con PP.
Métodos:Estudio retrospectivo de las pacientes con PP y estadio ≥ B2 de Tanner controladas en nuestro centro los últimos 4 años.
Resultados:Se recogen 124 casos (37 con BPN), 61 han presentado menarquia y 44 han finalizado el crecimiento. Al diagnóstico de pubarquia la edad media (EM) cronológica es de 7,5 ± 0,8años y la EMósea de 8,5 ± 1,1años. La EM al inicio de la pubarquia, telarquia y menarquia es, respectivamente: 6,8 ± 0,8; 9,5 ± 0,9 y 11,7 ± 0,9años (edad de menarquia similar a la materna [12,3 ± 1,6 años] y a la de la población general [12,3 ± 0,7 años]). La estatura media en la pubarquia, telarquia, menarquia y final es, por este orden: 127,9 ± 7,1; 140 ± 7; 153,8 ± 6,9 y 160,1 ± 6,5 cm. No se hallan diferencias significativas en la correlación entre PN normalizado (SDS) y edad de presentación de la pubarquia, edad ósea a su diagnóstico, telarquia y menarquia. Se observa una tendencia a un desarrollo puberal más rápido en las niñas con BPN y una correlación positiva entre PN y talla en la pubarquia (r = 0,33; p < 0,001), la telarquia (r = 0,33; p < 0,001), la menarquia (r = 0,34; p = 0,03) y en la final (r = 0,44; p < 0,005). Todas las pacientes alcanzan su talla diana pero sólo las de adecuado PN superan la talla materna.
Comentarios:La PP no parece tener efectos adversos sobre el inicio y la progresión de la pubertad y/o sobre la talla final con excepción de las niñas con antecedente de BPN en las que si se relacionaría con un curso más rápido del desarrollo puberal y una talla final más baja. Así pues, la PP en las niñas con antecedente de BPN precisa un control periódico.
P91RESISTENCIA A LA INSULINA Y ADRENARQUIA EN LOS NIÑOS CON BAJO PESO AL NACER
Victoria Trenchs Sáinzde la Maza, Ana Isabel Curcoy Barcenilla, Lourdes Ibáñez Toda y Francisco Rodríguez Hierro
Hospital San Joan de Deu, Barcelona.
Objetivos:Determinar en niños con bajo peso al nacer (BPN) asintomáticos y en edad prepuberal la prevalencia de hiperinsulinemia en ayunas y de adrenarquia exagerada. Comparar la insulinemia media en ayunas y la concentración media de DHEAS de estos pacientes con niños con adecuado peso al nacimiento.
Método:Se estudia la insulinemia y glucemia y/o concentración de dehidroepiandrosterona-sulfato (DHEAS) de niños entre 6 y 9 años de edad con BPN. Se excluyen los afectos de: síndromes específicos con retraso de crecimiento intrauterino, cromosomopatías o malformaciones, pubarquia prematura, pubertad precoz, hiperplasia suprarrenal y diabetes mellitus.
Resultados:Se incluyen 46 pacientes con una edad de 7,5 ± 1,3 años, 26 son niñas. El PN es de 2450,6 ± 386,5g en las niñas y 2359,8 ± 438,5g en los niños, con un SDS de 2,1 ± 0,6 (rango 3,64 a 1,02) y una edad gestacional de 39 ± 2,4semanas. Presentan un IMC de 16,4 ± 2,7 las niñas y de 16,1 ± 3 los niños; SDS 0,1 ± 1,6. En 3 casos no consta determinación de DHEAS y en otros 2 de insulina y glucosa. Se observa una concentración de DHEAS de 44,2 ± 20,8 μg/dl en los niños, 49,9 ± 34,6 μg/dl en las niñas, con un SDS de 0,6 ± 1,8; una de insulinemia de 6 ± 3 mU/l en los niños, 7,1 ± 6,1 mU/l en ls niñas con un SDS de 0 ± 2; y una de glucemia de 90,6 ± 6,5 mg/dl en los niños, 87,5 ± 6,5 mg/dl en las niñas, con un SDS de 0 ± 0,7. La prevalencia de adrenarquia exagerada es del 23,6 % y de hiperinsulinemia del 16 %. El 4,5 % presentan una relación glucemia-insulinemia < 6. No se halla una correlación estadísticamente significativa entre los SDS de DHEAS y PN, insulinemia y PN, ni insulinemia e IMC.
Comentarios:1. La insulinemia y la concentración de DHEAS promedio de los niños con BPN y los de la población general resultan similares. 2.Pese a las limitaciones del método utilizado, se detecta una prevalencia elevada de hiperinsulinismo y adrenarquia exagerada en los niños con BPN.
P92DIABETES TIPO MODY EN DOS FAMILIAS VALLISOLETANAS
Juan Gonzalo Santos García, María Jesús Gallego Fuentes y Florinda Hermoso López
Hospital Universitario de Valladolid.
Introducción:La diabetes del adulto de inicio en la juventud (MODY) presenta una prevalencia en la población desconocida, pero infravalorada a la luz de las nuevas investigaciones. De herencia autosómica dominante presenta diferencias con las diabetes tipo 1 y 2 en herencia, inicio y formas de presentación y tratamiento.
Caso clínico:Presentamos 2 familias de nuestro área de influencia. La primera hay 4 miembros afectos en 3 generaciones sucesivas afectos de diabetes tipo MODY-2 por déficit de Glucokinasa, portadores de la mutación Val200Leu: afectos el caso indice enviado a nuestro hospital por hiperglucemia; su hermano detectado en el estudio familiar; la madre afecta de diabetes gestacional; y la abuela materna afecta de diabetes tipo 2. La segunda presenta 6 miembros afectos: el caso índice es una mujer de 13 años con debut reciente con clínica típica de diabetes con inmunidad negativa; su padre diagnosticado de diabetes a los 18 años en tratamiento con insulina y afecto de complicaciones vasculares; 2 tías rama paterna en tratamiento con insulina y complicaciones vasculares; un primo rama paterna con diagnóstico a los 18 años y en tratamiento con insulina; y la abuela paterna en tratamiento con antidiabéticos orales.
Discusión:La diabetes tipo MODY se caracteriza por su comienzo a edad temprana, la herencia autosómica dominante y ser acusada por defectos primarios en la secreción de insulina en ausencia de anticuerpos presentes en la diabetes tipo 1. Actualmente se definen 7 tipos de diabetes tipo MODY: MODY-1 (por déficit del factor hepático nuclear-4α), MODY-2 (déficit de glucokinasa), MODY-3 (déficit HNF-1α), MODY-4 (alteración en IPF-1), MODY-5 (alteración en HNF-1β), MODY-6 (alteración en Neuro D1 o BETA-1) y MODY-X (casos en los que no se ha detectado una anomalia). Las más frecuentes son MODY-2 y MODY-3. la MODY-2 es leve, se controla con dieta y ejercicio físico. La MODY-3 es progresiva, potencialmente grave, con complicaciones vasculares frecuentes precisando habitualmente tratamiento con insulina. Es posible que la curva de secreción de insulina en la Sobrecarga Oral de Glucosa puede orientar a la diferenciación entre MODY-2 y MODY-3, importante dada la diferente evolución de estas 2 formas, las más frecuentes.
P93 FUNCION TIROIDEA EN PREMATUROS Y SU RELACION CON EL TIPO DE ALIMENTACION
José Luis Gómez Llorente, Fco. Javier Aguirre Rodríguez, M. del Rosario Jiménez Liria, Moisés Leyva Carmona, M. Ángeles Llamas Guisado, Pedro Cortés Mora, Raúl Sánchez Pérez, Emilio José García García y Juan López Muñoz
Hospital Torrecárdenas del SAS, Almería.
Objetivos: 1.Determinar la concentración de yodo en la leche materna que reciben los recién nacidos prematuros de menos de 32 semanas de edad gestacional de nuestra provincia. 2. Comparar la función tiroidea en los prematuros alimentados con lactancia materna y con fórmula.
Pacientes y métodos:Estudio prospectivo de los recién nacidos prematuros de menos de 32 semanas de edad gestacional nacidos en nuestra provincia durante el año 2001. Se excluyeron aquellos con factores de riesgo de alteraciones tiroideas. Se determinaron T4 libre, T3 libre y TSH en plasma y yodo en orina de los recién nacidos los días 7.º y 14.º de vida y yodo en leche materna. Se utilizó fórmula de prematuros "Alprem" con una concentración de yodo de 29 μg/100 kcal. Los resultados se expresan como media (desviación estándar).
Resultados:Se incluyeron en el estudio 44 prematuros, 20 recibieron lactancia materna (12 exclusiva y 8 además fórmula) procedente de 15 madres. La concentración de yodo en leche materna fue 15,0 (12,7) μg/dl, rango 7,0-47,6. En 10/15 muestras fue menor de la recomendada en estos niños (20 μg/dl). No se encontraron diferencias significativas en el aporte medio diario de yodo y las determinaciones hormonales y de yoduria del día 7.º de vida en los prematuros alimentados con leche materna con los alimentados con fórmula artificial.
Conclusiones:1. El aporte de yodo en la leche materna en nuestra provincia es menor que el recomendado para los prematuros.2. No existen diferencias en la función tiroidea entre los prematuros alimentados con leche materna y con fórmula.
GASTROENTEROLOGIA
P94 SINDROME DE DUMPING EN EL NIÑO
M. del Carmen Torres Torres, M.ª José González García, M.ª José López Rodríguez, Amparo López Lafuente y Valentín Carretero Díaz
Hospital San Pedro de Alcántara, Cáceres.
Introducción:El Síndrome de Dumping (SD) es una complicación de la Funduplicatura de Nissen, técnica quirúrgica empleada en el tratamiento del Reflujo Gastroesofágico (RGE) severo. El aumento del peristaltismo y el rápido vaciamiento gástrico se consideran los factores más importantes en su patogenia. Los síntomas que se incluyen en esta patología son rechazo de las tomas, irritabilidad, naúseas, palidez y somnolencia. El método más fiable para establecer el diagnóstico es el Test de Sobrecarga oral de Glucosa. El Tratamiento es principalmente dietético.
Caso clínico:Niña de 11 años que es remitida por dolor abdominal y dificultad para la alimentación desde hace 3años. AF: sin interés. AP: embarazo: normal. Parto: ventosa. PRN: 3 kg PN: normal. Enfermedades anteriores: Parálisis Cerebral Infantil diagnosticada a los 3 meses; infecciones urinarias de repetición por reflujo vésicoureteral diagnosticado al año de edad; estrabismo corregido con lentes. Historia digestiva:Regurgitadora frecuente desde el nacimiento, es diagnosticada por pH-metría de RGE a los 5 años de edad. Precisa corrección quirúrgica del RGE (técnica de Nissen) a los 7 años de edad. Desde este momento objetivan que come menos cantidad, tiene sensación temprana de plenitud, aerofagia,palidez y sudoración. Hábito intestinal: 1 deposición cada 48 horas.
Pruebas complementarias:Hematimetría y Bioquímica basal: normal. Curva de sobrecarga oral de glucosa: glucemia basal: 63 mg/dl, a los 309: 206 mg/dl, a los 609:126 mg/dl, a los 909: 21 mg/dl, a los 1209: 55 mg/dl. Curva insulinemia: insulina basal: 2,1 mcUI/ml, a los 309: 124,4; a los 609: 68,1; a los 909: 3,6 y a los 1209: 1,1. HbA1c: 4,9 %. Tránsito digestivo superior: estómago distendido por importante aerofagia, observándose vaciamiento gástrico precoz, sin otras alteraciones anatómicas.
Tratamiento:Dieta rica en fibra, con tomas fraccionadas y frecuentes.
Comentarios:1. El SD es una complicación bien conocida en la cirugía gástrica de adultos, pero poco frecuente en la infancia. 2. Debe sospecharse cuando ocurren dificultades para la alimentación después de una funduplicatura de Nissen. 3. El Tratamiento es dietético y debe estar orientado a evitar las crisis de hipoglucemia.
P95DERMATITIS PERIANAL STREPTOCOCCICA
Beatriz Guillot Roselló, Raquel Escrig Fernández, Amalia Pérez Payá, Carmen Martínez Serrano y Antonio Pereda Pérez
Hospital Universitario La Fe, Valencia.
Objetivo:La Dermatitis Perianal Streptocóccica es una entidad escasamente diagnosticada, probablemente debido a un bajo índice de sospecha, que motiva una perpetuación de la sintomatología con múltiples consultas, tratamientos incorrectos y las consiguientes molestias para los pacientes. Su diagnóstico es sencillo y su tratamiento eficaz. Comunicamos nuestra experiencia en el último año.
Casos clínicos:Se trata de 4 pacientes (de 3,4,6 y 7 años, dos de ellos hermanos) remitidos a C. ext. de gastroenterología, 2 por deposiciones sanguinolentas y 2 por molestias anales, no resueltas con distintos ttos. Los 4 presentaban defecaciones dolorosas e inquietud, apreciándose a la exploración clínica lesión cutánea perianal bien delimitada, rojo brillante y muy friable. En los 4 casos el tacto rectal fue normal. Se realizó cultivo de frotis perianal, identificándose Streptococo β hemolítico del grupo A (SBHGA), asociándose en 1 de los casos a Staphilococo Aureus (S.A.). En dos pacientes el antibiograma presentó resistencia del germen a la Eritromicina. En tres de los pacientes se realizó frotis faríngeo que resultó negativo para el SBHGA.
Se instauró tto con Amoxicilina-Ác. Clavulánico v.o. a dosis de 40 mg/kg/día de Amoxicilina, durante 8-14 días y tto local con mupirocina en un caso y con neomicina, triamcinolona y nistatina en los otros 3. Previamente al diagnóstico 2 habían recibido tto local exclusivo, sin mejoría. Todos los pacientes quedaron asintomáticos tras el tto, con resolución de la lesión.
Conclusiones:1. El diagnóstico de sospecha es fácil si se piensa en él. 2.Es más frecuente en varones (rel. 8:1), menores de 10 años.3. El germen habitual es el SBHGA, pero puede asociar otros gérmenes como el S.A. principalmente que pueden dificultar el tto (importancia del cultivo con ATBiograma).4. Dos de nuestros pacientes presentaban resistencia a la Eritromicina, que se postula como 1 alternativa al tto en la bibliografía. 5.No hemos podido detectar portadores faríngeos (65 % de los pacientes con afectación perianal según distintas series, siendo sólo un 6 % de los portadores faríngeos los que presentan esta patología).6. No se han detectado recidivas en el seguimiento entre 1-6 meses a pesar de que según la bibliografía son frecuentes.
P96APLICACION DE LA TALIDOMIDA EN LA ENFERMEDAD DE CRÖHN FISTULIZANTE
Raúl Cabrera Rodríguez, Honorio Miguel Armas Ramos, Roque Abián Montesdeoca Melián, Raquel Perera Soler y José Luis Aparicio Sánchez
Hospital Universitario de Canarias, La Laguna.
Introducción:La talidomida se ha usado satisfactoriamente en el control de patologías de carácter inmune como la artritis reumatoide, enfermedad de Bechet, úlceras aftosas y enfermedad de Cröhn en población adulta. Sin embargo, aún son muy limitados los estudios en población pediátrica.
Objetivo:Se presenta un caso clínico de un paciente con una fístula perianal tratada con talidomida.
Caso clínico:Paciente mujer de 14 años diagnosticada de enfermedad de Cröhn a los 10 años es de edad. El brote inicial se caracterizó por diarrea intermitente, dolor abdominal recidivante y anemia ferropénica controlándose a los 3 meses con el uso de nutrición enteral, mesalazina y glucocorticoides. La primera recaida se produjo a los 16 meses. En la tercera recaida ocurrida a los 30 meses del debut se inició una fisura perianal que aumentó progresivamente de tamaño a pesar del tratamiento anterior al que se le añadió metronidazol. La lesión inicial se convirtió en un trayecto fistuloso de difícil terapeutica por lo que se decidió iniciar tratamiento con talidomida (2 mg/kg/día) tras consentimiento informado. A los 5 meses se consigue un cierre casi total de la lesión ulcerosa, no presentándose nuevos brotes en este tiempo. Se decide suspender el tratamiento al observarse en una exploración oftalmológica de rutina una hemorragia papilar en la cabeza del nervio óptico izquierdo, clínicamente asintomática y ligeras parestesias en la extremidad superior izquierda que cedieron tras suspensión del tratamiento.
Conclusiones:En la literatura y en nuestro caso, se constata buenos resultados en el cierre de las fístulas asociadas a la enfermedad de Cröhn, por lo que la talidomida representa una opción terapeutica en las lesiones refractarias al tratamiento convencional. Sin embargo son necesario estudios más amplios en la población pediátrica para valorar la eficacia y la seguridad de la misma.
P97TRATAMIENTO CON INFLIXIMAB EN EL BROTE AGUDO DE LA ENFERMEDAD DE CRÖHN
Raúl Cabrera Rodríguez, Honorio Miguel Armas Ramos, Roque Abián Montesdeoca Melián, Begoña Martínez Pineda, Cristina León Quintana y M. Teresa Herráiz Culebras
Hospital Universitario de Canarias, La Laguna.
Introducción:La Enfermedad de Cröhn puede presentar brotes graves amenazantes para la vida. Los tratamientos clásicos para éstos incluyen glucocorticoides, mesalazina, inmunosupresores y cirugía en los casos refractarios. Recientemente se han introducido los antagonistas del TNFα como tratamiento prometedor aunque la experiencia pediátrica aún es limitada.
Caso clínico:Paciente varón de 11 años diagnosticado de enfermedad de Cröhn un año antes y asintomático tras controlarse el brote inicial con glucocorticoides y mesalazina. Comienza a presentar deposiciones diarreicas con sangre (8-10/día), vómitos, anorexia y dolor abdominal. A la exploración física se observa gran decaimiento general y dolor abdominal en región infraumbilical. En las pruebas complementarias destaca: reactantes de fase aguda elevados, ecografía abdominal con gran dilatación del colon ascendente y gammagrafía con leucocitos marcados que muestra migración patológica de leucocitos en el colon ascendente. En la colonoscopia se visualiza válvula ileocecal edematosa y deformada, ciego con úlceras mayores a 10 mm y colon ascendente con múltiples ulceraciones de menor tamaño. Tras ingreso hospitalario se instaura tratamiento con glucocorticoides, mesalazina, azatioprina, esteroides rectales y nutrición parenteral a pesar de lo cual presenta 3 sangrados masivos con compriso hemodinámico que requirieron transfusión de concentrado de hematíes e ingreso en U.C.I.P (los días 5.º, 8.º y 16.º). Se decide el uso de infliximab como tratamiento previo a la resección quirúrgica pa (5 mg/kg/dosis ev) consiguiéndose la mejoría clínica al 5.º día. Se han administrado 2 nuevas dosis en la segunda y sexta semana, permaneciendo asintomático en el seguimiento actual de 6 meses.
Conclusiones:En este caso se ha observado una excelente respuesta al tratamiento con infliximab. Aunque la literatura que lo apoya aún es limitada, debe ser una opción a considerar en los brotes graves corticorresistentes, especialmente en la enfermedad de Cröhn donde la resección quirúrgica es solo paliativa, siendo necesario estudios más amplios para generalizar su uso.
P98INFECCION POR CITOMEGALOVIRUS Y ENTEROPATIA PIERDE PROTEINAS
Javier Barberán Pérez, Montserrat Roqueta Sureda, José Manuel Gómez López, Gloria Ruiz Aragón, Joaquín Martorell Aymerich, Anna Cabot Dalmau, Manuel Casado Toda, Lourdes García Rodríguez y Rosario Díez Martín
Consorci Sanitari del Maresme, Mataró.
La enfermedad de Menetrier es rara en la edad pediatrica. Se caracteriza por engrosamiento de pliegues gástricos, perdida de proteínas por tubo digestivo con hipoalbuminemia y edemas. Presentamos un caso. Paciente de 2 años que inicia astenia y anorexia dos semanas antes del ingreso. Dos días antes inicia dolor abdominal y el día del ingreso acude con edemas palpebrales y pretibiales. En la exploración física destaca la existencia de edemas palpebrales, pretibiales con fóvea y en pies. No ascitis. El resto de exploración física es anodina, estando afebril. Exploraciones complementarias: VSG 7 Hb 13,5 Leucocitos (S/35 L/52 M/6 Eos/3 Bas/1 Luc/3) Proteinas totales 35,7 Albumina 13,39 IgA 0,36 IgG 1,72 IgM 0,39. IgE 7,6 T. Protrombina 1,11. Proteinuria: negativa. Sangre oculta en heces: negativa. Grasa en heces: negativa. Alfa 1 antitripsina en heces: 12 mg/g de heces. Ac. Antiendomisio negativos. Ac antitransglutaminasa: negativos. Test Ureasa: negativo. IgE especifica leche de vaca < 0,35 Ac Citomegalovirus IgM 1,26 (VR < 1). Cultivo de orina: citomegalovirus. Transito esofagogastroduodenal: esofago de calibre y peristaltismo normal. Pliegues mucosos fundicos, de curvatura mayor y preantral engrosados e irregulares. Ecografía abdominal: engrosamiento de la pared gastrica a nivel de curvatura mayor y en region preantral (15 mm). A los 17 días del ingreso: grosor maximo de pared gastrica: 7 mm. A las 5 semans del ingreso: ecografia normal.
Conclusiones:La enfermedad de Menetrier es infrecuente en la edad pediatrica. A diferencia del adulto el curso suele ser autolimitado, siendo la duración aproximada de 5 semanas, requiriendo únicamente tratamiento de soporte y corrección de la hipoalbuminemia. Dentro del diagnostico diferencial de la enteropatia pierde proteínas debe incluirse la enfermedad de Menetrier, en especial cuando faltan otros síntomas de enfermedad intestinal, tal como diarrea. La etiología más frecuente es la infecciosa, y dentro de esta, la infección por citomegalovirus. Mostramos el seguimiento ecografico del grosor de los pliegues gástricos que se correlacionó bien con los hallazgos del transito esofagogastroduodenal.
P99 HIPOPROTEINEMIA SIN PROTEINURIA: GASTROENTEROPATIA PIERDE PROTEINAS
Adelaida Cardona Barberán, Antonio Sorni Hubrecht, Montserrat Pech Solá, Jaume Rosal Roig, Anna M.ª Hostalot I Abas, Josep Merce Gratacós y Javier Acuña González
Hospital de Tortosa Verge de la Cinta, Tortosa.
Introducción:La enfermedad de Menetrier infantil, se caracteriza por la hipertrofia de la capa mucosa gástrica e hipertrofia de sus pliegues, con o sin dilataciones quísticas en los componentes glandulares. Una infección, la mayoría de veces vírica, provocaría la disregulación del crecimiento y función del epitelio gástrico.
Caso clínico:Varón de 15 meses de edad que acudió al servicio de urgencias por vómitos, aumento de peso y edemas en el dorso de los pies. En la exploración física destacaba exantema macular puntiforme palmo plantar, edemas palpebrales bilaterales que progresan en las siguientes horas afectando extremidades inferiores y escroto, distensión abdominal y hepatoesplenomegália de 2 cm. En las exploraciones complementarias destacaba la presencia de linfocitosis con linfocitos estimulados, hiponatremia, hipoproteinemia e hipertransaminasemia. La coagulación y la función renal fueron normales. No proteinuria. En la ecografía abdominal aparecía un engrosamiento de la pared gástrica con hiperaflujo vascular a expensas de la capa mucosa y submucosa, afectando a cuerpo y fundus gástrico. Las IgG e IgA para citomegalovirus (CMV) fueron positivas, y la Biopsia gástrica mostró gastritis crónica y células infectadas por CMV. La evolución clínica fue favorable, con resoluciónad integrum tanto clínica como eco gráficamente a la semana, recibiendo tratamiento exclusivamente sintomático.
Conclusiones: 1.Hay que sospechar enfermedad de Menetrier ante todo niño con vómitos, hipoproteinemia y edemas generalizados sin causa renal ni hepática que lo justifique. 2. La ecografía, dada la buena correlación con la clínica, es una técnica útil en el diagnóstico y seguimiento de estos pacientes.3. El CMV es el agente más frecuentemente implicado seguido por Helicobacter Pylori, Herpes simple y Mycoplasma.4. La evolución de la enfermedad, al contrario de lo que ocurre en los adultos, es autolimitada, tendiendo a la resolución espontánea en el curso de semanas.5. Son necesarios más estudios (virológicos, serológicos e investigaciones clínicas) para definir el papel de las infecciones, sobretodo del CMV en esta enfermedad.
P100 PICA COMO SINTOMA DE PRESENTACION DE ENFERMEDAD CELIACA
M.ª José González García, M. del Carmen Torres Torres, M.ª José López Rodríguez, Amparo López Lafuente y Valentín Carretero Díaz
Hospital San Pedro de Alcántara, Cáceres.
Introducción:La enfermedad celiaca se define por una intolerancia permanente a la gliadina del gluten, que provoca una lesión anatómica del intestino delgado superior. Como consecuencia se produce una malabsorción de nutrientes con repercusión clínica y funcional variable. En los últimos años se está produciendo un cambio en la forma de presentación, aumentando las formas monosintomáticas y atípicas, lo que puede llevar a un retraso en el diagnóstico.
Material y métodos:Presentamos el caso de una niña de 9 años inmigrante marroquí, que consulta por ingesta frecuente de tierra en los últimos 3-4 años, además de episodios diarreicos repetidos con distensión abdominal. Siempre curva de peso lenta y escaso apetito. Lactancia materna exclusiva durante el primer año de vida, posteriormente alimentación complementaria normal.
La exploración física es normal, salvo una palidez cutánea; talla y peso por debajo del percentil 3.
Los valores analíticos muestran una anemia microcítica e hipocrómica (hematíes 5.420.000/mm3; Hb 8,4 g/l; Hto. 28,6 %; VCM 5,2 fl; Fe 18 mg/dl; ferritina 3,2 ng/ml; ác. fólico 0,01 ng/ml; Vit. B12 148 pg/ml; Hb en heces negativo; AGA IgA +; EMA +; anti-tTG +; inmunoglobulinas normales; biopsia intestinal: atrofia subtotal de las vellosidades; HLA: DR3, DQ2 (DQA1: 0501, DQB1: 0201).
Con el diagnóstico de EC se inicia dieta exenta en gluten.
Al año del diagnóstico la niña está asintomática, la analítica se ha normalizado y ha aumentado el percentil de la talla y el peso.
Conclusiones:La pica es un síntoma característico de la anemia ferropénica, a su vez, este tipo de anemia es una manifestación común de la enfermedad celiaca.
La clínica de la enfermedad celiaca es muy variable, desde cuadros muy floridos a formas monosintomáticas y atípicas, incluso con síntomas gastrointestinales mínimos o nulos.
P101TRASPLANTE HEPATICO EN FIBROSIS QUISTICA: ¿LA OPCION TERAPÉUTICA?
Mercedes Serrano Gimaré, M. del Carmen Díaz Fernández, Ángela de la Vega Bueno, M. Loreto Hierro Llanillo, M. del Carmen Camarena Grande, Esteban Frauca Remacha, M. del Carmen Antelo Landeira, Rosa A. Lama More y Paloma Jara Vega
Hospital Universitario La Paz, Madrid.
Antecedentes y objetivo:La fibrosis quística (FQ) es una enfermedad frecuente en la que la afectación hepática se desarrolla en un 2-25 %. El riesgo de muerte prematura por complicaciones respiratorias aumenta con la enfermedad hepática y la función pulmonar empeora paralelamente al incremento de la hipertensión portal. Se describe tras el trasplante hepático una mejoría subjetiva, en la clínica respiratoria, y objetiva en los parámetros de función pulmonar, así como un progreso favorable en el estado nutricional. Aportamos 5 nuevos casos de trasplante hepático en FQ con el fin de añadir evidencia en esta opción terapéutica como salida satisfactoria para nuestros pacientes.
Métodos:Cinco pacientes en edad pediátrica con cirrosis hepática e hipertensión portal en el contexto de FQ fueron trasplantados del modo habitual en cuanto a técnica quirúrgica. Tras el trasplante se evaluaron las complicaciones postquirúrgicas,la función del injerto (función hepática sintética, transaminasas), la función respiratoria (espirometría y sintomatología) y el estado nutricional (peso, talla, índice de masa corporal y perímetro braquial) comparándolos con los parámetros previos al trasplante.
Resultados:Las complicaciones en el postoperatorio inmediato no diferían de las presentadas por pacientes trasplantados por otros motivos. La función hepática normal se alcanzó a los 36,5 días del trasplante como media. La función respiratoria valorada por espirometría mejoró en la mayoría y se objetivó una disminución de los síntomas subjetivos: tos, tolerancia al ejercicio, producción de esputo... El estado nutricional evolucionó a la mejoría o estabilización. Los inmunosupresores fueron bien tolerados.
Conclusiones:El trasplante hepático aislado es una opción con resultados satisfactorios para los pacientes con FQ y enfermedad pulmonar leve que presenten hipertensión portal severa o cirrosis. Cuando la enfermedad pulmonar o cardíaca es severa, se debe considerar el trasplante multiorgánico. Las mejorías en la función pulmonar y en el estado nutricional se observan con frecuencia tras el trasplante hepático y los inmunosupresores no parecen tener efecto deletéreo sobre la función pulmonar.
P102 HIPOBETALIPOPROTEINEMIA FAMILIAR: DISTINTAS FORMAS DE PRESENTACION CLINICA
M. Blanca Herrero Mendoza, M. Belén Robles García, Ana Díaz Moro, Carmen Villafañe Landeira, M. Teresa Fernández Castaño y José Manuel Marugán de Miguelsanz
Hospital de Léon.
Introducción:La hipobetalipoproteinemia es un trastorno genético infrecuente transmitido según un patrón autonómico codominante, producido por mutaciones en el gen de la apo-B (apoproteína-B). Se caracteriza por niveles bajos de LDL-colesterol, apo-B y colesterol total. La forma homocigota presenta un cuadro clínico similar al de la abetalipoproteinemia, mientras que la heterocigota suele ser asintomática. Se han revisado los pacientes diagnosticados en el Servicio de Pediatría de nuestro Centro de hipobetalipoproteinemia.
Casos clínicos: 1.Niño de 7 años que en un ingreso por neumonía se detectan niveles bajos de colesterol y TG (triglicéridos). Analítica: colesterol 75 mg/dl (100-220 mg/dl); TG 14 mg/dl (50-170 mg/dl); apo-B 19 mg/dl (60-150 mg/dl); LDL-col 18 mg/dl; HDL-col 86 (40-65 mg/dl). Diarrea crónica sin repercusión nutricional. Biopsia yeyunal normal. En el estudio familiar la madre presenta déficit ligero de apo-B. 2. Niña de 10 años remitida a Digestivo por hipertransaminasemia desde el primer año de vida sin sintomatología asociada. Analítica: GOT 39 UI/l (0-37 UI/l); GPT 87 UI/l (0-40 UI/l); Colesterol 88 mg/dl; TG 22 mg/dl; apo-B 25 mg/dl; LDL-col 23 mg/dl; HDL-col 60 mg/dl. Biopsia hepática: esteatosis. En el estudio familiar el padre presenta niveles bajos de TG (28 mg/dl) y de apo-B (30 mg/dl).3. Niño de 10 años que en un estudio por urticaria se obtienen los siguientes resultados en la analítica: colesterol 90 mg/dl; TG 66 mg/dl; apo-B 54 mg/dl; LDL-col 52 mg/dl; HDL-col 24 mg/dl. Forma leve asintomática.
Discusión:Los tres casos corresponden a la forma heterocigota de hipobetalipoproteinemia familiar, y, de acuerdo con lo descrito en la literatura, son clínicamente asintomáticos, aunque es importante su diagnóstico ya que en ocasiones se asocian a acantocitosis, retinitis pigmentaria, esteatosis hepática (como en el 2.º caso), diabetes mellitus, malabsorción de grasas, hipotiroidismo, alteraciones neurológicas y retraso pondo-estatural. Estas manifestaciones son típicas y más frecuentes entre los pacientes diagnosticados de hipobetalipoproteinemia en su forma homocigoto o bien de abetalipoproteinemia.
P103BACTERIEMIA POR SALMONELLA NO TIFOIDEA
Elena Balmaseda Serrano, Carolina Gutiérrez Junquera, C. Sáinz de Baranda, Isabel Cuartero del Pozo y Alberto Vidal Company
Complejo Hospitalario de Albacete. Hospital General.
Objetivo:La incidencia de bacteriemia por Salmonella no tifoidea varía, según los estudios, de 5,2 a 13,7 %. Los serotipos más invasivos son Salmonella typhimurium, Salmonella choleraesuis y Salmonella virchow. El objetivo de nuestro estudio es determinar la incidencia y características clínico-bacteriológicas de la bacteriemia por Salmonella en los niños ingresados en el servicio de pediatría del Hospital General Universitario de Albacete en los últimos cinco años.
Resultados:Durante los años 1998 a 2002 fueron ingresados con el diagnóstico de gastroenteritis aguda (GEA) por Salmonella 311 niños, de los cuales 10 (3,2 %) tuvieron hemocultivo positivo para este gérmen. El serotipo más frecuente fue Salmonella enteritidis (70 %), seguido de Salmonella typhimurium (30 %). La edad media fue 11,2 meses (rango 2-36 meses). Solo dos casos presentaban factores predisponentes: malnutrición asociada a cardiopatía (drenaje venoso pulmonar anómalo) y déficit selectivo de IgA. La duración media del ingreso fue 7,8 días. La duración media de la fiebre fue 5,8 días, con media de temperatura máxima de 39,3 °C. Las deposiciones diarreicas fueron en el 50 % de características enteroinvasivas, con una duración media de 6 días y una frecuencia media de 6,4 deposiciones/día. El 20 % de los pacientes presentaron una deshidratación, que fue leve-moderada. Ninguno de los niños presentó datos clínicos compatibles con sepsis. Ocho niños recibieron tratamiento antibiótico parenteral (cefotaxima o ceftriaxona), un paciente fue tratado con amoxicilina-clavulánico oral y en otro caso la bacteriemia se resolvió sin tratamiento antibiótico. La evolución fue favorable en todos los casos, no presentando complicaciones metastásicas secundarias.
Conclusiones:1. La incidencia de bacteriemia por Salmonella en nuestro medio ha sido de 3,2 %, siendo el serotipo más frecuente Salmonella enteritidis. 2. La mayoría de los pacientes que desarrollaron bacteriemia secundaria eran menores de tres años, no presentando factores predisponentes, por lo que es recomendable la realización sistemática de hemocultivos en gastroenteritis invasivas febriles en este rango de edad.
P104SINDROME DE SOTOS ASOCIADO A TIROIDITIS LINFOCITARIA CRONICA Y ENFERMEDAD CELIACA
Elena Balmaseda Serrano, Carolina Gutiérrez Junquera, Rafael Ruiz Cano, Ignacio Onsurbe Ramírez y Ester Gil Pons
Complejo Hospitalario de Albacete. Hospital General.
Introducción:El espectro clínico de la enfermedad celíaca abarca desde síndrome de malabsorción clásico hasta formas paucisintomáticas y asintomáticas, pudiendo detectarse en personas con nutrición normal. Describimos el caso de una paciente con hipercrecimiento en el contexto de un síndrome de Sotos (SS) que desarrolló en su evolución clínica tiroiditis linfocitaria crónica (TLC) y enfermedad celiaca (EC) silente.
Caso clínico:Adolescente de 15 años con parámetros somatométricos > > P97 desde el nacimiento, facies tosca, macrocefalia e hipopsiquismo. Cariotipo 46 XX. RNM craneal: ampliación de ventrículos laterales sin signos de reabsorción transependimaria (megalencefalia) y malformación de Arnold-Chiari I. Es diagnosticada de SS a los 8 meses de vida. A los 10 años presenta bocio Ib sin clínica tiroidea, con aumento de hormonas tiroideas, supresión de TSH, ATA microsomal y tiroglobulina positivos, TSI negativa, ecografía tiroidea: tiroides aumentado de tamaño con imágenes nodulares. Se diagnostica de TLC hipertiroidea y se pauta tratamiento con antitiroideos y levotiroxina. Normalización de valores hormonales con suspensión de tratamiento a los 20 meses. A los 14 años presenta en analítica de despistaje anticuerpos antiendomisio IgA 1/160, antigliadina IgG 15 U/ml, antirreticulina IgG 1/320. Se encuentra asintomática desde el punto de vista digestivo, manteniendo talla y peso por encima del percentil 97. Biopsia intestinal: atrofia vellositaria grado III, hiperplasia críptica con infiltrado linfo-plasmocitario y permeación linfocitaria del epitelio superficial. Se diagnostica de EC comenzando dieta exenta de gluten sin apreciarse cambios clínicos significativos ni en los parámetros somatométricos.
Comentarios:El hipercrecimiento constituye una de las características principales del SS. En nuestro caso, la patología tiroidea autoinmune que presentó la paciente condujo al despistaje activo de EC que se confirmó mediante biopsia intestinal. Sin embargo, no existía clínica digestiva ni repercusión evidente en los parámetros somatométricos lo que es concordante con EC silente. Destacamos la necesidad de despistaje de EC en la patología tiroidea autoinmune y la posible existencia de la misma en niños con parámetros antropométricos por encima del percentil 97 para su edad.
INFECTOLOGIA
P105 EVALUACION DEL PROGRAMA DE PREVENCION DEL SIDA PEDIATRICO EN CUBA
Ida González Núñez, Manuel Díaz Jidy, Jorge Pérez Ávila y Patricia Díaz González
Instituto Pedro Kourí, La Habana (Cuba).
Introducción:Los casos pediátricos han aumentado con respecto a la transmisión vertical como resultado de la diseminación VIH/SIDA entre mujeres en edad reproductiva. En Cuba, al existir pocos casos infectados por el VIH y a la intervención temprana epidemiológica se ha podido controlar de cierta manera el nacimiento de niños provenientes de padres seropositivos.
Objetivos:Evaluar la reducción de la transmisión perinatal del VIH en los hijos de madres seropositivas al VIH/sida.
Material y método:Un programa controlado ha sido implantado en el país. La política de salud incluye la interrupción del embarazo siempre y cuando la pareja lo acepte y de su autorización. El proyecto incluye que todas las mujeres seropositivas que deseen tener su hijo, a partir de las 14 semanas del embarazo reciban 500 mg de AZT hasta el parto que la mayoría de las veces es por cesárea. El niño después de las 8 horas de nacido recibe 2 mg/kg/dosis de AZT en jarabe cada 6 horas durante las primeras 6 semanas de vida. Estos niños son seguidos en las consultas de pediatría del IPK para determinar si están infectados o no por el VIH, además se les atiende cualquier patología que puedan presentar.
Resultados:Desde el 1ero de Enero de 1986 al 30 de Noviembre del 2002 han parido el 11,6 % de las mujeres seropositivas reportadas en el país (104/896), hay 4 madres que han parido en dos ocasiones. De estos 108 niños nacidos de madres seropositivas al VIH, 12 niños resultaron seropositivos que han desarrollado el SIDA (11,1 %) 5 están con tratamiento antirretroviral, 1 asintomático, y han fallecido 6 por esta causa. Además hay 14 casos, uno por transfusión de sangre, ya fallecido (que ocurrió antes del pesquisaje masivo de la sangre en Cuba), 2 casos por hemofília A severa, por transfusión de crioprecipitado que también están con tratamiento antirretroviral, así como 11 adolescentes que lo adquirieron por vía sexual, que hacen un total de 26 niños VIH/SIDA en el país. Actualmente hay 66 niños negativos al VIH (61,1 %) y 30 niños que aún se encuentran en estudio (27,7 %).
Conclusiones:De acuerdo a los factores presentados aquí podemos considerar que el programa de control y prevención establecido en el país ha sido efectivo, pues el número de niños infectados permanece bajo cuando lo comparamos con otros países.
P106HOSPITALIZACIONES POR VARICELA COMPLICADA EN MENORES DE 15 AÑOS (1993-2002)
José Javier Uriz Monaut, Ángeles M. Ruiz Benito, Leonor Arranz Aranda, Ainhoa Sarasua Miranda, Maribel Mendiburu, Beatriz Azkunaga Santibañez, Jesús Alustiza Martínez y Eduardo González Pérez-Yarza
Grupo Varicela Gipuzkoa, Hospital Bidasoa, Hospital Donostia, Hospital Mendaro y Hospital Zumárraga.
Objetivo:Cuantificar y cualificar las complicaciones por varicela que requieren hospitalización.
Material y métodos:Estudio multicéntrico, retrospectivo, observacional, por revisión de historias clínicas codificadas como varicela (CMBD, ICD-9, códigos 052,0-052,9) del 1-1-1993 a 31-12-2002, en niños menores de 0-15 años ingresados más de 24 horas, en los hospitales del Bidasoa, Donostia, Mendaro y Zumárraga, de Osakidetza-Servicio vasco de salud. Se han analizado las variables: edad, sexo, antecedentes personales, estado inmunológico, tiempo entre inicio del exantema e ingreso, tratamiento previo y fiebre antes del ingreso, exámenes complementarios, duración del ingreso y tratamiento, diagnóstico, complicaciones y secuelas. Los datos poblacionales se han obtenido del padrón municipal (1996) y de las tablas de mortalidad (1997-2000) y de fecundidad (1996-2000) del Eustat.
Resultados:La cobertura poblacional de menores de 15 años del conjunto de hospitales ha sido 54.999 (rango 54.419-56.136) niños. Se han recogido 71 casos (edad media 2,91 ± 2,53 años), 80 % menores de 5 años, distribuidos en 42 niños (59 %) y 29 niñas (41 %). Inmunocompententes, 68 casos. No-inmunocompetentes, 3 casos (2, enfermedad de Hodgkin; 1, déficit de IL-8). Ningún caso estaba vacunado. La incidencia anual ha sido 12,9/100.000/año. Representa 0,03 % de los ingresos anuales. Los diagnósticos han sido: sepsis aStaphilococcus aureus (2), celulitis (20), varicela hemorrágica (2) y úlcero-necrótica (1), laringitis (1), crisis asmática (2), neumonía (18), bronconeumonía (1), neumonía intersticial (2), edema pulmonar no-cardiogénico (1), meningitis (1), cerebelitis (11), vómitos-intolerancia oral (4), gastroenteritis aguda (1), hepatitis (1), otomastoiditis (2) y artritis (1). Se realizaron 57 hemocultivos, siendo positivos en 10 casos:Streptococcus pyogenes (3),Streptococcus epidermidis (2),Steptococcus pneumoniae (2)y Staphilococcus aureus (3). La estancia media total fue 6,50 ± 5,15 días (rango 1-29) y 5 casos requirieron ingreso en UCIP. No ha habido secuelas ni mortalidad.
Conclusiones:La tasa de incidencia de complicaciones por varicela que precisan hospitalización y los tipos de complicaciones observadas, son similares a otros estudios nacionales e internacionales.
P107ERITEMA MIGRATORIO (ENFERMEDAD DE LYME) TRAS MANIPULACION DE AVES DE CAZA
Gloria López Lois, María Penín Antón, Pilar Sevilla Ramos, M. Luisa Murga Sierra, Kamal Kamal Kayali, Eva Vegas Muñoz, Mercedes García Rodríguez y José Enrique García de Frías
Hospital Príncipe de Asturias, Alcalá de Henares y Centro de Atención Primaria Daganzo.
La enfermedad de Lyme (EL) ha sido descrita en amplias zonal de la geografía española. Es producida por varias genoespecies de Borrelia burgdorferi sensu lato y su vector principal es la garrapata Ixodes ricinus que es huesped de diferentes vertebrados (mamíferos y aves). Varios estudios han destacado la importancia epidemiológica de la aves y sus movimientos migratorios como portadoras del vector o incluso como reservorio de la enfermedad. El eritema migratorio es la manifestación más frecuente de la EL en la infancia; su diagnóstico se basa en la características clínicas y el antecedente de exposición ambiental ya que las pruebas serológicas tienen baja sensibilidad en la fase precoz de EL.
Caso clínico:Paciente de 7 años que consulta en el mes de Agosto por una lesión cutánea torácica, discretamente pruriginosa y de crecimiento centrífugo, que ha aparecido alrededor de una picadura de insecto detectada 5 días antes; asocia astenia, cefalea y anorexia. Antecedentes personales y familiares: el padre participó en una cacería de aves en la provincia de Cuenca una semana antes y la paciente se encargó del desplume. Exploración física: lesión anular 15 x5 cm de diámetro con bordes eritematosos y aclaramiento central en región pectoral derecha. Pruebas complementarias: hemograma, bioquímica y proteina C reactiva normales; biopsia cutánea con patrón ezcematoso y escaso infiltrado eosinófilo; la serología al diagnóstico no fue concluyente (EIA Ig total positivo, Western blot IgG negativo). La paciente fue tratada con Amoxicilina oral durante 21 días, desapareciendo la lesión cutánea en la segunda semana de tratamiento, sin aparecer manifestaciones tardías de EL.
Conclusiones: 1.El diagnóstico precoz del eritema migratorio es importante para evitar complicaciones tardías. 2. La investigación epidemiológica cuidadosa puede descubrir exposición ambiental inaparente. 3. El contacto estrecho con aves de caza debe considerarse factor de riesgo de EL a cualquier edad.
P108
ABSCESO CEREBRAL INFANTIL: SERIE DE 5 CASOS
Raúl Roberto Borrego Domínguez, M. Luisa Navarro Gómez, José Antonio Gómez Campderá, Jesús Carreras, Gema Arriola Pereda, Rocío Chacón Aguilar, Cristina Menéndez Hernando, Sonia Villar Castro y Kay Boris Brandstrup Azuero
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción:El absceso cerebral es una colección purulenta intracerebral. Tiene una incidencia global de 4/1.000.000 de habitantes y año. Es más frecuente entre 4 y 8 años y, en el sexo masculino. Los microorganismos mayormente implicados son: estreptococos, estafilococos, enterobacterias, anaerobios. La etiología más común es: embolización por cardiopatía congénitas y los procesos infecciosos de oído, senos paranasales, mastoides, partes blandas de cara y cuero cabelludo y focos dentales. Su mortalidad aunque ha disminuido con la introducción de los antibióticos y el TAC, sigue siendo importante (5-10 %). Hasta un 50 % presentan secuelas permanentes como hermiparesia, crisis epilépticas, hidrocefalia, anomalías de pares craneales y problemas de comportamiento y aprendizaje.
Casos:Presentamos una serie de 5 casos clínicos vistos en nuestro hospital desde 1996 hasta 2002. Las edades comprendidas de los pacientes son entre 8 y 15 años, siendo 3 de ellos niños y 2 niñas. La etiología presentada es: sinusitis en 3 casos, meningitis en otro y, cardiopatía congénita cianógena con foco dentario en el otro. Todos fueron tratados mediante drenaje quirúrgico y antibióticos. Los 5 pacientes se trataron con cefotaxima y metronidazol, y 4 de ellos asociaron vancomicina. En tres de los pacientes los cultivos del material obtenido por drenaje fueron estériles y, en los otros 2 pacientes se detectaron: estreptococo viridans, anaerobios y gram negativos. Todos los pacientes sobrevivieron. Todos ellos están con medicación anticomicial. Uno de ellos presenta crisis comiciales secundarias a cicatriz cortical y otro ligera pérdida de fuerza en hemicuerpo derecho de predominio braquial.
Conclusiones:1. Aunque es una patología poco frecuente presenta una extrema gravedad, debiendose realizar un diagnóstico precoz basado en la clínica (cefalea, vómitos y fiebre) y el TAC.2. No encontramos diferencia en cuanto al sexo y, la edad de nuestros pacientes es mayor que la referida en la literatura como la más frecuente. 3.El tratamiento se basa en antibioterapia empírica durante 4-6 semanas con: cefalosporina de 3.ª generación, metronidazol y vancomicina. El tratamiento quirúrgico está indicado en los abscesos mayores de 2,5 cm, clínica de hipertensión intracraneal o mala evolución clínica.4. Las secuelas son frecuentes precisando habitualmente tratamiento anticomicial a largo plazo.
P109CARACTERIZACION DEL SISTEMA INMUNE TRAS UN LARGO PERIODO CON CARGA VIRAL INDETECTABLE EN NIÑOS INFECTADOS POR EL VIH-1
Salvador Resino García, Isabel Galán Carrillo, Alicia Pérez Arroyo, José M.ª Bellón Cano, M. Luisa Navarro Gómez, Juan Antonio León Leal y M. Ángeles Muñoz Fernández
Hospital General Universitario Gregorio Marañón, Madrid y Hospital Universitario Virgen del Rocío, Sevilla.
Objetivo:Valorar el sistema inmune en niños infectados verticalmente por el VIH-1, que llevan más de 18 meses con carga viral indetectable y tienen > 500 células TCD4+/mm3.
Materiales y métodos:En un estudio longitudinal retrospectivo, se evaluaron 32 niños VIH, sometidos a terapia antirretroviral de gran actividad (TARGA), de acuerdo con sus niveles de carga viral (CV) durante todo el período de seguimiento (> 18 meses): a) grupo uVL: 15 niños con CV por debajo de 400 copias/ml; b) grupo dVL:17 niños con CV más alta.
Resultados:El grupo uVLmostró mayor número de células T memoria (CD4 + CD45RO +) que el grupo dVL,y mayor número de células de memoria activadas (CD4 + CD45RO + HLADR +) que el grupo Control(niños sanos no infectados), mientras que el número de CD4 + CD45RAhi + CD62L + fue similar. Sin embargo, los TRECs fueron superiores en el grupo uVLque en el grupo dVL.El grupo uVLmostraron un número más alto de CD8 + CD45RO + y CD8 + CD45RO + CD38 + que el grupo Control, pero más bajo que el grupo dVL. Los porcentajes de CD8 + CD45RAhi + CD62L +, CD8 + CD45RA +, CD8 + CD62L +, y CD8 + CD28 + fueron superiores en el grupo uVLque en el grupo dVL, pero inferiores que en elgrupo Control. Elgrupo uVL tenía mayor número de células T CD4+ activadas (HLADR + CD38 +, HLADR +, HLADR + CD38) y porcentajes más bajos de CD4 + HLADRCD38 +, que el grupo dVL. En células T CD8+ activadas, el grupo uVLpresentaba menor número de CD8 + HLADR + CD38 +, CD8 + HLADR +, y CD8 + CD38 + que el grupo dVL. Las células T pre-efectoras (CD8 + CD57CD28 y CD8 + CD45RACD62L) fueron menores en el grupo uVL que en elgrupo dVL.
Conclusiones:Los niños infectados por el VIH que responden a TARGA tuvieron reconstitución de los TRECs, disminución de activación inmune y menor número de células T CD8+ efectoras. Además, una TARGA de éxito llevó a un incremento en la células T CD4+ activadas.
P110EFECTIVIDAD DE LA TERAPIA ANTIRRETROVIRAL POTENTE SOBRE LOS MARCADORES DE SEGUIMIENTO EN NIÑOS INFECTADOS VERTICALMENTE POR EL VIH
Salvador Resino García, José M.ª Bellón Cano, Rosa Resino García, José Tomás Ramos Amador, M. Dolores Gurbindo Gutiérrez, María José Mellado, M. Isabel de José Gómez y M. Ángeles Muñoz Fernández
Hospital General Universitario Gregorio Marañón, Madrid y Hospital 12 de Octubre, Madrid.
Objetivo:Evaluar la efectividad de la terapia antiretroviral potente (TAP) sobre las células T CD4+ y la carga viral plasmática (CV) de niños infectados por el VIH-1 a nivel población (efectividad en la "vida real") en niños con y sin SIDA.
Pacientes y métodos:Estudio retrospectivo entre 1994 y 2001. Se dividió el tiempo de seguimiento en 3 períodos según los protocolos de tratamiento antiretroviral (ART) usados:a) 94-96: terapia combinada (TC) con dos analogos de nucleosidos inhibidores de la transcriptasa inversa (NRTI).b) 97-98: TAP con tres fármacos: NRTI, y/o inhibidor de proteasas (IP), y/o no análogo de nucleosidos inhibidores de la transcriptasa inversa (NNRTI).c) 99-01: TAP pero con un uso predominante de nelfinavir y efavirenz que permitió que más del 60 % de los niños tuvieran TAP. Se calculó la media de las células de T CD4+ y CV por año de calendario como la medida más representantiva de cada niño por año y además hemos estratificado a los niños según la presencia o ausencia de SIDA para comparar los años que cierran los periodos descritos (96-96 [1996], 97-98 [1998], y 99-01 [2001]).
Resultados:Nosotros encontramos un aumento de los valores medios de células T CD4+ y disminución de la CV con la incorporación de las nuevas estrategias terapéuticas. Estos cambios son estadísticamente significativos tanto en los niños con SIDA como sin SIDA, pero a pesar del beneficio de TAP, los niños con SIDA no alcanzaron valores de células T CD4+ similares a los niños sin SIDA. Sin embargo, la CV de los niños con SIDA si que disminuye hasta los valores de los niños sin SIDA después de 1999.
Conclusiones:La efectividad de TAP a nivel población es elevada tanto en los niños con SIDA y sin SIDA.
P111HIMENOLEPIOSIS INTESTINAL EN LA INFANCIA
Elena Lucas Sáez, Jesús Fleta Zaragozano, Mercedes Gracia Casanova, Margarita Vázquez Olivares, Santiago Gallego Vela, Antonio Clavel Parrilla y M. Teresa Llorente Cereza
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción:La himenolepiosis es una infección parasitaria producida por tenias del géneroHymenolepis, las cuales pueden producir manifestaciones clínicas, preferentemente digestivas. De las dos especies patógenas para el hombre, la H. nanaes la más frecuente. La infección es rara en nuestro país, pero su incidencia en niños se ha incrementado, debido a casos observados procedentes de países en vías en desarrollo y de ambientes con deficiente nivel higiénico. Presentamos a continuación los casos de 13 niños parasitados porH. nana.
Casos clínicos:Se han observado 13 casos de infección porH. nana, 7 mujeres y 6 varones, de 2,6 a 12,5 años de edad, ingresados en el hospital por otra patología, y en familiares de éstos, desde 1995 hasta 2002. Todos los pacientes procedían de Portugal y de países en vías de desarrollo, eran de categoría socioeconómica baja y 4 de ellos convivían habitualmente con ratas. No existe en nuestra serie preferencia por edad ni por sexo y todos residían en España desde hacia un año o menos.
Diez (76,9 %) de los 13 pacientes estaban asintomáticos, dos (15,3 %) ingresaron en el hospital por convulsiones y uno por paludismo. En 6 pacientes (46,1 %) se aislaron otros agentes infecciosos patógenos: 2 especies de bacterias (Campylobactery Haemophylus influenzae) y 4 de parásitos (Plasmodium vivax, Giardia lamblia, Cryptosporidium parvuny Strongyloides stercolaris). Uno de los pacientes tenía, además, tres agentes infecciosos patógenos, dos pacientes dos agentes y tres pacientes un agente. En el resto de los casos no se encontraron otros parásitos, bacterias ni virus. Fue estudiada toda la familia del paciente del primer caso, encontrando 5 personas parasitadas asintomáticas y una con convulsiones.
Conclusiones:La parasitación por H. nana no es infrecuetne en niños procedentes de países en vías de desarrollo o de ambientes higiénicos muy deficitarios, por lo que debe ser investigada en nuestro medio por la facilidad de transmisión. No se excluye la posibilidad de que esta tenia pueda producir patología digestiva o convulsiones, como en dos de los casos estudiados por nosotros.
P112FIEBRE PERIODICA, ESTOMATITIS AFTOSA, FARINGITIS Y ADENITIS CERVICAL (PFAPA): APORTACION DE DOS CASOS
Antonio Madrid Madrid, Marta Cruz Cañete, David Moreno Pérez, Elisa Márquez Gámez, Olga M. Escobosa Sánchez, Vanesa Alonso, José Manuel Jiménez, Rosario Vázquez López, Miguel Galán Xarrié y Antonio Jurado Ortiz
Hospital General Carlos Haya, Málaga.
Introducción:El síndrome PFAPA es poco conocido. Criterios diagnósticos: 1.º fiebre periódica que comienza antes de los 5 años, 2.º síntomas constitucionales sin signos de infección respiratoria superior, con al menos uno de los siguientes hallazgos: estomatitis aftosa, faringitis, adenopatías cervicales, 3.º exclusión de neutropenia cíclica, 4.º intervalo asintomático entre episodios, 5.º desarrollo y crecimiento normales.
Los casos de PFAPA publicados en nuestro país, son muy escasos.
Casos clínicos:Niño de 2 años y 7 meses (Caso 1) y niña de 2 años (Caso 2), que durante un año de seguimiento, presentan un episodio mensual, de 4-5 días (Caso 1) y 2-7 días (Caso 2) de duración, de fiebre alta que cede con antitérmicos. Todos los episodios asocian síntomas constitucionales con adenopatías cervicales (de 3 cm en el Caso 1) y algunos además estomatitis aftosa minor y/o faringitis.
Pruebas complementarias en los episodios: hemograma y frotis sanguíneo con leucocitosis y neutrofilia, VSG y PCR elevadas en Caso 1, normales en el Caso 2. La función hepática, inmunoglobulinas, subpoblaciones linfocitarias, cultivo faríngeo, hemocultivos, Mantoux, serologías de brucella, citomegalovirus, herpes simple, Epstein-Barr, toxoplasma, y VIH, factor reumatoide, ANA, y radiografías del tórax, eran normales o negativos en ambos casos.
Evolución: el cuadro clínico cede espontáneamente, normalizándose los hallazgos analíticos, existe intervalo asintomático entre episodios y no afecta al desarrollo ni al crecimiento.
Comentarios:Nuestros casos cumplen criterios de síndrome PFAPA.
El reconocimiento de este síndrome, ayuda a un diagnóstico precoz, evitando estudios e ingresos hospitalarios innecesarios.
P113PREVALENCIA DE ETILOGIA ESTREPTOCOCCICA EN EL DIAGNOSTICO DE FARINGOAMIGDALITIS EN LA POBLACION PEDIATRICA DE CASTELAR
Ernesto Mónaco, Enrique Castiglioni y Miriam Medina
Instituto Pediátrico Castelar, Buenos Aires (Argentina).
Introducción:La faringoamigdalitis aguda es causada por diferentes microorganismos. La mayoria es de etilogia viral. El diagnóstico de faringoamigdalitis por estreptococo beta hemolítico del grupo A (EBHGA) es fundamental para iniciar el tratamiento antibiótico a fin de reducir el contagio, prevenir la fiebre reumática, glomerulonefritis y las complicaciones supurativas.
Objetivo general:Definir la prevalencia de los distintos serogrupos de estreptococos beta hemolíticos en los diferentes grupos etáreos de la población estudiada durante el año 2000.
Objetivos específicos:1. Identificar la frecuencia de infecciones estreptocóccicas en período estival e invernal.2. evaluar la sensibildad del test rápido para EBHGA.
Material y método:Análisis retrospectivo descriptivo. Se procesaron 1643 muestars de exudados de fauces para cultivo en agar sangre de carnero al 5 %. Se utilizó disco de Bacitracina a las colonias beta hemolíticas. Se realizó tipificación en grupos A,C,G,F. Conjuntamente se realizaron estudios de serología rápida para EBHGA con el dispositivo Strep A with OBC II a 1566 de las 1643 muestras totales.
Resultados:De 1643 muestras, 32 % fueron positivas para colonias de estreptococos beta hemolíticos. De las 1566 muestras para serología rápida 343 fueron positivas. De los cultivos positivos para estreptococos el 91,08 % fueron del grupo A; 7 % del grupo C; 1,55 % grupo G y 0,37 % grupo F. La sensibilidad del test rápido para EBHGA fue del 85 %. EBHGA predominó en edad escolar (4 a 12 años) con un pico maximo entre los 4 y 6 años y con descenso a partir de los 12 años. De los estreptococos beta hemoliticos no A, predominó el grupo C en mayores de 16 años. Prevalencia estacional: Abril-Mayo y Octubre-Noviembre.
Conclusiones:Los resultados hallados se corresponden con diferentes datos de distintas partes del mundo. La prevalencia más alta de infección por EBHGA se halló en los niños en edad escolar. La presencia de estreptococo del grupo C en adolescentes se convirtió en la segunda causa de faringoamigdalitis estreptocóccica luego de la producida por EBHGA. La docimentación bacterilógica permite el uso racional de antibióticos.
P114EMPIEMA EPIDURAL POR S. AUREUS TRAS INTERVENCION POR LAPAROSCOPIA DE ESTENOSIS HIPERTROFICA DE PILORO
Miguel Ángel Marín Gabriel, Pedro Gómez de Quero Masia, Sylvia Belda Hofheinz, María Baro Fernández, M. del Carmen García Miranda, M. Victoria Ramos Casado e Ignacio Sánchez
Hospital 12 de Octubre, Madrid.
Lactante previamente sano con empiema epidural, absceso prevertebral y pulmonar tras cirugía de estenosis hipertrófica de píloro (EHP):
Paciente de 1 mes y 7 días, previamente sano y sin anomalías en la exploración física, que fue intervenido de EHP mediante técnica de laparoscopia con acceso umbilical mediante anestesia general, y que presenta fiebre e hiporexia. Hemograma con 24.400 leucocitos (48 % segmentados y 16 % cayados), PCR 38,79 mg/dl, ecografía abdominal sin alteraciones, punción lumbar con salida de pus franco y cultivos de sangre y LCR positivos para S. Aureus, motivo por el que ingresa en UCIP, inicialmente con vancomicina y cefotaxima y finalmente con cloxacilina y rifampicina, presentando en todo momento un excelente estado general. Se realiza ecografía del raquis, observándose contenido ecogénico del espacio extraaxial posterior que ocupa la práctica totalidad del canal raquídeo. Se practica una resonancia magnética donde se observa acúmulo de líquido hiperintenso en zona epidural en todo el raquis, absceso pulmonar izquierdo de 2 * 2 cm y colección prevertebral de 4 cm, decidiéndose realizar abordaje quirúrgico. En el quirófano, y a las 24 horas de la RM, se practica una ecografía apreciándose una clara disminución de la colección, por lo que se decide realizar tratamiento conservador con los antibióticos citados, presentado una total resolución del cuadro sin presencia de secuelas neurológicas en la actualidad. Se realiza gammagrafía ósea sin alteraciones y estudio inmunológico con inmunoglobulinas normales y test de bioluminiscencia alterado pendiente de confirmación dado que se realizó en fase aguda.
Según la bibliografía consultada es el primer caso de empiema epidural que presenta como antecedente una intervención por laparoscopia con abordaje umbilical, siendo ésta la posible vía de entrada del patógeno. Así mismo destaca la excelente evolución y la ausencia de secuelas neurológicas mediante tratamiento oral exclusivo.
P115 MENINGITIS TUBERCULOSA CONCURRENTE CON MENINGOENCEFALITIS POR HERPES VIRUS TIPO 1
Jordi Martínez-Baylach, Gloria M. Fraga Rodríguez, Nuria Rabella García, M. Adela Retana Castán y Josep Cubells Rieró
Hospital de la Santa Creu y Sant Pau, Barcelona.
Introducción:La meningitis tuberculosa (MT) es la forma más grave de tuberculosis. Es una enfermedad muy poco frecuente en nuestro medio. El diagnóstico precoz y el inicio de las medidas terapéuticas en una fase temprena son necesarios para la buena evolución. En el mismo sentido, la encefalitis herpética puede ocasionar graves secuelas de no iniciarse un tratamiento en las primeras fases de la enfermedad.
Presentamos el caso de un paciente que fue diagnosticado de meningoencefalitis y en la que se detectó coinfección por M. tuberculosis y herpes simple tipo 1.
Caso clínico:Varón de 20 meses de edad que acudió a Urgencias por fiebre intermitente de unos 12 días de evolución. Antecedentes de reflujo vesicoureteral bilateral grado II-III. Exploración física normal, sin petequias ni signos meningeos ni focalidad neurológica. Analítica: Hb 90 g/l, leucocitos 12.100/mm3 (35 % neutrófilos, 55 % linfocitos). Plaquetas 360.000/mm3. PCR 7,5 mg/l. Ionograma, bioquímica y equilibrio ácido-básico normales. Urinocultivo positivo para E. coli. Mantoux negativo. Se diagnostica de infección del tracto urinario y se ingresa bajo tratamiento antibiótico con ceftriaxone.
A los 5 días de vida persiste fiebre y se añade empeoramiento del estado general, tendencia al sueño y somnolencia. Punción lumbar: líquido claro y normotenso con hipoglucorraquia (1,3 mmol/dl), hiperproteinorraquia (1.240 g/l) y 213 células de predominio linfocítico. A las pocas horas realiza convulsión de hemicuerpo izquierdo con desviación de la mirada, que cede con diazepan, quedando el paciente comatoso y con midriasis derecha reactiva. Se conecta el paciente a ventilación mecánica. TAC craneal: hidrocefalia moderada. Se inicia tratamiento con aciclovir y cuatro tuberculostáticos (ridampicina, isoniazida, pirazinamida y etambutol). Empeoramiento clínico y radiológico siendo el paciente éxitus en 48 horas. En el LCR se aisla M. tuberculosis y se detecta virus herpes tipo 1 por técnicas de PCR.
Comentario:La infección del SNC por M. tuberculosis es muy grave. Pensamos que la coinfección con virus herpes tipo 1 desencadenara o al menos pudiera tener algun papel en la mala evolución del paciente pese a recibir el tratamiento etiológico adecuado. No se encuentra en la literatura ningún otro caso de coinfección del SNC por estos dos gérmenes.
P116 FALLO MULTIORGANICO POR INFECCION POR VIRUS HERPES TIPO 6 EN PACIENTE INMUNOCOMPROMETIDO
Jordi Martínez-Baylach, Nuria Rabella García, Gloria M. Fraga Rodríguez, Isabel Badell Serra, Gemma Arca Díaz, M. Adela Retana Castán y Josep Cubells Rieró
Hospital de la Santa Creu y Sant Pau, Barcelona.
Introducción:La infección por virus herpes tipo 6 se asocia a una gran variedad de sintomatología clínica, generalmente de tipo banal. Sin embargo, en pacientes inmunocomprometidos, la infección o reactivación del virus latente pueda dar lugar a un proceso de mayor gravedad.
Presentamos el caso de una paciente diagnosticada de artritis crónica juvenil (ACJ) con tratamiento corticoideo prolongado que presentó fallo multiorgánico.
Caso clínico:Mujer de 12 años que fue diagnosticada de ACJ, con factor reumatoide de 81 UI/ml y ANA con patrón moteado 1/80. Función hepática normal. Precisa de prednisona a 1 mg/kg/48 horas para mantenerse asintomática.
A los dos meses del diagnóstico de ACJ consulta por ictericia cutáneo-mucosa y astenia. En la exploración física destaca además, hepatomegalia franca de cuatro traveses. La analítica muestra alteración del funcionalismo hepático (AST 858 U/l, ALT 2.120 U/l, GGT 158 U/l, bilirrubina total 167 umol/l). Ecografía abdominal: hepatomegalia homogenea. Serologías para virus hepatotropos negativas. Estudio de hepatitis autoinmune negativo. La biopsia hepática muestra signos de hepatitis inespecífica. Se inicia prednisona a 1 mg/kg/día mejorando tanto clínicamente como en el funcionalismo hepático (a los 30 días: AST 127 U/l, ALT 418 U/l). Cinco días más tarde, presenta empeoramiento progresivo con fallo multiorgánico: bicitopenia arregenerativa (Hb 66 g/l, plaquetas 41.000, mielograma con eritroblastopenia y hemofagocitosis), insuficienca cardíaca grave (hipocontractilidad biventricular con insuficiencia mitral y fracción de eyección de 35 %) y coagulopatía que requiere de vitamina K y plasma.
La necropsia muestra miocarditis con focos de necrosis extensa, congestión pulmonar aguda y crónica, necrosis centrolobulillar hepática y médula osea con aumento de la celularidad de la serie blanca y disminución de los eritroblastos. El estudio virológico de las muestras de la necropsia evidenció la presencia de virus herpes tipo 6 por técnicas de PCR.
Comentario:La infección por virus herpes tipo 6 puede desencadenar en pacientes inmunoomprometidos un proceso grave, que puede dar lugar a fallo multiorgánico y muerte.
P117 DENGUE IMPORTADO
Nagore García de Andoain Barandiarán, Alberto González García, Itziar Sota Busselo, Gustavo Cilla, Manuel Palacio Pina y Yon Albisu Andrade
Hospital Donostia, San Sebastián.
Introducción:El Dengue es una enfermedad limitada y benigna caracterizada por fiebre ocasionalmente bifásica, exantema, mialgias, artralgias, cefalea, adenopatías, leucopenia y náuseas o vómitos. La Fiebre Hemorrágica Dengue es su variante grave con alteraciones de la hemostasia y shock.
Existen 4 serotipos del Virus Dengue (familia flaviviridae), cuyo principal vector de transmisión es el mosquitoAedes aegypti. El periodo de incubación es de 1-7 días. Es una enfermedad endémica en zonas tropicales donde afecta principalmente a niños y viajeros. Aparecen brotes epidémicos en áreas urbanas infectadas por A. Aegypti por propagación a través de seres humanos virémicos.
Al diagnóstico se llega por cultivo del virus con recogida de sangre antes del 5.º día de enfermedad, o por serología, entre los 6 días y 6 semanas.
El tratamiento es sintomático, siendo raras las complicaciones. La prevención consiste en evitar la exposición a las picaduras de mosquitos en las zonas endémicas y destrucción de los criaderos de A. aegypti. La frecuencia y rapidez de los viajes aéreos hacen que pueda presentarse esta patología en nuestro medio.
Caso clínico:Varón de 9 años con cuadro de fiebre acompañado de artralgias, mialgias, cefalea, astenia y vómitos. A las 48 horas aparición de un exantema que motiva su ingreso. Antecedentes:la sintomatología se inició a las 48 horas de su llegada de Nicaragua, donde su madre padeció Dengue 15 días antes.Exploración física: buen estado general. Exantema tenue maculopapuloso generalizado. Resto sin hallazgos significativos.Exámenes complementarios: hemograma, bioquímica, PCR y pruebas de coagulación normales.Examen gota gruesa y hemocultivo:negativos. Serología IgM virus Dengue (4.º día de enfermedad) negativo (15.º día) positivo. Evolución:tratamiento de sostén. Al 5.º día desaparece la fiebre y atenuándose el exantema hasta desaparecer.
Conclusiones:El dengue debe considerarse entre los posibles diagnósticos, en todo paciente con exantema febril y estancia reciente en un país tropical.
La confirmación serológica se realiza mediante positividad IgM entre el 5.º día y 6.ª semana.
Habitualmente sólo es necesario tratamiento de sostén.
P118TUBERCULOSE PULMONAR: UM CASO DE TUBERCULOSE CONGÉNITA EM LACTENTE
Ariane Brand, Loio Pedro y Maria dos Anjos Bispo
Hospital de S. Francisco Xavier, Lisboa (Portugal).
Introdução:A tuberculose congénita é uma entidade clínica rara com menos de 300 casos documentados na literatura. Em apenas 50 % dos casos o diagnóstico materno é feito até á data do parto.
Caso clínico:RN com 32 semanas de idade gestacional com gravidez sem problemas, nascido por forceps, após ruptura de membranas de 48 horas. I. Apgar 8/10, PN 2.050 gramas. Nas primeiras 48 horas teve síndrome de dificuldade respiratória com radiografia com padrão intersticial bilateral interpretado como doença das membranas hialinas-I, fez O2 e antibioterapia que suspendeu às 48 horas por ausência de parâmetros de infecção. Ao 15.º dia de vida iniciou febre, hepatomegalia e dificuldade respiratória com hipoxemia, radiografia com hipotranparência da base dta. Iniciou CPAP seguida de ventilação assistida por ISMV, do 16.º ao 18.º dia de vida agravou o quadro respiratório passando à VAF. Os radiogramas seguintes mostraram hipotransparência difusa bilateral. Ao 19.º dia surgiu hipoxémia refractária necessitando ventilação mais agressiva e utilização de óxido nítrico. No 23.º dia é isolado Mycobacterium tuberculosis nas secreções brônquicas sendo a estirpe sensível a todos os tuberculestáticos, iniciando terapêutica específica. A hipoxémia manteve-se refractária e, após dois pneumotóraces, faleceu ao 27.º dia. A autopsia revelou tuberculose pulmonar com focos caseosos multiplos, sem envolvimento nem ganglionar nem de outros orgãos, nomeadamente hepático.
Na sequência do diagnóstico do RN foi observada a mãe tendo-lhe sido diagnosticada tuberculose urogenital (urocultura positiva e fluxo menstrual positivo dois meses depois). A caracterização genética das bactérias isoladas das secreções maternas e do RN pelos métodos de PCR e RFLP (Restriction fragment length polymorfism) permitiu comprovar a sua sobreponibilidade genética e distinção da estirpe vacinal.
Conclusões:Discute-se a via e a altura da contaminação do RN com manifestações precoces de doença em que se observam granulomas pulmonares permitindo o diagnóstico de tuberculose congénita apesar de não preencher todos os critérios de Cantwell.
P119 ARTRITIS MENINGOCOCICA PRIMARIA COMO PRESENTACION DE ENFERMEDAD INVASORA INSOSPECHADA
M.ª Verísima Barajas Sánchez, Cristina Ruiz Serrano, M. Elena Fernández Villalba, Miriam Blanco Rodríguez, José Manuel Sánchez Granados, Nieves Domínguez Garrido y Mercedes Bernacer Borja
Fundación Jiménez Díaz, Madrid.
Introducción:La artritis séptica es inhabitual en mayores de 5 años. El microorganismo causal más frecuente en todas las edades es elS. aureus, seguido de estreptococo beta hemolitico del grupo A.Neisseria meningitidis es un patógeno poco documentado en la artritis séptica. Presentamos el caso de una artritis meningocócica como forma de presentación de enfermedad invasora.
Anamnesis:Paciente de 10 años que consulta por fiebre elevada de una semana de evolución. Refiere dolor en la rodilla izquierda en los últimos dos días, que la paciente atribuye a un traumatismo previo. Al inicio del cuadro presentó un exantema máculo-violáceo en tronco y extremidades asociado a vómitos y diarrea autolimitada.
Antecedentes personales:No alergias conocidas. Sana. Calendario vacunal correcto (incluida vacuna de polisacáridos A + C en 1997).
Exploración física:T.ª 37,1°, TA 102/56. BEG, bien perfundida e hidratada. No aspecto séptico. No exantemas ni petequias. ACP: normal. Abdomen: normal. Neurológico: no rigidez de nuca, meningeos negativos. Aparato locomotor: derrame articular en la rodilla izquierda con signos inflamatorios y limitación de la movilidad. No escoriaciones
Exploraciones complementarias: Hemograma: leucocitos 15640 (N 64 %, L 21 %, Mo 13,5 %), serie roja y plaquetaria normales. VSG 40 mm/h. PCR 6,3 mg/dl. Bioquimica del liquido articular: glucosa 3 mg/dl, proteínas totales 5,5 g/dl, LDH 5.533 U/l. Gram en liquido articular: diplococos gram negativos. Cultivo: meningococo C. Hemocultivo negativo. RX tórax normal. Inmunoglobulinas y complemento normales.
Conclusiones: 1. N. meningitidis es causa infrecuente de artritis séptica. 2. La infección meningocócica debe ser considerada en presencia de fiebre con síntomas articulares (incluida artritis) con o sin exantema.
P120 REVISON DE VARICELA COMPLICADA DURANTE LOS AÑOS 2001-2002
Kay Boris Brandstrup Azuero, Susana Elena Zeballos Sarrato, Cristina Serrano, Piedad Isabel Dobon Westphal, Mar Roncero, Patricia Aparicio, José Antonio Gómez Campderá, M. Luisa Navarro Gómez, Rosa Rodríguez y Carlos Merello Rodino
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción:La varicela es una enfermedad exantemática viral muy frecuente en la infancia. Considerada como una enfermedad benigna, puede presentar complicaciones severas.
Material y método:Presentamos la serie de casos de varicela que acudieron a la urgencia de nuestro hospital infantil analizando aquellas que precisaron ingreso durante los años 2001-2002.
Resultados:Durante los años 2002 el número de casos de varicela que acudió al servicio de urgencias fue de 411 lo que supone un 0,78 % del total de urgencias atendidas y un incremento del 27,6 % con respecto a los pacientes asistidos con este diagnóstico durante el año anterior (322 pacientes que fueron un 0,64 % de las urgencias totales). Se pudo apreciar también una mayor severidad de la infección durante el año 2002, precisando ingreso 20 pacientes (4,8 % del total de varicelas), mientras que en el año 2001 fueron solo 5 pacientes los ingresados, lo que supone un 400 % de incremento. El 60 % de los pacientes ingresados eran menores de 2 años. No se objetivaron diferencias en la incidencia en cuanto al sexo. La estancia media hospitalaria fue de 7 dias. La complicación más frecuente fue la sobfreinfección bacteriana por Streptococcus pyogenes (generalmente cutáneas). Uno de los pacientes precisó ingreso en UCIP por un cuadro de shock estreptocócico. Dos de los pacientes atendidos durante el año 2002 y que precisaban ingreso tuvieron que ser trasladados a otro hospital.
Conclusiones: 1.En el año 2002 se ha objetivado un importante aumento tanto de la frecuencia como de la severidad de la infección por virus de la varicela.2. La infección por varicela puede producir complicaciones muy severas, siendo el grupo de mayor riesgo los niños menores de 2 años. 3. La varicela supone una importante carga social, familiar, económica y sanitaria.4. Consideramos que la vacunación sistemática de la población infantil frente a este virus podría estar justificada.
P121 INFECCION POR VIRUS INFLUENZA EN NIÑOS HOSPITALIZADOS EN EL PERIODO 2001-2002
Carmen Ruiz de la Cuesta Martín, Itziar Echevarría Matía, M. del Carmen Marín, Margarita Bouthelier Moreno, Manuel Omeñaca, Segundo Rite Gracia y Fernando de Juan Martín
Hospital Miguel Servet, Zaragoza.
Introducción:Las infecciones por virus Influenza A y B son una causa importante de ingreso en niños durante los meses de invierno. El objetivo del estudio es analizar los hallazgos epidemiológicos y clínicos más relevantes.
Material y métodos:Estudio retrospectivo del periodo diciembre 2001-marzo 2002. El aislamiento del virus Influenza A o B se realizó por inmunofluorescencia y cultivo de aspirado nasofaríngeo. Las diferencias encontradas entre los afectados por el virus A o B se analizaron mediante test de la U de Mann Whitney. Se analizó mediante test de correlación de Spearman la asociación entre las distintas variables.
Resultados:Durante dicho periodo se aislaron 91 casos de Influenza, de los cales 70 correspondían al tipo A (77 %) frente a 21 casos con el tipo B (23 %). La distribución por meses fue: Diciembre (5,5 %), Enero (48,3 %), Febrero (42,8 %) y Marzo (3,3 %). La totalidad de los casos se confirmó mediante cultivo. La inmunofluorescencia detectó un 83,9 % del tipo A y un 42,8 % del tipo B (p = 0,000). El tipo B fue más frecuente durante los meses de febrero y marzo (p = 0,000). La distribución por edad: < 1 año (41,7 %), 1-3 años (41,7 %), > 3 años (16,6 %). Las causas de ingreso fueron afectación del estado feneral, fiebre y síntomas respiratorios (25,9 %), crisis convulsiva febril (25,9 %), gastroenteritis aguda (16,4 %), sospecha de bacteriemia/meningitis (3,3 %), miositis aguda (3,3 %) y neumonía (5,5 %). La radiología durante el ingreso estuvo alterada en un 24,1 %; para el Influenza A en un 28,5 % y para el B en un 9,5 %; si bien estas diferencias no fueron significativas (p = 0,075). Un 65,9 % de los niños habían recibido antibioterapia pervia al ingreso. En un 18,6 % se asociaron otros virus respiratorios: virus respiratorio sincitial (VRS) (15,4 %) y adenovirus (21,9 %). No se encontró asociación entre la edad y el tipo de Influenza con la existencia de virus asociados. Sin embargo la existencia de neumonía presenta una correlación significativa con el VRS: r = 0,26; p = 0,024 y adenovirus: r = 0,26; p = 0,01.
Conclusiones:Las manifestaciones clínicas de la infección por Influenza son fundamentalmente respiratorias. La inmunofluorescencia mostró una escasa sensibilidad en los casos afectos por el tipo B. Un porcentaje elevado desarrollaron neumonía, siendo más frecuente en los que asociaban infección por otros virus respiratorios. Llama la atención el elevado uso de antibióticos en el tratamiento.
P122OTOMASTOIDITIS TUBERCULOSA
David Mora Navarro, Sara Aparicio Palomino, Irene Ibáñez Godoy, M. Ángeles Delgado Rioja, Lucía González Vila, Luis F. Nieto Hernández, José Nieto Fernández y José M. Cordoncillo Prieto
Hospital Juan Ramón Jiménez, Huelva.
Introducción:La tuberculosis continua siendo una infección relativamente frecuente en nuestro medio, a pesar de los avances existentes. La otitis media tuberculosa es una forma de presentación de la enfermedad tuberculosa muy poco frecuente. Se produce por el paso secreciones pulmonares infectadas al oído medio, más frecuente en niños pequeños, o por diseminación hematógena. Clínicamente se manifiesta como otorrea unilateral indolora, con acúfenos e hipoacusia, asociándose frecuentemente a parálisis de nervio facial. Se puede acompañar de inflamación de ganglios preauriculares o de cadena cervical anterior. El diagnóstico es difícil porque es una entidad en la que no se suele pensar.
Caso clínico:Niño de 8 meses de edad que ingresa para completar estudio de otorrea izquierda de cuatro meses de evolución resistente a tratamiento y tumoración otomastoidea izquierda informada por anatomía patológica como granuloma necrotizante Antecedentes familiares: nacionalidad ecuatoriana, resto sin interés. Antecentes personales: sin interés. Exploración:Buen estado general. Dermatits atópica. Pápula indurada en rodilla derecha. Tumefacción retro y prearicular izquierda sin signos inflamatorios con desplazamiento hacia fuera y delante del pabellón auricular. Otorrea purulenta ipsilateral con hiperemia y abombamiento timpánico apreciándose perforación en pars flácida. Resto por aparatos sin hallazgos patológicos. En los examenes complementarios realizados destacan VSG 74 con hemograma normal. Mantoux positivo. BAAR jugo gástrico negativo. Rx de Torax sin alteraciones significativas. TAC Torácico: condensación inflamatoria en lóbulo superior derecho con múltiples adenopatías hiliares y mediastínicas algunas con necrosis caseosa. TAC mastoide y oído medio izquierdo: masa de tejido blando mastoideo y epitímpanico que ocupa y destruye celdillas mastoideas, cadena osicular y oído medio. Lowestein exudado ótico crecimiento de M. Tuberculosis. Mantoux a la madre positivo, 20 mm de induración (vaunada BCG). Rx torax de la madre normal. Se inicia tratamiento antituberculoso con buena respuesta.
Conclusiones:Pensar en esta entidad ante otorrea crónica para realizar un diagnostico y tratamiento precoz y así evitar complicaciones y secuelas. Importancia de TAC torácica como método diagnóstico de imagen en la tuberculosis infantil.
P123MENINGITIS Y SEPSIS POR E. COLI EN UN LACTANTE CON URETEROHIDRONEFROSIS TRAS CISTOGRAFIA PREVIA Y PROFILAXIS ANTIBIOTICA INCOMPLETA
Elena García-Zarza Martínez, Florencio Jiménez Fernández, M.ª Elisa Corrales del Río, M. Pilar Gutiérrez Díez, Cristina Ferrero Martín y Juana Herrera Montes
Hospital Universitario de Getafe.
Antecedentes y objetivos:La profilaxis antibiótica adecuada en pacientes con anomalías del tracto urinario es esencial para prevenir las infecciones urinarias de repetición, precisando además la adecuada cumplimentación a dosis terapéuticas en los procedimientos diagnósticos de riesgo como es la cistografía, a fin de evitar, sobre todo en lactantes, infecciones graves como sepsis y meningitis.
Material y métodos:Lactante de un mes y ocho dias que presenta fiebre y rechazo del alimento de doce horas de evolución, tras habersele realizado una cistografía cuarenta y ocho horas antes bajo profilaxis antibiótica incompleta. Destacan: sedimento de orina: leucocitos: campos llenos, con leucocitosis: 23.000 leucocitos (56 % segmentados, 12 % cayados, 20 % linfocitos, 12 % monocitos) con hemocultivo positivo para E. coli, con urocultivo: > 100.000 UFC E. coli; LCR: 30 células, Ag en LCR para E. coli: positivo, ecografia renal: ureterohidronefrosis izquierda, estudio gammagráfico con MAG-3: ureterohidronefrosis izquierda con obstrucción parcial, secundario a obstrucción distal del uréter. Recibió tratamiento intravenoso con ampicilina y cefuroxima con buena respuesta terapéutica. Así mismo mediante la realización de técnicas de detección de Antígenos en líquido cefalorraquideo se pudo objetivar la presencia de meningitis por E. coli. Durante el seguimiento realizado, los potenciales evocados auditivos han sido normales.
Conclusiones:Señalar la gran importancia de realizar la profilaxis antibiótica a las dosis adecuadas en aquellos procedimientos considerados de riesgo, a fin de poder evitar complicaciones graves, así como la relevancia en aquellos casos parcialmente tratados con antibióticos de las técnicas de detección de antígenos en liquido cefalorraquideo que nos permiten el diagnóstico de los mismos.
P124
É POSSIVEL BAIXAR SIGNIFICATIVAMENTE A TAXA DE TRANSMISSÃO VERTICAL DO VIH CUMPRINDO UM PROGRAMA DE ACÇÃO COMUNITARIA
Eduarda Reis Monteiro, Fátima Figueira, Rosario Lobo, Antonio Lucas y José Palminha
Hospital de S. Francisco Xavier, Lisboa (Portugal).
Introdução:A infecção VIH é actualmente um dos maiores problemas mundiais de saúde pública. A SIDA, sendo uma doença devastadora dos paises pobres, é nos paises desenvolvidos uma patologia sobretudo das famílias disfuncionais. Este aspecto coloca complexas dificuldades no seguimento destas crianças/famílias.
A via de infecção do VIH na criança é quase exclusivamente materno-fetal. O protocolo ACTG076, criado em 1994, constitui um marco histórico na luta contra a transmissão vertical do VIH. No HSFX em Lisboa, organizamos em 1996 um grupo de trabalho (GTTV) com os objectivos de fazer cumprir o ACTG 076, tratar as crianças infectadas e apoiar estas famílias de risco bio-psico-social através de visitação domiciliária e de estreita articulação com os centros de saúde e outros recursos na comunidade.
Material e métodos:Apresenta-se um estudo prospectivo das crianças filhas de mães seropositivas para o VIH nascidas no HSFX entre Fevereiro de 1996 e Dezembro de 2002.
Foram analisados os seguintes parâmetros maternos: idade, raça, tipo de vírus, paridade, comportamentos de risco, ocupação, nível sócio-económico, altura do diagnóstico, vigilância da gravidez, e cumprimento do protocolo ACTG076.
Das crianças foram analisados: tempo de gestação, tipo de parto, peso à nascença, status infeccioso e cumprimento das consultas.
Resultados:Seguidas 116 famílias,117 crianças (1 parto gemelar). Das mães 87,9 % foram VIH 1, 6,9 % eram adolescentes, 42,2 % eram de raça negra, 13,7 % multíparas, 46,5 % desempregadas/domésticas, 40,5 % toxicodependentes e/ou prostitutas, 89,6 % eram socialmente desfavorecidas, em 62,9 % o diagnóstico foi feito na sequência da gravidez, 67,2 % foram vigiadas na gravidez, 43,5 % fizeram o ACTG076 completo.
Das crianças 13,6 % foram prematuras, 21,3 % nasceram por cesariana electiva, 9,4 % eram leves para a idade gestacional, foram infectadas 7,6 %. Em 3 % houve abandono das consultas.
Conclusões:Conseguiu-se uma diminuição da taxa de transmissão vertical de 19 % para 4,7 % em 2002 e o cumprimentocompleto do ACTG076 aumentou de 25 % para 52 %. A percentagem de cesarianas electivas nestas mulheres aumentou significativamente. O abandono das consultas diminuiu de 60 % antes da formação do GTTV para 3 % nos últimos seis anos e meio; sugerindo a eficácia deste modelo de intervenção comunitária.
P125INFECCIONES POR VIRUS RESPIRATORIOS EN NIÑOS INMUNODEPRIMIDOS: REVISION DE CASOS EN NIÑOS EN TRATAMIENTO ONCOLOGICO O CON INFECCION POR VIH
M. del Carmen Mendoza Sánchez, Jesús Ruiz Contreras, Francisco Fernández Carrión, Begoña Manso García y José Antonio Salinas Sánchez
Hospital 12 de Octubre, Madrid.
Antecedentes y objetivos:Los virus respiratorios constituyen la causa más frecuente de infecciones febriles en niños sanos. Las situaciones de inmunodeficiencia aumentan el riesgo de infecciones graves. Sin embargo, la mayoría de estudios se han dirigido hacia la etiología bacteriana, existiendo poca información sobre el impacto de infecciones por virus respiratorios comunes en inmunodeprimidos. Los objetivos de nuestro estudio son evaluar la prevalencia de infección por virus respiratorios en niños con infección por VIH o en tratamiento oncológico en nuestro hospital, en los últimos 15 años, valorar la significación clínica de estas infecciones en este grupo de riesgo, determinar la proporción debida a infección nosocomial, estudiar las consecuencias de la infección y evaluar el tratamiento administrado.
Métodos:Estudio retrospectivo de la base de datos de niños infectados por VIH en nuestro hospital y del archivo de historias de niños oncológicos tratados en nuestro centro en los últmos 15 años.
Resultados:Se revisaron en total 129 pacientes con infección por VIH, y 218 niños en tratamiento oncológico, encontrándose resultados positivos en 26 y 27 casos respectivamente. Un 43 % de los casos fueron debidos a VRS, seguido de virus Influenza A (26 %). Un 90 % de los casos se presentó con síntomas catarrales y la fiebre fue un signo también común (65 %). Casi un 40 % de los casos se debieron a transmisión nosocomial. Más de la mitad de los pacientes ingresaron, con una mediana de una semana. Un 15 % de ellos precisó ingreso en una unidad de cuidados intensivos. La quimioterapia se retrasó en la mitad de los pacientes oncológicos, una media de 6 días. Dos de nuestros pacientes fallecieron. Dos de los pacientes con infección por Influenza A presentaron sobreinfección bacteriana, y otro desarrolló un cuadro de sepsis por meningococo grupo B a las 24 horas de aislarse el virus Influenza. En 9 casos se usaron antivirales en el tratamiento, en 6 casos ribavirina y en 3 casos amantadina, sin poderse obtener conclusiones de eficacia en este estudio.
Conclusiones:Los virus respiratorios de la comunidad pueden causar infecciones graves en inmunodeprimidos. Es muy importante la detección precoz y prevención para disminuir su transmisión nosocomial. Se precisan más datos epidemiológicos, y estudios sobre nuevos agentes antivirales y terapia antiviral combinada.
P126SINDROME FEBRIL PROLONGADO POR YERSINIA
Alejandro López Escobar, Ana Siles Sánchez-Manjavacas, Yolanda Lage Alfranca, Cristina Calvo Rey y Beatriz García Cuartero
Hospital Severo Ochoa, Leganés.
La yersinia enterocolítica es habitualmente causante de cuadros gastrointestinales Muy raramente se ha descrito como causante de cuadros febriles prolongados.
Caso clínico:Varón de 14 años que acude por fiebre de hasta 38,5 °C de 12 días de evolución. Refería dolor abdominal leve e inespecífico las primeras 48 horas de fiebre y astenia y anorexia durante toda la evolución sin otra sintomatología.
Entre los antecedentes destaca un retraso ponderoestatural, una fractura de radio izquierdo un mes y medio antes del ingreso y otitis medias de repetición.
La exploración por aparatos fue completamente normal.
En las pruebas complementarias que se realizan destacaban:
Una discreta leucocitosis (17.290/mm3) con neutrofilia y eosinofilia (76 y 5 % respectivamente), anemia microcítica e hipocroma (Hb 11,5 g/dl, Hto. 34,4 %, VCM 85,8) y VSG en la primera hora de 86 mm.
En la bioquímica: discreta hiperproteinemia (8,9 g/dl) con aumento de la IgG (1690 mg/dl) y una PCR de 156 mg/l. Ferritina: 176 ng/ml. Resto de parámetros de sangre y orina, coagulación y autoinmunidad normales.
Hemocultivo, urocultivo, mantoux y frotis faríngeo negativos. El coprocultivo y las muestras de parásitos no se procesaron por no ser heces diarreicas.
Serología para CMV, VEB, Brucella, Toxoplasma, Coxiella, Lyme, Parvovirus y Fiebre botonosa: negativos. Serología para Yersinia enterocolítica: positiva.
Radiografía de tórax, ecografía abdominal y ecocardiograma: normales.
Tras permanecer 6 días ingresado persistiendo picos febriles de hasta 39 °C y con todos los resultados de las pruebas complementarias descritos salvo la serología, se pensó inicialmente en un debut de AIJ sin otra sintomatología, por lo que se inició tratamiento con Ibuprofeno a dosis de 20 mg/kg/día desapareciendo la fiebre. Durante toda la evolución permaneció asintomático, siendo siempre la exploración completamente normal. Se le controló ambulatoriamente en consultas, y tras la llegada de los resultados de la serología se inició la retirada del ibuprofeno sin reiniciar la fiebre.
Comentarios:Pese a ser una causa extremadamenta rara de síndrome febril prolongado, la yersinia siempre debe incluirse en el diagnóstico diferencial de esta entidad.
P127MENINGITE BACTERIANA EM PEDIATRIA. ESTUDO RETROSPECTIVO DE 1990-2002
Marisol Anselmo, Fernando Tapadinhas, Luís Gonçalves,
Manuela Calha y Margarida Silva
Hospital Distrital de Faro, Portugal.
A meningite bacteriana é uma doença de extrema importância para os Pediatras, tanto pela sua frequência, como pela sua potencial gravidade.
Objectivo:Analisar a epidemiologia da doença; avaliar a actuação das novas vacinas anti: Haemophilus influenza tipo b, S. pneumonea e Neisseria meningitidis C, nesta população infantil.
Material e método:Estudo retrospectivo de 1990 a 2002 que incluiu todas as crianças com idades entre 1 mês e 12 anos, internadas no Serviço de Pediatria do Hospital Distrital de Faro, com o diagnóstico de meningite bacteriana.
Parâmetros analisados:Idade, sexo, residência, data do internamento, vacinação (incluíndo as novas vacinas), terapêutica antibiótica prévia, tempo decorrido desde o inicio dos sintomas e o internamento, quadro clínico, agente infeccioso, terapêutica, evolução clínica, morbilidade e mortalidade.
Resultados:Verificaram-se 128 casos, com uma média de 10 casos/ano, a maior incidência ocorreu no sexo masculino (61 %) e no grupo etário com idade inferior a 2 anos (56,2 %). A Neisseria meningitidis foi o agente etiológico mais frequente (45 %). A ceftriaxona foi o antibiótico de eleição na maioria dos casos, traduzindo a sua eficácia através da evolução clínica globalmente favorável.
Conclusão:Os resultados obtidos nomeadamente no que respeita à frequência, grupo etário, sazonalidade e agentes infecciosos foram sobreponíveis às outras séries encontradas na literatura. Não obstante as novas vacinas mantiveram-se o número de casos ao longo dos anos, verificando-se uma diminuição efectiva do número de meningites bacterianas porHaemophilus influenza tipo b após a vacinação.
P128 INFECCIONES GRAVES POR PSEUDOMONAS EN INMUNODEPRIMIDOS
Laura San Feliciano Martín, Dorotea Raquel Fernández Álvarez, Manuela Muriel Ramos, M. Dolores García García, Aránzazu Hernández Fabián y G. Mateos
Hospital Universitario de Salamanca. Hosp. Clín. y Hosp. Virgen de la Vega.
El número de pacientes inmunodeprimidos ha experimentado un importante aumento en las últimas décadas, debido en parte, a los avances en tratamientos oncohematológicos, la enfermedad por el virus de la inmunodeficiencia humana, el uso de corticoides, inmunosupresores...
Las infecciones constituyen la primera causa de morbimortalidad en los enfermos inmunodeprimidos.
Pseudomonas aeruginosa es un patógeno bacteriano gram negativo aerobio que causa infecciones importantes en estos pacientes.
Presentamos una revisión de infecciones producidas por pseudomonas en niños inmunodeprimidos con cinco casos clínicos: meningitisen un niño de 4 años afecto de leucemia aguda linfoide en tratamiento de mantenimiento; sepsisen niño de 9 años con linfoma no Hodgkin de células B con tratamiento quimioterápico; neumonía recurrente en una paciente con inmunodeficiencia variable común en tratamiento con corticoides;neumonía con atelectasia en una niña diagnosticada de miastenia gravis en tratamiento con corticoides; ypioderma gangrenoso en una niña de 4 años afecta de leucemia aguda linfoide en tratamiento quimioterápico.
Comentarios:Debemos considerar a Pseudomonas Aeruginosa como posible agente causal de enfermedades diversas y graves en el inmunodeprimido, sobretodo en situaciones de tratamientos y procedimientos invasivos y aerosolterapia.
Es necesario administrar el tratamiento antibiótico adecuado lo más precozmente posible para evitar complicaciones.
P129NEUROCISTICERCOSE
Juan Calviño Cabezas, Filipa Vasconcelos Espada, Jorge Quintas, Natalina Miguel, António Trindade, Rui Chorao, Jorge Amil, Fátima Dias y José Arêlo Manso
Hospital de São João, O Porto (Portugal) y Centro Hospitalar Vila Real, Portugal.
A cisticercose é consequência da infecção pela larva daTaenia solium. A clínica é pleomorfa, embora atinja mais frequentemente o músculo esquelético e SNC, falando-se neste último caso de Neurocisticercose. Chega-se ao diagnóstico fundamentalmente por uma correcta interpretação dos exames imunológicos e de neuroimagem, em contexto clínico e epidemiológico compatíveis. Entre as drogas cestocidas destacam-se o Albendazol e o Praziquantel, associados quando necessário a tratamento sintomático (antiepilépticos, corticosteroides, analgésicos, antieméticos, etc.). Em casos de hidrocefalia ou cisticercos ventriculares deverá considerar-se a possibilidade de tratamento cirúrgico.
Os AA apresentam o caso de um adolescente do sexo masculino, 12 anos de idade e residente em meio rural, admitido por cefaleias e hemianopsia homónima esquerda com 6 dias de evolução, e posteriormente aparecimento de febre, vómitos e rigidez da nuca.
A investigação analítica inicial foi inconclusiva, mas a TAC revelou uma imagem compatível com abcesso cerebral esquerdo. A positividade da serologia para cisticerco e a RMN cerebral confirmaram como diagnóstico provável Cisticerco Gigante em estadio coloidal.
Realizou Albendazol, durante 7 dias, em associação com Dexametasona, e teve alta clínica, melhorado.
É seguido na Consulta Externa de Pediatria. Fez TAC de controlo, 2 meses depois da alta, sem alterações. Actualmente manten-se assintomático.
P130 GLOMERULONEFRITIS AGUDA CON HEMATURIA MACROSCOPICA SECUNDARIA A VIRUS DE EPSTEIN-BARR
Pilar Sevilla Ramos, María Penín Antón, Joaquín Ramírez Fernández, Ana M. Carrasco Torres, Cristina Olivas López de Soria, Mercedes Gómez Manchón, M. Luisa Murga Sierra y José Enrique García de Frías
Hospital Príncipe de Asturias, Alcalá de Henares.
Introducción:La infección por el virus de Epstein-Barr es una enfermedad frecuente que posee un amplio espectro clínico. La afectación renal en la mononucleosis es poco frecuente oscilando en la literatura desde el 0 al 13 %.
Caso clínico:Niña de 11 años que acude a urgencias por presentar orinas oscuras y edema palpebral bilateral de 24h de evolución. Refería fiebre desde hacía 4 días y estaba en tratamiento con amoxicilina por faringoamigdalitis.
Exploración física:TA: 122/79 mmHg (P90). BEG, adenopatías laterocervicales rodaderas menores de 1 cm, edema palpebral bilateral leve, faringe levemente hiperémica sin exudados, auscultación cardiopulmonar normal, abdomen blando no doloroso sin masas ni megalias, neurológico normal.
Pruebas complementarias:Sedimento: 3 leucocitos, hematuria macroscópica. Hemograma 10100 leucocitos/mm3 (16N, 65l, 16M), Hb13,2 g/dl, plaquetas 162000/mm3. Coagulación: normal. Bioquímica: Cr 0,6 mg/dl, Alb 4,2 g/dl, GOT 211 U/l, GPT 624 U/l, GGT 205 U/l, BiT 1,5 mg/dl. C3c 97,8 mg/dl (N 45-120), C4 40 mg/dl (N15-45), ASLO < 62,2, PCR 6 mg/l. Inmunoglobulinas: normales. Bioquímica en orina 24h: ClCr: 73 ml/min/1,73 m2, RTP 94 %, EFNa 0,25 %, proteinuria 100 mg/dl. Mantoux: negativo. Frotis faringeo: negativo. Hemocultivo y urocultivo estériles. Serología VHB, VHA, VHC, VIH, CMV, Toxoplasma: negativa. Serología VEB: IgM VCA positiva. Latex VEB positivo.
Evolución:Se mantuvo ingresada durante 8 días con TA y diuresis dentro de rangos normales, proteinuria negativa. Se apreció aumento de las adenopatías laterocervicales con exudado amigdalar. La fiebre desapareció a los 20 días. Al alta persistía microhematuria que se normalizó a las 3 semanas. A los 3 meses TA normales y control analítico a con bioquímica en orina de 24h: ClCr 65 ml/min/1,73 m2,RTP 90 %, EFNA 0,68, proteinuria negativo.
Comentario:La manifestación renal más frecuente de la mononucleosis es la hematuria macroscópica. Con menos frecuencia también pueden aparecer edemas, oliguria e hipertensión arterial. La proteinuria es rara aunque se han descrito casos de síndrome nefrótico. Creemos que la mononucleosis infecciosa debería considerarse en el diagnostico diferencial de las macrohematurias.
P131 RECIÉN NACIDO CON EXANTEMA MACULOPAPULAR AL NACIMIENTO
Amaia Bilbao Garitagoitia, Inés Esteban Díez, Pilar García Sánchez, Rosa M. Escriba Polo y Félix Omeñaca Teres
Hospital Universitario La Paz, Madrid.
Introducción:La forma de candidiasis cutánea neonatal más frecuente es la connatal en la que las lesiones se localizan en zona perineal y orofaríngea y aparecen a partir de la primera semana de vida. Por el contrario, la candidiasis cutánea congénita es poco frecuente. A pesar de que un 20-25 % de las mujeres embarazadas presentan vulvovaginitis por Candida spp. aún están por determinar la razón por la que algunos recién nacidos adquieren la infección intraútero. Presentamos uno de los infrecuentes casos de candidiasis cutánea congénita que se ha dado en nuestro hospital.
Caso clínico:Una niña nació a término, por parto eutócico, de una madre sana, sin antecedentes personales de interés. Presentó un Apgar de 8/9; Peso:3360g; Talla:51cm; Perímetro cefálico: 33cm; Los percentiles antropométricos eran normales para su edad gestacional de 40 semanas. La amniorrexis fue 3 horas previas al parto.
Al nacimiento presentaba un exantema generalizado maculpapular con áreas exudativas en cabeza, tórax y espalda, con participación palmoplantar. Estaba afebril y con buen estado general. Resto de la exploración normal. Ante la sospecha de una infección congénita se instauró antibioterapia intravenosa empírica tras toma de frotis de superficie, hemocultivo y urocultivo.
La madre no refería procesos intercurrentes de interés durante el embarazo.
La evolución del exantema fue favorable. A las 48 horas, el laboratorio de microbiología informó de crecimiento de Candida albicans en frotis umbilical, ótico y faríngeo. Se suspendió la antibioterapia y se inició tratamiento con nistatina suspensión oral y miconazol tópico que se mantuvieron hasta completar 10 días. Las lesiones curaron al quinto día de vida.
Comentarios:La candidiasis cutánea congénita es un cuadro poco frecuente que hay que tener presente en el diagnóstico diferencial de un exantema maculopapular congénito. El frotis de superficie tiene un importante valor diagnóstico. Es importante diferenciar la forma localizada que tiene una evolución benigna de la forma sistémica en la que el pronóstico es fatal si no se trata con antifúngicos intravenosos de manera inmediata.
P132BRUCELOSE. ESTUDO RETROSPECTIVO (1992-2002)
Marisa Sousa, Natalina Miguel, Fátima Dias y José Arêlo Manso
Centro Hospitalar Vila Real, Portugal.
Introdução:A Brucelose é uma zoonose causada por microorganismos do géneroBrucella. Esta continua a ser um importante problema de saúde pública em países da Bacia Mediterrânea (principalmente na Grécia, Portugal, Espanha, Itália e países do Norte de África), onde a infecção permanece por irradicar no hospedeiro animal.
Objectivos:O objectivo principal foi caracterizar o grupo de crianças internadas no serviço de Pediatria do Centro Hospitalar Vila Real Peso da Régua com o diagnóstico de Brucelose nos últimos 11 anos (1992-2002). Analisaram-se os parâmetros epidemiológicos, laboratorias e terapêuticos.
Material e métodos:Realizou-se estudo retrospectivo dos processos clínicos das crianças internadas entre 01/01/1992 e 31/12/2002 com o diagnóstico de Brucelose. O diagnóstico foi considerado perante uma clínica sugestiva, R. de Wright > 1/80, R. Rosa de Bengala positiva e/ou hemocultura positiva paraBrucella. Foram analisados os seguintes parâmetros: sexo, idade, incidência anual, distribuição sazonal, história familiar de Brucelose, fonte de contágio, tempo de evolução da doença, clínica de apresentação, estudo analítico, terapêutica instituída, complicações e evolução da doença.
Resultados:Analisaram-se os processos clínicos de 84 crianças, entre os 28 meses e os 14 anos de idade. Verificou-se maior incidência nos anos de 1994 e 1997 (13/84) e nos meses de Maio a Agosto. Houve predomínio no sexo masculino e 40,47 % das crianças tinham mais de 10 anos. Em 27 crianças havia história familiar de Brucelose e na maioria o contágio foi por contacto com animais infectados. O tempo de evolução da doença foi em 36,90 % inferior a 7 dias. Os principais sintomas foram artralgias, febre, anorexia e astenia. Foi isolada a bactéria em 20,23 % dos casos. Os fármacos utilizados foram Doxiciclina, Cotrimoxazol, Estreptomicina, Rifampicina e Gentamicina. Diagnosticaram-se 2 casos de Neurobrucelose. A evolução foi na maioria dos casos favorável mas ocorreram 3 recidivas e 2 casos de reinfecção.
Comentários:A Brucelose continua a ser um importante problema de saúde pública na nossa região, atingindo a população pediátrica sobretudo pelo contacto directo com animais infectados. Só um controlo sanitário mais adequado e erradicação da doença no hospedeiro animal poderá evitar a infecção humana.
P133ESTUDIO COSTE-EFICACIA DE UNA ESTRATEGIA PREVENTIVA EN PREMATUROS
M. Belén Robles García, Ana Díaz Moro, Javier Mallo Castaño, Carmen Villafañe Landeira, M. Blanca Herrero Mendoza, Santiago Lapeña López de Armentia, Juan Pablo Martínez Badas y Leticia Castañón López
Hospital de Léon.
Objetivo:Evaluar el hipotético impacto económico de la aplicación de una estrategia asistencial con palivizumab en la prevención de bronquiolitis.
Metodología:Se incluye a los neonatos nacidos en el hospital de León desde enero de 1995 a diciembre de 1998 ingresados por bronquiolitis. Aplicando datos del estudioImpact-RSV se realiza un estudio de coste-eficacia que incluye el coste directo de la prevención, el coste de hospitalización y el coste de ingresos no evitados. Se completa el estudio con la relación entre el hipotético coste en el grupo tratado y el coste en el grupo no tratado
Resultados:De 7.766 neonatos, hay 56 con edad gestacional ≤ < 32 semanas y en 8 se diagnostica bronquiolitis. En los ≤ 32 semanas en el grupo no tratado, el coste de la hospitalización sería de 15.673 €(3.134 €/año, 55,98 €/neonato), y en el grupo tratado de 88.490 €(17.698 €/año, 316,04 €/neonato) (fig. 1). Tras la hipotética profilaxis los mejores resultados se obtienen en el grupo de ≤ 30 semanas con 3,8 veces más de gasto que el actual (fig. 1).

Figura 1. Coste en euros y ratio por neonato, en grupo tratado y no tratado, según semanas de gestación.
Conclusiones:El estudio coste-eficacia ofrece un método adecuado para decidir recomendaciones en este programa preventivo.
P134PIOMIOSITE DO OMBRO
Georgeta Oliveira, Eduarda Cruz, Filipa Vasconcelos Espada, Sandra Cristina Alves, Carlos Abel y Armanda Rainho
Hospital Pedro Hispano, Matosinhos (Portugal).
A piomiosite primária é um processo infeccioso raro que envolve o músculo estriado, sem fonte local ou adjacente óbvia de infecção. A natureza exacta da piomiosite primária é ainda pouco clara, mas pensa-se que resulta da disseminação hematogénea de uma fonte oculta e/ou de um traumatismo prévio que cause agressão muscular. Apesar de ter sido inicialmente descrita em regiões tropicais, tem-se verificado um aumento do n.º de casos registados noutras áreas geográficas. A infecção focal tipicamente forma um abcesso que, quando diagnosticado precocemente, em regra responde a uma cobertura antibiótica adequada. Os autores apresentam um caso clínico de piomiosite do ombro esquerdo numa criança do sexo masculino, de 4 anos, que se apresentou no serviço de urgência com um quadro de febre, vómitos e dor á mobilização do ombro esquerdo, sem sinais inflamatórios ou de abcedação evidentes. O estudo imagiológico efectuado (RMN/TC) revelou miosite dos músculos peitoral e esternocleidomastoideu esquerdos, com extenso infiltrado inflamatório dos tecidos moles adjacentes. Após imediato tratamento com flucloxacilina e.v., verificou-se uma atempada melhoria clínica, sem qualquer deficiência funcional residual. O estudo imunológico da criança revelou-se normal. Esta é, portanto, uma patologia que deve ser incluida nos diagnósticos diferenciais a considerar em qualquer criança com síndrome febril e queixas dolorosas musculares ou articulares.
P135 NEUROCISTICERCOSIS PEDIATRICA: PRESENTACION DE UN CASO
Ana Gómez Zamora, Lara Olazar Gómez, Ramón Velázquez Fragua, María Jesús García de Miguel, M. Isabel de José Gómez, Francisco Martínez Cortés, Clementina Borque Andrés y Fernando Castillo
Hospital Universitario La Paz, Madrid.
La neurocisticercosis es una enfermedad endémica producida por la larva deTaenia Solium que alcanza el SNC vía circulación sistémica, tras ser ingerida a partir de alimentos contaminados. Es más frecuente en el Tercer Mundo.
Caso clínico:Niña hindú de 3 años que presenta desde su llegada a España 6 episodios de crisis parciales generalizadas con sintomatología vasovagal y sensación previa de angustia. Se objetiva en TAC una imagen compatible con absceso en región occipital derecha.
A.P.:Niña adoptada procedente de la India residente en España desde hace 8 meses. Vacunación según calendario.E.F.: normal.P. complementarias: Hemograma: Hb: 12,9 g/dl. Hto: 37,7 %. Leucocitos: 16,8 x 103/mm3 (N: 57,2; E: 2,1) Plaquetas: 339 x 103/mm3. Rx tórax: normal. EEG: focalidad de ondas lentas sobre areas posteriores del hemisferio derecho. TAC craneal: tumoración intraaxial occipital derecha. RMN craneal: lesiones nodulares occipitales corticales derechas, abundante edema y discreto efecto masa que sugiere una naturaleza abscesificada de origen bacteriano o parasitario. Ecografia abdominal: normal. Rx miembros inferiores: normal. Microbiologia: parasitos en heces: negativo. Serología: toxoplasmosis: (). hidatidosis: (+) baja, no valorable, posible reacción cruzada. Amebiasis (). Cisticercosis: ELISA IgG (+) F. Vesicular (+). ELISA D. antígeno ().Tratamiento y evolución: Se inicia tratamiento antibiótico con cefotaxima y metronidazol, continuando con oxcarbamacepina y dexametasona para el control de las crisis convulsivas. Se confirma serologicamente el diagnóstico de neurocisticercosis y se instaura tratamiento con albendazol (15 mg/kg/día) cada 8 horas que se mantiene un total de 15 días. Durante su ingreso se mantiene afebril, con buen estado general, sin presentar crisis convulsivas. En la RMN de control se objetiva una disminución del edema occipital perilesional y se siguen visualizando lesiones en anillo.
Conclusiones:A pesar de que la neurocisticercosis es infrecuente en menores de 5 años, es un diagnostico a tener en cuenta ante todo niño inmigrante procedente de zonas tropicales endémicas que presente crisis epilépticas.
El desencadenante de estas crisis suele ser el edema perilesional secundario a la muerte del parásito que se produce en el transcurso de la enfermedad.
P136VIGENCIA DEL N-METILGLUCAMINE EL TRATAMIENTO DE LEISHMANIASIS CUTANEA EN LOS NIÑOS
Amando Martín Peña, Pedro Navarro, Elinor Garrido, Yesica García, Virginia Coraspe y Olinda Delgado
Hospital Universitario de Caracas, Venezuela y Universidad Central de Venezuela. Instituto de Médicina Tropical, Caracas (Venezuela).
La leishmaniasis es frecuentemente una afección de la piel en los niños de las áreas rurales de Venezuela. Al Hospital Universitario de Caracas. Departamento de Pediatría: Sección de Pediatría Medica Infecciosas y en el Instituto de Medicina Tropical de la Universidad Central de Venezuela refieren niños con úlceras cutáneas y de mucosa nasal, procedentes del medio rural o niños que viven en la ciudad pero visitan áreas endémicas de la parasitosis, con elementos clínicos sugestivos.
Se les práctica diagnóstico clínico-presuntivo, inmunológicos (determinación de anticuerpos fluorescentes ante-leishmania y prueba intradérmica de leishmanina), parasicológicos por frotis por aposición (coloraciones especiales). Evaluaciones: química sanguínea (funcionalismo hepático y renal), hematológicas, uroanálisis y control por la sección de cardiología infantil; al ingreso del paciente y en los días finales del tratamiento. Se incluyeron 193 niños con los criterios diagnósticos, en edades comprendidas entre ocho meses y doce años de edad. El 75 % de los casos procedían de áreas periféricas a Caracas. En los miembros inferiores se ubicó el 51 % de las úlceras y en los brazos otro 27 %, en el 23 % de los casos presentaban lesiones múltiples. Fueron tratados con N-metilglucamina a la dosis de 100mg/kg/día por 10 días; se controlaban en el día siete y quince de su egreso y se decidía repetir un nuevo ciclo de acuerdo al curso clínico y esto fue necesario en 60 % de los niños del estudio. No existió alteración cardio-pulmonar evaluable. El 19 % de los pacientes presentaron en sedimento urinario + o + + de proteinuria y en el 17 % elevación de transaminasas hepáticas; en todos revirtió los efectos mencionados. Ningún paciente fue retirado del estudio por efectos adversos atribuibles al antimonial. Las úlceras presentaron curación total al final de la terapéutica. El N-metilglucamina demuestra en esta evaluación que continúa siendo un medicamento seguro y eficaz en leishmaniasis, tiene en contra la necesidad del seguimiento supervisado que requieren los pacientes que lo reciben. Se necesitan más controles para poder usar este antimonial en consultas ambulatorias como se realiza en pacientes adultos.
La leishmaniasis es frecuentemente una afección de la piel en los niños de las áreas rurales de Venezuela. Al Hospital Universitario de Caracas. Departamento de Pediatría: Sección de Pediatría Medica Infecciosas y en el Instituto de Medicina Tropical de la Universidad Central de Venezuela refieren niños con úlceras cutáneas y de mucosa nasal, procedentes del medio rural o niños que viven en la ciudad pero visitan áreas endémicas de la parasitosis, con elementos clínicos sugestivos.
Se les práctica diagnóstico clínico-presuntivo, inmunológicos (determinación de anticuerpos fluorescentes ante-leishmania y prueba intradérmica de leishmanina), parasicológicos por frotis por aposición (coloraciones especiales). Evaluaciones: química sanguínea (funcionalismo hepático y renal), hematológicas, uroanálisis y control por la sección de cardiología infantil; al ingreso del paciente y en los días finales del tratamiento. Se incluyeron 193 niños con los criterios diagnósticos, en edades comprendidas entre ocho meses y doce años de edad. El 75 % de los casos procedían de áreas periféricas a Caracas. En los miembros inferiores se ubicó el 51 % de las úlceras y en los brazos otro 27 %, en el 23 % de los casos presentaban lesiones múltiples. Fueron tratados con N-metilglucamina a la dosis de 100mg/kg/día por 10 días; se controlaban en el día siete y quince de su egreso y se decidía repetir un nuevo ciclo de acuerdo al curso clínico y esto fue necesario en 60 % de los niños del estudio. No existió alteración cardio-pulmonar evaluable. El 19 % de los pacientes presentaron en sedimento urinario + o + + de proteinuria y en el 17 % elevación de transaminasas hepáticas; en todos revirtió los efectos mencionados. Ningún paciente fue retirado del estudio por efectos adversos atribuibles al antimonial. Las úlceras presentaron curación total al final de la terapéutica. El N-metilglucamina demuestra en esta evaluación que continúa siendo un medicamento seguro y eficaz en leishmaniasis, tiene en contra la necesidad del seguimiento supervisado que requieren los pacientes que lo reciben. Se necesitan más controles para poder usar este antimonial en consultas ambulatorias como se realiza en pacientes adultos.
P137CELULITIS PERIORBITARIA: REVISION DE NUESTRA CASUISTICA
M. Luisa Herreros Fernández, Alfonso González Laguillo, Juana Barja Tur, M. Teresa de Santos Moreno, Ana M. Pastor Gómez y Sonia Ramírez Parenteau
Clínica Moncloa, Madrid.
Objetivo:Valorar la epidemiología, presentación clínica, diagnóstico, tratamiento y evolución de las celulitis periorbitaria en nuestro medio hospitalario.
Material y métodos:Revisión retrospectiva de las historias clínicas de los niños ingresados con el diagnóstico de celulitis periorbitaria desde enero de 1997 a enero de 2003.
Resultados:Analizamos un total de 64 pacientes, representando el 1,2 % de los ingresos realizados en el área de pediatría. El 63 % fueron varones. Las edades oscilaron entre un día y trece años, con una media de 44 meses. En 28 casos se evidenció un antecedente de conjuntivitis y en 11 de traumatismo local. El ojo derecho se vió afectado en el 62 % de los casos y un solo caso fue bilateral. Se realizó radiografía de senos en 17 pacientes, y TAC craneal en 23. Se obtuvo hemocultivo en el 64 % de los pacientes, que fue negativo en todos los casos y cultivo de secrección ocular en 11 casos siendo positivo en 5. Como diagnóstico secundario encontramos conjuntivitis en 25 casos, sinusitis en 21, celulitis frontal en 3 y dacriocistitis en 3. Existieron complicaciones en 5 casos (2 celulitis orbitarias, un absceso subperióstico, un empiema epidural y un absceso local). El tratamiento fue intravenoso con cefotaxima ± cloxacilina, cefuroxima o amoxicina clavulánico, y en un caso se asoció cefotaxima, vancomicina y metronidazol. La estancia media fue de 5,3 días y la evolución buena en todos los casos, sin precisar ninguno de ellos tratamiento quirúrgico.
Conclusiones:La celulitis periorbitaria representa el 1,2 % de los ingresos pediátricos en nuestro medio, siendo en la mayoría de los casos secundaria a un proceso infeccioso local. A pesar que observamos complicaciones en un 7,8 % de los pacientes, la evolución fue favorable en todos los casos con tratamiento médico exclusivo.
P138LEISHMANIOSE VISCERAL. 14 ANOS DE EXPERIENCIA
Ana Sarmento, Nilza Ferreira, Susana Sousa y José Arêlo Manso
Centro Hospitalar Vila Real, Portugal.
ALeishmaniase visceral infantil é tida como mais frequente entre o primeiro e terceiro ano de vida. Não tratada, é fatal em 75-95 % dos casos. Em Portugal encontra-se difundida esporadicamente de Norte a Sul, com especial destaque para o foco endémico delimitado à região do Douro. A maioria dos casos tem início insidioso, sendo comum, à data do diagnóstico a tríada clássica: febre, palidez e esplenomegalia. O diagnóstico definitivo é feito pela identificação do parasita. Os sais de antimónio foram a terapêutica de eleição e as diaminas aromáticas e Anfotericina B, as drogas alternativas mais utilizadas.
Hoje a Anfotericina B lipossomica conjuga eficácia e maior tolerabilidade, mas a custo muito elevado. Em utilização recente está a miltefosine a primeira droga anti-leishmania por via oral.
Tendo por objectivo conhecer e divulgar a nossa realidade, bem como a evolução epidemiológica, ao longo dos últimos 14 anos procedeu-se ao estudo retrospectivo dos casos tratados e orientados no Serviço de Pediatria do Hospital de S. Pedro. Analisaram-se os processos de 101crianças internadas com o diagnóstico de Leishmaniase Visceral no período de Abril de 1988 a Dezembro de 2002.
Os resultados apontam para uma clara diminuição do número de casos/ano, maior incidência no sexo feminino e no primeiro ano de vida, bem como uma resposta favorável à N-metil-glucamina. Esta foi utilizada durante 14 dias, perfazendo uma dose diária de antimónio ligeiramente superior à estipulada pela OMS, com reduzido número de recidivas e/ou complicações.
A evolução recente da doença, na maioria dos casos, e o início precoce da terapêutica pensamos ter condicionado favoravelmente os resultados obtidos.
P139MASTOIDITIS AGUDA. REVISION DE CASOS ENTRE 1993-2002
Nagore García de Andoain Barandiarán, Itziar Sota Busselo, Guillermo Unanue Tejera, Francisco Javier Mintegui Aramburu, Esperanza Pérez Ruiz y Ángeles M. Ruiz Benito
Hospital Donostia, San Sebastián.
Objetivos:Analizar la frecuencia y características clínicas de los niños con Mastoiditis aguda ingresados en el Hospital Donostia en los últimos 10 años.
Métodos:Estudio retrospectivo de los casos de mastoiditis entre 1993 y 2002.
Resultados:Se han estudiado 36 casos. 7 (19,4 %) fueron en invierno, 6 (16,7 %) en primavera, 10 (27,7 %) en verano y 13 (36,1 %) en otoño. El n.º de casos por año fue respectivamente desde 1993 hasta 2002: 0, 1, 1, 2, 10, 2, 8, 5, 2 y 5.
Edad: 20 casos (55,5 %) en menores de 3 años, 8 (22,2 %) entre 3 y 10 años, y 8 (22,2 %) entre 10 y 14 años. Niños 21 (58,3 %) y niñas 15 (41,6 %).
Mastoiditis de lado derecho 23 (63,9 %) e izquierdo 13 (36,1 %). Sintomatología: otitis 31 pacientes (86,1 %), supuración ótica 19 (52,7 %), fiebre 26 (72,2 %) y otitis más fiebre 24 (66,7 %). En 1 caso el único antecedente fue un traumatismo.
Analítica al ingreso: valor medio de PCR: 35,2 y mediana: 76 mg/l; cifra media de leucocitos y neutrófilos: 14.012 y 8.181/μl, y la mediana: 14.200 y 9.665/μl.
Se realizó Rx simple de mastoides en 10 pacientes, 4 mostraron signos de mastoiditis. En 16 se practicó TC, 11 en el momento agudo apreciándose signos de mastoiditis y 5 después del alta siendo normales. RM en 1 caso descubriendo trombosis venosa. El hemocultivo fue negativo en todos. Únicamente se aisló germen en cultivo de exudado ótico en 15 casos (41,6 %). Los gérmenes más frecuentes fueron:P. aeruginosa en 7 casos (46,7 %);S. viridans en 3 (20 %);S. aureus en 2 (13,3 %). 19 pacientes (52,7 %) recibían tratamiento antibiótico previo. Al ingreso se instauró tratamiento con cefalosporina iv. Al alta se continuó tratamiento con b -Lactámico vía oral. Recibieron tratamiento durante una media de 16,6 días. Días ingreso: media 8,4 días. Complicaciones en 5 casos (13,9 %): 2 recidivaron en menos de 2 meses, 1 absceso mastoideo que requirió drenaje quirúrgico, 1 celulitis preauricular y 1 trombosis de seno transverso y sigmoideo acompañado de absceso retroauricular.
Conclusiones: 1.En la última década hay un aumento en la incidencia de las mastoiditis aguda, generalmente como complicación de una otitis que solo en la mitad de los casos recibían tratamiento previo.2. El grupo de edad más afectado son los menores de 3 años, no debiéndose despreciar los casos secundarios a una otitis por P. Aeruginosaen niños mayores durante los meses de verano.
INMUNOLOGIA Y ALERGIA
P140ANALISIS DE PACIENTES MONOSENSIBILIZADOS EN LA PROVINCIA DE GRANADA
Carina Llopis Baño, Ana M. Martínez-Cañavate Burgos, Francisco Girón, Concepción de Linares, Ana Rojo, Cristina García y Luis Padilla
Universidad de Granada, Granada; Hospital Virgen de las Nieves, Granada y Hospital Clínico Universitario San Cecilio, Granada.
Objetivo:Analizar las características de los pacientes monosensibilizados frente a aeroalergenos de nuestra zona de influencia, y su relación con el análisis de los niveles polínicos y de esporas en nuestra provincia.
Material y métodos:Se han revisado 825 historias clínicas (entre 1999-2000) de pacientes diagnósticados de asma extrínseco, rinoconjuntivitis, urticaria y/o dermatitis atópica. Se ha realizado ha clínica completa, test cutáneos (Prick) e IgE CAP/RAST a aeroalergenos (olivo, gramíneas, ciprés, artemisia, parietaria, chenopodium, alternaria, cladosporium). Se analizaron los niveles de polen y esporas registrados, utilizando captador volumétrico tipo Hirst "Burkard Spore-trap modelo 7-day recorder".
Resultados:

Conclusión:El mayor porcentaje de monosensibilizados lo encontramos entre los 5-10 años, siendo menos frecuente en los de más edad.
El olivo es el alergeno más frecuente en nuestra zona, con una marcada correlación con lo detectado en la atmósfera.
La alternaria es un importante aeroalergeno, encontrándose una alta monosensibilización sobre todo en pacientes asmáticos procedentes del cinturón.
P141INMUNODEFICIENCIA COMUN VARIABLE EN NIÑOS
M. del Carmen Zárate, Jazmín El-Hakeh, José M. Beltrán, Isabel Caragol, Manuel Hernández y Teresa Español Boren
Hospital Vall D'Hebrón, Barcelona.
Antecedentes:La inmunodeficiencia común variable (IDCV) es un síndrome heterogéneo, de etiología desconocida, caracterizado por hipogammaglobulinemia de 2 o más isotipos de inmunoglobulinas e infecciones respiratorias de repetición. La diversidad de las manifestaciones puede ser tan amplia como gastrointestinales, autoinmunes, otras infecciones, alergia, y riesgo de cáncer, esto puede dar lugar a un porcentaje de error al establecer su diagnóstico, solapando otros síndromes de etiología conocida. Su tratamiento consiste en el reemplazo con gammaglobulina de por vida.
Material y métodos:Se estudiaron 16 pacientes entre edades 2-18 años, que acuden al hospital de día de inmunología para tratamiento con gammaglobulina endovenosa. Se revisó el expediente clínicopara determinar las manifestaciones clínicas más frecuentes, y por medio del método de Western Blot, descartamos otros síndromes de etiología conocida, como la agammaglobulinemia ligada a x(XLA), síndrome linfoproliferativo ligado a x (XLP), y el déficit de Lck.
Resultados:Fueron 13 pacientes del sexo masculino y 3 del sexo femenino. Las manifestaciones respiratorias fueron las más frecuentes en un 93,7 %. El 37,5 % tenían bronquiectasias al diagnóstico. La aparición de los primeros síntomas, fue antes de los 10 años de edad y un 56,25 % de los pacientes presentó clínica antes de los 2 años. El intervalo de tiempo entre el inicio de síntomas y el diagnóstico vario entre 1 mes y 10 años (3,2 años de media). Un paciente presento datos de insuficiencia respiratoria al diagnóstico. Todos los pacientes expresaron las tres proteínas estudiadas.
Conclusiones:Aunque no se haya descrito ningún defecto de la expresión de Btk, Lck y SAP, en los 16 pacientes estudiados, consideramos que es un método de screening adecuado y rápido, como diagnóstico diferencial de la IDCV.
P142SINDROME HIPER IgM Y ASOCIACION CHARGE
M. Pilar Bahillo Curieses, M. Teresa Cantero Tejedor, Pilar Solís Sánchez, M. Paz Aragón García, Samuel Gómez García, Rosario Cambronero Martínez y Fco. Javier Ballesteros Gómez
Hospital Universitario de Valladolid, y Hospital Universitario La Paz, Madrid.
Introducción:La asociación Charge y el síndrome hiper IgM siempre han sido consideradas dos entidades no relacionadas. La asociación Charge es un conjunto de anomalías congénitas cuyo diagnóstico debe ser considerado en todo recién nacido con coloboma, atresia de coanas, parálisis facial asimétrica o anomalías del oído/sordera, en combinación con otras anomalías. El síndrome hiper IgM es una inmunodeficiencia primaria rara, caracterizada por infecciones piogénicas de repetición.
Caso clínico:Niña de 3 años de edad sin antecedentes familiares de interés ni consanguinidad. En el periodo neonatal presenta retraso de crecimiento intrauterino, arteria umbilical única y coloboma de iris. Durante el primer año de vida es diagnosticada de asociación Charge, en base a la existencia de coloboma de iris, disfunción de pares craneales (VIII par, discinesia de Marcus Gunn), facies peculiar, hipocrecimiento pónderoestatural y sordera neurosensorial. A los 8 meses de edad comienza con infecciones óticas de repetición. A los 3 años de edad una de estas infecciones se complica con neumonía y septicemia por Streptococo pneumoniae y Corinebacterium sp. El estudio inmunológico realizado muestra IgG < 4,6 mg/dl, IgA < 4,6 mg/dl e IgM 570 mg/dl, con normalidad del resto de exploraciones inmunológicas. Se diagnostica de síndrome de hiper IgM y se inicia tratamiento sustitutivo con inmunoglobulina intravenosa, con buena evolución.
Conclusiones:Presentamos un síndrome de hiper Ig M no ligado a xen una niña diagnosticada de asociación Charge no clásica. No encontramos explicación a la coincidencia de estas dos entidades en la misma paciente.
P143 HIPOGAMMAGLOBULINEMIA TRANSITORIA DE LA INFANCIA
Amalia Pérez Payá, Beatriz Guillot Roselló, Antonio Pereda Pérez y Juan Ferrer Calvete
Hospital Universitario La Fe, Valencia.
Antecedentes y objetivo:El hallazgo de cifras bajas de inmunoglobulinas en los niños de nuestra consulta es un hecho que se observa ocasionalmente. Nuestro objetivo ha sido estudiar si estos niños presentaban inmunodeficiencia y cual ha sido su evolución.
Métodos:Estudio retrospectivo. Revisión de historias clínicas de pacientes de la consulta de Gastroenterología desde Febrero de 1997 hasta Diciembre de 2002 que presentaron cifras de una o varias clases de Igs por debajo del intervalo de referencia para la edad.
Resultados:Encontramos 26 casos (13 niños y 13 niñas). Los diagnósticos finales fueron hipogammaglobulinemia transitoria de la infancia en 23 casos, deficiencia de IgA en 2 casos e inmmunodeficiencia común variable en un caso. La media de edad al diagnóstico fue de 21,11 meses (rango 6-72 meses).
Los motivos de estudio fueron intolerancia a las proteinas de leche de vaca en 12 niños, desmedro en 6 niños, gastroenteritis de repetición en 3 niños, diarrea crónica en 2 niños y dolor abdominal crónico, hipertransaminasemia y broncoespasmo en un niño respectivamente. Los pacientes diagnosticados de inmununodeficiencia transitoria presentaron al inicio de su estudio disminución de IgG y de IgA en 13 casos y de IgG en 10 casos. Con el fin de descartar otras inmunodeficiencias se efectuaron, segun los casos, poblaciones linfocitarias, pruebas de estimulación linfocitaria, estudio de anticuerpos específicos y subclases de IgG. En dos casos se observó deficiencia aislada de IgG4 sin clínica especial.
El tiempo medio hasta la normalización de la IgG fue de 37,5 meses (23,8 DE). Durante el periodo de seguimiento no hubieron procesos infecciosos relevantes.
Conclusiones:La hipogammaglobulinemia en la primera infancia es un hallazgo relativamente frecuente. En su diagnostico hemos de descartar otras inmunodeficiencias graves mediante la clínica y estudios de laboratorio.
En nuestros pacientes encontramos un porcentaje bajo de deficit de IgA aislado debido a que los niños con este deficit estaban previamente diagnosticados y no entraron en el grupo de estudio.
La hipogammaglobulinemia transitoria se ha comportado como un proceso benigno, tal como se refiere en la literatura.
P144 BUENA RESPUESTA TRAS LA ADMINISTRACION DE ALTAS DOSIS DE GAMMAGLOBULINA INTRAVENOSA EN NECROLISIS EPIDÉRMICA TOXICA (NET) POR LAMOTRIGINA. ESTUDIO INMUNOLOGICO
José Manuel Jiménez, José Luis Corzo Higueras, Olga M. Escobosa Sánchez, Francisco Jesús García, Cristina Antúnez, José Antonio Cornejo-García, M. José Torres, Cristobalina Mayorga, Miguel Blanca y Antonio Jurado Ortiz
Hospital General Carlos Haya, Málaga.
Caso clínico:Niño de tres años que desarrolla un cuadro de NET (60 % del área corporal) tras lamotrigina ingresa en el Departamento de Pediatría. Se suspende la lamotrigina, comienza tratamiento con dexametasona que se suspende en 48 horas sin mejoría. Se administra inmunoglobulina IV (IGIV) (2 g/kg) durante 24 horas con gran mejoría.
Métodos:Se analizaron por RT-PCR y citometría de flujo muestras seriadas de sangre y líquido de ampolla, tras consentimiento paterno.
Resultados:Inicialmente en el fluido de las ampollas estaban invertidos los niveles de CD4/CD8 y la predominancia de los niveles de células T memoria y activadas. Estas células expresan CLA con mayor porcentaje que las células sanguíneas. En el séptimo día, hay un incremento de las células T memoria y las células T activadas, con un alto porcentaje expresando CLA en sangre periférica. En el día quince todos los marcadores comienzan a retornar a sus valores normales. Asimismo en los primeros días el patrón de citoquinas es Th 1 que tiende a retornar a Th0 con la mejoría del paciente. La expresión de la perforina y el Fas-lingand se observó solo antes de IVIG.
Conclusión:En nuestro caso se demuestra la utilidad de altas dosis de IGIV en el tratamiento de la NET. Esto se observa tanto en la evolución clínica como inmunológica. Encontramos una negativización en la expresión de marcadores apoptóticos inmediatamente después de la administración de la IGIV.
P145DIFERENCIAS INMUNOLOGICAS ENTRE REACCIONES ALÉRGICAS A FARMACOS E INFECCIONES VIRICAS EN PEDIATRIA
Olga M. Escobosa Sánchez, M. Esther Rojas Gracia, M. José Torres, Cristina Antúnez, José Antonio Cornejo-García, Antonio Jurado Ortiz, Cristobalina Mayorga, Miguel Blanca, Sebastián Ramos González y José Luis Corzo Higueras
Hospital General Carlos Haya, Málaga y Centro de Salud Ciudad Jardín, Málaga.
Objetivos:Establecer diferencias entre reacciones cutáneas inducidas por fármacos (Grupo 1) o virus (Grupo 2) en los mecanismos inmunológicos mediante el estudio de marcadores linfocitarios y patrón de citoquinas en niños.
Material y métodos:Determinación por citometría de flujo de marcadores linfocitarios (CD3, CD4, CD8, CLA, CD25, CD69, CD45RO Y CD45RA) y por RT-PCR citoquinas (IL 2, IL 4, IFN-γ, TNF-α, IL 10, Perforina, Granzima B y Fas- ligando. Niños sin enfermedad cutánea se usaron como controles (Grupo 3).
Resultados:El grupo 1 (n = 8) se diagnóstico por la historia, negatividad del estudio viral y mejora al suspender el fármaco y el Grupo 2 (n = 10) por serología positiva viral. Se encontraron diferencias estadísticamente significativas entre los grupos en la expresión del receptor de "homing" cutáneo CLA (p < 0,001), el marcador de activación temprana CD69 (p < 0,001), y las subpoblaciones memoria (CD3 +, CD45RO +) (p < 0,02), y virgen (CD3 + CD45RA +) (p < 0,03) usando el test de Kruskall-Wallis. En el Grupo 1 se expresan niveles más elevados de CLA, CD69 y células memoria que en el Grupo 2 y un patrón de citoquinas Th1 frente a un patrón Th0 del Grupo 2.
Conclusiones:Los niños con reacciones cutáneas no inmediatas a fármacos, tienen una respuesta de células T más potente que los niños con infecciones virales con afectación cutánea. Asimismo los primeros expresan un patrón Th1 frente a los segundos con un patrón Th0. Todo esto puede ayudar a discriminar rápidamente entre reacciones cutáneas inducidas por virus o por fármacos.
P146EVOLUCION A LARGO PLAZO DE NIVELES Y ESPECIFICIDAD DE ANTICUERPOS IgE E IgG EN NIÑOS CON REACCIONES ALÉRGICAS INMEDIATAS A TOXOIDE TETANICO
M. Esther Rojas Gracia, Francisco Canca Doblas, Antonio Jurado Ortiz, José Luis Corzo Higueras, Olga M. Escobosa Sánchez, Miguel Blanca, José Antonio Cornejo-García, Cristina Antúnez, Cristobalina Mayorga y M. José Torres
Hospital General Carlos Haya, Málaga y Centro de Salud Torremolinos.
Objetivos:Determinar los niveles de anticuerpos IgE e IgG específicos y valorar su especificidad frente a los componentes de la vacuna en cinco niños con reacción alérgica inmediata a toxoide tetánico (TT).
Métodos:Pruebas cutáneas con: TT, timerosal y fosfato de aluminio. Los anticuerpos IgE e IgG específicos a TT se determinaron mediante RAST y ELISA respectivamente. Las proteínas antigénicas de la vacuna se estudiaron mediante SDS-PAGE y las especificidades de los anticuerpos por inhibición del RAST e immunoblotting. Se realizaron en sueros obtenidos en los meses siguientes a la reacción y cinco años después.
Resultados:Todos los pacientes tuvieron pruebas cutáneas positivas a TT y negativas al timerosal y fosfato de aluminio. Las pruebas cutáneas fueron negativas en los dos grupos controles: sujetos inmunizados sin reacción e inmunizados con reacción local. Cinco años tras la reacción hubo una disminución en los niveles de IgG específica. En el inmunoblotting observamos que los anticuerpos IgG reconocían dos diferentes proteínas del TT, de 220 kDa y 60 kDa. Este patrón de reconocimiento fue observado en el suero de todos los pacientes estudiados. A los cinco años de la realización del primer estudio los niveles de anticuerpos IgE específicos mostraron una disminución de casi un 50 % para cada suero individual. Esta disminución fue también detectada mediante immunobloting y el patrón de bandas reconocidas fue idéntica a la de los anticuerpos IgG.
Conclusión:Aunque no es un hecho frecuente, la vacuna del tétanos, induce anticuerpos IgE específicos que se asocian a la aparición de una reacción alérgica. Dichos anticuerpos pueden permanecer durante un largo periodo tiempo lo que debe de ser tenido en cuenta para posteriores inmunizaciones.
P147 ESTUDIO DE LAS INFLUENCIAS DE LAS VARIANTES GENOTIPICAS DEL RECEPTOR β2-ADRENÉRGICO EN LA RESPUESTA AL TRATAMIENTO DEL ASMA
Ignacio Díez López, Juan José Telleria Orriols, Sara Muntion Olave, Alicia Armentia Medina y Alfredo Blanco Quirós
Hospital Universitario de Valladolid, Hospital Universitario del Río Hortega, Valladolid y Facultad de Medicina. Universidad de Valladolid.
El asma es un desorden fenotípicamente heterogéneo, afectando en nuestra región al 8 % de la población. Se involucran factores genéticos y ambientales, presentando un rasgo de herencia complejo. El receptor b2-adrenérgico es una proteína transmembrana con actividad de estimulación intracelular, codificada en el cromosoma 5q31-q32. Se han descrito 9 polimorfismos, siendo los más frecuentes en los aa16 y 27. Trabajos previos han demostrado que la frecuencia de aparición de estos en la población asmática y no asmática es similar, pero bien pudieran influir en la expresión clínica del asma y su respuesta al tratamiento.Presentamos el estudio de las variantes polimórficas del receptor β2-adrenérgico (variantes alélicas aa16 y aa27) y su respuesta al tratamiento (aparición o no de taquifilaxia) en una población de pacientes diagnosticados de asma en la edad pediátrica frente a un grupo control de la población general. Se recogen otros datos referentes a edad, sexo, tipo de fármaco utilizado y respuesta terapeútica. Se analizan 80 pacientes asmáticos y 64 casos de población general, con los siguientes resultados:

No existen diferencia estadística (p > 0,05) entre los hallazgos de las variantes polimórficas respecto al grupo asma y el no asma. Es significativa (p < 0,05) las diferencias aparecidas en las variantes polimórficas aa16 y aa27 dentro del grupo asma entre aquellos individuos que llegan a desarrollar taquifilaxia y los que no. Un solo individuo con la variante Glu/Glu en el aa27 presentó taquifilaxia (p = 0,03).
Conclusiones:En nuestro medio la distribución de las variantes alélicas del receptor β2-adrenérgico es semejante entre la población asmática y la general, no siendo un factor predictivo de padecimiento o no de asma. Existe un posible papel protector en el desarrollo de taquifilaxia en aquellos individuos afectos de asma: la presencia de la variante del aa27Glu/Glu. Este hecho pudiera facilitar el desarrollo de futuras estrategias terapéuticas en estos pacientes.
P148 PERITONITE BACTERIANA PRIMARIA E IMUNODEFICIENCIA
Filipa Vasconcelos Espada, Juan Calviño Cabezas, António Trindade, Fernando Próspero, Isabel Soares, Julia Vasconcelos, Natalina Miguel y Fátima Dias
Centro Hospitalar Vila Real, Portugal y Hospital Geral de Santo António, Porto (Portugal).
Introdução:A Peritonite bacteriana primária é uma entidade pouco comum em idade pediátrica, existindo poucos casos descritos na literatura. É uma infecção bacteriana da cavidade peritoneal sem foco intra-abdominal evidente, constituindo uma urgência que necessita de hospitalização e pronta instituição terapêutica. O agente etiológico mais comum é o Streptococcus pneumonaie e a ascite secundária a síndrome nefrótico ou a cirrose predispõem ao desenvolvimento de peritonite bacteriana primária.
Caso clínico:Descreve-se o caso clínico de uma criança do sexo masculino com 9 anos, sem antecedentes patológicos relevantes que recorreu ao Serviço de Urgência do Centro Hospitalar Vila Real - Peso da Régua por dor abdominal, febre e vómitos com 6 h de evolução. À admissão apresentava-se hemodinamicamente estável, com dissociação térmica (temperatura axilar-37 °C, temperatura rectal-39,2 °C), defesa e contractura abdominal generalizada e ausência de ruídos intestinais audíveis. Do estudo efectuado há a revelar: radiografia abdominal com níveis hidroaéreos, leucocitose com neutrofilia [(Leucócitos 21300/ml (N-90 %)] e PCR elevada [26 mg/dl (N < 0,49 mg/dl)]. Foi submetido a laparotomia exploradora que demonstrou a existência de pus na cavidade peritoneal, sem evidência de infecção ou perfuração de víscera e realizada lavagem da cavidade abdominal. Face ao diagnóstico de Peritonite bacteriana primária instituíu-se cefotaxime e netilmicina durante 14 dias. No exudato purulento peritoneal foi isolada uma estirpe deStreptococcus pneumoniae. Teve alta sem sinais clínicos ou analíticos de infecção. A investigação exaustiva realizada em ambulatório, na procura de uma causa, revelou a existência de um défice de C2.
Comentários:A deficiência de componentes de complemento da via clássica está associada ao risco aumentado de infecções. O défice de C2 é a deficiência de complemento hereditária mais comum. Embora possa ser detectada em indivíduos assintomáticos, está associada ao desenvolvimento de doenças auto-imunes ou a infecções piogénicas recorrentes, principalmente, devidas a bactérias encapsuladas como oStreptococcus pneumoniae.
Neste caso, esta imunodeficiência facilitou a disseminação por via hematogénea da bactéria, com atingimento peritonial.
P149ASMA BRONQUICA. QUAIS OS FACTORES PRECIPITANTES DE AGUDIZAÇÃO?
Sandra Manuela Silva Mesquita, Eva Gomes e Inês Lopes
Hospital de Crianças Maria Pia, Porto (Portugal).
Introdução:A asma caracteriza-se por uma hiperreactividade brônquica que predispõe o doente a desenvolver episódios repetidos de dificuldade respiratória, dispneia e tosse. Estes episódios associam-se muitas vezes a factores precipitantes conhecidos como infecções respiratórias, exercício e exposição a irritantes e alergenos.
Objectivo:Definir os factores precipitantes/agravantes das crises espontaneamente referidos por dois grupos de asmáticos com idades até aos 5 e superiores a 5 anos.
Material e métodos:Estudo retrospectivo com vertente exploratória abrangendo 187 crianças asmáticas observadas na Consulta de Imunoalergologia entre 7 e 14 de Fevereiro de 2003. Variáveis estudadas: sexo, idade, patologias associadas, factores precipitantes/agravantes de crise apontados e tipo de sensibilização encontrada. Tratamento de dados feito no programa de SPSS.
Resultados:Idades entre 1 e 18 anos (média 7,9), 80 (42,8 %) do sexo feminino. Noventa e seis por cento dos doentes (96,3 %) identificaram pelo menos um factor precipitante das crises, sendo o exercício (60,6 %) e as intercorrências infecciosas (52,8 %) os mais referidos. Outros factores apontados incluíram: alterações climatéricas, expressão de emoções e exposição a poluentes. Os testes cutâneos para alergenos inalantes comuns (n = 177) foram positivos em 73,8 % dos casos, a sensibilização a ácaros do pó doméstico foi a mais frequente (67,9 %).
Por comparação entre os dois grupos etários: ≤ 5 anos (n = 64)versus > 5 anos (n = 123) verificamos que as intercorrências infecciosas, como factor precipitante, predominam no 1.º grupo (59,4 %) e o exercício físico no 2.º grupo (67,2 %). A diferença entre os dois grupos é estatisticamente significativa em relação ao exercício (p < 0,05). A prevalência de sensibilização é maior no 2.º grupo (50 %/86,2 %) (p < 0,001). Tinham rinite associada 49,2 % das crianças sendo maior no grupo com mais de 5 anos (p < 0,001).
Conclusões:Neste estudo o factor precipitante mais vezes apontado foi o exercício físico sobretudo nas crianças mais velhas. As infecções respiratórias foram associadas ao aparecimento de agudizações numa percentagem elevada de casos. A prevalência de sensibilização aumenta com a idade assim como associação entre asma e rinite.
NEONATOLOGIA
P150VOLVULO GASTRICO INTRATORACICO IMITANDO ESTENOSIS DE PILORO
Jorge Muñoz, Tim Malpas y Anthony Mansul
Hospital General de Jersey, Reino Unido.
Un varon de cinco semanas de edad se presento con historia de vomito violento no bilioso durante 24 horas. El infante habia estado previamente bien sin otros sintomas asociados aparte de una tos seca de 48 horas de duracion. Dicho bebe habia nacido espontaneamente a las 37 semanas de gestacion con un peso al nacer de 2.800 g. Ecografias antenatales a 16 y 36 semanas de gestacion no mostraron patologia alguna y la examinacion al nacer se valoro normal.
Durante la examinacion clinica el paciente estaba vigoroso aunque moderadamente desidratado, sin sintomas respiratorios. Durante la ingesta de leche no se palpo tumor pilorico alguno. La gasometria venosa mostro un pH de 7.403, pCO2 de 6,64 kPa, pO2 de 4,78 kPa, bicarbonato de 30,4 mmol/l y un exceso de base de 4,5 mmol/l. La bioquimica fue normal y se instauro dextrosalino intravenoso. Se realizo una ecografia abdominal siendo el resultado negativo para piloro hypertrofico. A la manana siguiente el pequeño continuaba con vomitos y la tos empeoro sin ningun otro sintoma respiratorio. Se realizo un aspirado nasofaringeo siendo positivo para Parainfluenza tipo 3. La radiografia de pecho mostro hernia diafragmatica izquierda con desvio del mediastino a la derecha. Papilla de Bario fue consistente con volvulo gastrico intratoracico. El bebe fue transferido a una unidad quirurgica donde se reparo el defecto congenito.
La presentacion de hernia diafragmatica congenita con vomito debido a volvulus gastrico intratoracico es extremadamente raro. Nuestro caso demuestra que un volvulo gastrico puede imitar estenosis de piloro, ya que ambas condiciones comparten caracteristicas comunes de presentacion clinica.
Por ultimo, la ausencia de patologia mediante ecografia abdominal demuestra que diagnosticos alternativos deberian ser activamente excluidos.
La conclusión a la que llegamos fue que el volvulo gastrico deberia formar parte del diagnostico diferencial de bebes que se presentan con vomito no bilioso y estudios radiologicos apropiados deberian llevarse a cabo.
P151EFICACIA Y LIMITES DE LA VENTILACION SINCRONIZADA EN EL NEONATO CON ENFERMEDAD DE MEMBRANA HIALINA
Giacomo Cavallaro, Italo Bernardo, Pietro Di Mónaco, Pascuale Laudiero, Vincenzo Della Monica, Antonella Ansalone, Elena Bullejos García, Annunziata de Lucia, Pietro Falco y Luigi Falco
Hospedale San Sebastiano, Caserta (Italia) y Hospital General Yagüe, Burgos.
Objetivos:Los autores describen el empleo de la ventilación mecánica "Triggered" (PTV), en su UCI Neonatal en los últimos cuatro años y comparan este método con la ventilación convencional (IPPV).
Métodos:Se han distribuido al azar, tras la obtención del consentimiento informado de los padres, todos los RN con Enfermedad de Membrana Hialina (EMH), ventilados con modalidad PTV y IPPV desde Enero 1999 hasta Diciembre 2000, y con peso al nacer entre 1000 y 2000 gr., que han necesitado ventilación mecánica. Fueron excluidos los RN con malformaciones, cardiopatías congénitas y síndrome de aspiración de meconio. Se incluyen 95 pacientes en el estudio; se excluyen 8 por necesitar cambio a Alta Frecuencia Oxilatoria (HFOV) y 6 porque no consiguien activar el trigger de manera eficaz. Por tanto, se incluyen en el estudio 81 pacientes; 44 en el grupo IPPV y 37 en el grupo PTV. En los dos grupos, los protocolos preveían estos parámetros del ventilador:1. Ti: 0,3-0,4 seg.2. PEEP: 4 cmH2O.3. PIP y FiO2 en función de la gasometría arterial. El Surfactante exógeno (Curosurf) se administró a todos los RN con EMH y todos los pacientes recibieron mínimo una dosis a 200 mg/kg.
Resultados:Se demostró una menor duración de los días de ventilación y el empleo de PIP y MAP inferiores en el grupo tratado en PTV. Las secuelas de exitus, broncodisplasia, hemorrágia intraventricular, leucomalacia periventricular y neumotórax no resultó significadamente diferente, aunque la incidencia de neumotórax ha sido inferior en el grupo PTV.
Conclusiones:El estudio demuestra la seguridad en el empleo de la ventilación "Triggered" en RN con EMH sin que haya un aumento de las complicaciones.

P152SINDROME DE HIPOVENTILACION CENTRAL CONGÉNITA EN NEONATO A TÉRMINO
Giacomo Cavallaro, Italo Bernardo, Gaetano Ausanio, Antonella Ansalone, Vincenzo Della Monica, Alessandro Piccirillo, Elena Bullejos García, Silvia Affinito, Pietro Falco y Luigi Falco
Hospedale San Sebastiano, Caserta (Italia) y Hospital General Yagüe, Burgos.
Objetivos:El síndrome de hipoventilación central congénita o maldición de Ondina, es una patología rara caracterizada por depresión respiratoria durante el sueño, con respiración normal en vigilia.
Métodos:RN a término, parto por cesárea de elección. Sin antecedentes familiares de interés. Apgar 4/7. A la exploración física destaca: peso 2700 gr, ausencia de malformaciones, distrés respiratorio, con hipopnea, bradipnea, hipoxia y hipercapnia, bradicardia extrema que ha necesitado el empleo de adrenalina i.v. Estas manifestaciones se presentaban sobretodo durante el sueño. Esas crisis aumentaron en número e intensidad en los primeros dos días de vida hasta necesitar intubación endotraqueal y ventilación mecánica. Las pruebas complementarias: hemogramas, hemocultivos, PCR, TORCH, Rx tórax y abdomen, Ecoabdominal, Eco, TAC y RMN cerebral, EMG y VCN han resultado normales. Ecocardio: foramen oval permeable, pequeña CIV. Polisomnografía con reducción del volumen tidal durante el sueño con progresivo aumento de la pCO2 y apneas centrales con hipoxia e hipercapnia. Tras el fallo de la terapia farmacológica con cafeína y posteriormente con teofilína, se ha recurrido a traqueotomía y ventilación asistida nocturna.
Conclusiones:La fisiopatología de este síndrome es desconocida, aunque hay numerosas hipótesis:1. Defecto de quimio-receptores centrales localizados en el área ventrolateral del bulbo. Esa hipótesis se basa en la evidencia de una mayor hipoventilación durante el sueño no-REM, y normalización en fase REM. 2. Alteraciones de ambos receptores centrales y periféricos.3. Defecto de la integración central de los estímulos de los receptores que resultan normales. A veces ese síndrome se asocia al Síndrome de Hirschsprung, el ganglioneuroma o el neuroblastoma, y por esas asociaciones se ha relacionado con un defecto común de las células de la cresta neural. El diagnóstico es sobretodo clínico basado en la observación de las crisis durante el sueño, ausencia de patología cardíaca, pulmonar, torácica y neuromuscular, resolución con la ventilación asistida, y avalada por presencia de polisomnografía patológica. El tratamiento se basa en el empleo de las xantinas como estimulante central, aunque con escaso éxito, y en los niños pequeños es necesario recurrir a la traqueotomía y ventilación asistida nocturna. En el niño mayor es útil la ventilación a presión positiva intermitente nasal durante el sueño. Controvertido es el empleo de marcapasos diafragmáticos.
P153HIPOSPADIAS: ¿ES NECESARIO SCREENING ECOGRAFICO RENAL?
Patricia Martín Cendón, Carlos Flores González, Simón Pedro Lubián López, M. Ester Guerrero Vega, Estefanía Romero Castillo, Enrique Robles Caballos y Juan Mena Romero
Hospital Puerta del Mar, Cádiz.
Antecedentes y objetivo:El hipospadias se define como la desembocadura anómala del meato uretral en la parte ventral del tallo del pene. Es relativamente frecuente con una incidencia de 5-9/1000 rn varones vivos. En la bibliografía se recomienda la realización sistemática de ecografía de vías urinarias con objeto de descartar malformaciones asociadas. Nuestro objetivo es determinar la incidencia de anomalías del tracto urinario diagnosticables ecográficamente en niños con hipospadias.
Métodos:Estudio retrospectivo de los casos de hipospadias nacidos en nuestro hospital en los últimos cuatro años. Siguiendo el protocolo de nuestra Unidad en todos los casos se realizó estudio ecográfico de vías urinarias.
Resultados:Presentaron hipospadias 73 casos de los 9.976 rn nacidos en el periodo 1998-2002 (7,32/1.000 rn). De los 73 casos, en 54 (80 %) se trataba de hipospadias distales. En un sólo caso se detectó ecográficamente una anomalía renal (riñón único), no observandose en ningún caso anomalía alguna de las vías urinarias.
Conclusión:1. La incidencia de hipospadias en nuestro medio es similar a la recogida en la bibliografía (7,32/1.000 rn vivos).2. El 80 % de los casos de hipospadias eran distales. 3. No se detectó en ningún caso malformación de las vías urinarias por lo que creemos que no es necesario el screening ecográfico sistemático en los casos de hipospadias distal.
P154 SEPSIS NEONATAL PRECOZ POR ESCHERICHIA COLI EN LA ERA DE LA PROFILAXIS ANTIBIOTICA INTRAPARTO DE STREPTOCOCCUSDEL GRUPO B
Ana Alarcón Allén, Félix Omeñaca Teres, Pilar Peña y Jesús Pérez
Hospital Universitario La Paz, Madrid.
Antecedentes:En 1996 fueron publicadas las primeras recomendaciones consensuadas para la prevención de la transmisión vertical deStreptococcus del grupo B (SGB). Con su aplicación, la incidencia de la sepsis neonatal precoz por SGB ha caído en~ 70 %. El efecto del uso aumentado de antibióticos intraparto sobre patógenos diferentes a SGB que causan sepsis neonatal precoz y su resistencia a antibióticos es desconocido.
Objetivos:Conocer la tendencia de la incidencia y la resistencia a antibióticos de la sepsis neonatal precoz porE. coli en la era de la profilaxis antibiótica intraparto.
Métodos:Estudio retrospectivo de los casos de sepsis neonatal precoz porE. coli ocurridos en un hospital terciario universitario entre 1992-2001, definidos por el aislamiento deE. coli en sangre o LCR antes de los 7 días de vida.
Resultados:Durante el periodo de estudio, el número de nacidos vivos fue de 74713. La incidencia de sepsis neonatal precoz porE. coli fue de: 0,56 ‰ en 1992-95; 0,29 ‰ en 1996-98; 0,69 ‰ en 1999-2001 (p =0,416). La proporción de prematuros en las infecciones precoces porE. coli fue de: 25 % en 1992-95; 50 % en 1996-98; 61,1 % en 1999-2001 (p = 0,141). La tasa de resistencia a ampicilina fue de: 6,7 % en 1992-95; 80 % en 1996-98; 82,3 % en 1999-2001 (p =0,052). Entre los casos de sepsis porE. coli en RNT, la proporción de infecciones resistentes a ampicilina fue de: 7/12 en 1992-95; 1/2 en 1996-98; 4/7 en 1999-2001 (p =0,899). En los RNPT dicha proporción fue de: 0/3 en 1992-95; 3/3 en 1996-98; 10/11 en 1999-2001 (p =0,002).
Conclusiones:La profilaxis intraparto de la infección perinatal por SGB no ha supuesto un incremento en la incidencia de sepsis neonatal precoz porE. coli. La proporción de infecciones porE. coli resistentes a ampicilina se ha mantenido estable en los RNT, mientras que ha aumentado en los RNPT.
Especulamos que el aumento de la resistencia a antibióticos deE. coli en RNPT puede estar en relación con su tendencia a estar expuestos a antibióticos maternos de mayor espectro, debido a la rotura prematura de las membranas y/o la sospecha de infección intraamniótica, lo cual puede favorecer la selección de patógenos resistentes.
P155LINFANGIECTASIA PULMONAR CONGÉNITA
Ainhoa Sarasua Miranda, Tatiana Menéndez Bada, Irune Ruiz Díaz, Maite San Vicente Leza y Luis Paisan Grisolia
Hospital Donostia, San Sebastián.
Recién nacido mujer de 39 semanas de edad gestacional y 3.725g de peso producto de una gestación de curso normal. Madre sana de 29 años, tercigesta, secundípara. Parto espontaneo y eutócico. En periodo neonatal inmediato presenta importante cianosis, hipotonía, pulsos periféricos débiles e hipoventilación generalizada. Tras maniobras de reanimación ingresa en la unidad de cuidados intensivos neonatales. En la Rx de tórax se observa un infiltrado bilateral y con sospecha de neumonía se inicia ventilación mecánica convencional con apoyo inotrópico y tratamiento antibiótico iv. Ante la mala evolución del cuadro se administra surfactante intratraqueal, ventilación de alta frecuencia y óxido nítrico no consiguiéndose resultados positivos y falleciendo a las 13 horas de vida.
En el estudio necrópsico se observa macroscópicamente una superficie pleural de aspecto microbulloso. La superficie de corte muestra aspecto de empedrado, sin alteraciones del árbol bronquial ni vascular. En el examen microscópico existe una marcada distensión de los linfáticos septales y subpleurales, colapsando el parénquima pulmonar adyacente.
El diagnóstico final es de linfangiectasia pulmonar congénita.
Comentarios:La linfangiectasia pulmonar congénita es una enfermedad poco frecuente caracterizada por el aumento del número y dilatación de los vasos linfáticos pulmonares que origina síntomas de disnea y cianosis refractarios al tratamiento en el recién nacido. Presentamos un caso de linfangiectasia pulmonar congénita tipo III según la clasificación de Noonan, sin otras malformaciones asociadas. La clínica y los exámenes complementarios en esta enfermedad suelen ser inespecíficos haciendo preciso el examen histológico para la confirmación diagnóstica. El pronóstico es malo, un 75 % de los casos fallece en periodo neonatal.
P156
ENFERMEDAD DE RITTER EN RECIÉN NACIDO HIJO DE MADRE VIH POSITIVO
M. Ester Guerrero Vega, M. Teresa de Benito Guerra, Simón Pedro Lubián López, Patricia Martín Cendón, Estefanía Romero Castillo, Ángeles Cervantes Orube y Rafael Duarte Cendán
Hospital Puerta del Mar, Cádiz.
El síndrome de la piel escaldada de origen estafilocócico abarca un grupo de trastornos ampollosos de la piel causados por toxinas epidermolíticas producidas por Staph aureus. Afecta predominantemente a recién nacidos y niños menores de 5 años. La forma generalizada o enfermedad de Ritter consiste en la descamación de grandes superficies de epidermis, inicialmente en los pliegues y luego en la mayor parte de la superficie corporal. La mortalidad es reducida (3 %) pero la morbilidad aumenta cuando surgen complicaciones sistémicas.
Caso clínico:Recién nacida de 7 días de vida que es remitida por presentar en las últimas 48 horas un cuadro de dermatitis exfoliativa generalizada con empeoramiento progresivo del estado general.
Antecedentes familiares:Madre en tratamiento con triple terapia antirretroviral durante el embarazo. Hermana: labio leporino.
Exploración:Mal estado general. Temperatura 35,5 °C. Mala perfusión periférica. Irritabilidad. Eritrodermia generalizada con lesiones vesículoampollosas confluentes, de predominio en flexuras y periorificial. Signo de Nikolsky positivo. Labio leporino. Paladar hendido.
Exámenes complementarios:Hemograma séptico. Hipocalcemia, hiponatremia e hipoproteinemia. Hemocultivo: se aisla Staph aureus. Biopsia cutánea: lesión vesiculoampollosa intraepidémica a nivel de la capa granular subcorneal.
Diagnóstico diferencial y conclusiones: 1.La enfermedad de Ritter es una patología no descrita en recién nacidos hijos de madre VIH positivo. 2. Dado el antecedente materno de la toma de fármacos y la presencia de malformación de la línea media, se plantea el diagnóstico diferencial con la necrólisis epidérmica tóxica (síndrome de Lyell), a pesar de ser ésta extremadamente rara en la época neonatal.3. Ante la sospecha de necrólisis epidérmica tóxica debe suspenderse inmediatamente la administración del fármaco hasta la confirmación del diagnóstico.4. El diagnóstico definitivo lo da la biopsia cutánea. 5.Resaltamos la importancia de la adecuada analgesia en el manejo de esta patología en el período neonatal.
P157 IMPACTO DE LA POBLACION INMIGRANTE EN NUESTRA SECCION DE NEONATOLOGIA. ANALISIS DESCRIPTIVO Y COMPARATIVO EN LA ULTIMA DÉCADA
Juan José Quesada López, Ángel B. Brea Lamas, Juan José Agüera Arenas, Carlos Pérez Cánovas, Arancha Escribano Muñoz y Vicente Bosch Jiménez
Hospital Universitario Virgen de la Arrixaca, El Palmar.
Introducción:La alta tasa de inmigración en nuestro medio es un hecho evidente; afecta a todas las estructuras sociales e irremediablemente al sistema sanitario. El consecuente aumento de la natalidad y el intento de racionalizar los recursos, nos hace plantearnos una serie de incógnitas que motivan este estudio.
Objetivos:Descripción de los pacientes ingresados y coste sanitario originados por la población inmigrante en la Sección de Neonatología del H.U.V. Arrixaca de Murcia a lo largo del año 2002, así como la comparación de estos resultados con los correspondientes al año 1992, diez años antes del presente estudio.
Métodos:Se obtuvieron datos a través de historias clínicas de pacientes ingresados hijos de inmigrantes (HI) durante 1992 y 2002, considerando variables como: procedencia, control o no del embarazo, gestaciones previas, patología prenatal, PN, causa que motivó el ingreso y estancia hospitalaria. Se tuvieron en cuenta datos epidemiológicos del área de salud dependiente de este hospital de referencia, así como el coste sanitario estimado por día de estancia.
Resultados:En el año 2002 ingresaron 1224 pacientes de los que 115 fueron HI (9,4 %). Diez años antes, este porcentaje fue del 1,4 %. Este incremento en el número de ingresos no tiene su correspondencia en igual medida con el aumento de la población inmigrante (de un 1,8 % en el 1992 a un 4,86 % en el 2002). De los 115 HI un 32 % (38) precisó ingreso previo en UCI, frente al 16,7 % de población no inmigrante. Según su procedencia,destacamos un 52 % (60) de origen sudamericano y un 37 % (42), norteafricano. El porcentaje de embarazos no controlados ascendió a un 34 % (40), muy superior al de la población española. Igualmente reseñamos un 25 % (29) de gestaciones con factores de riesgo infeccioso perinatales. No hemos encontrado diferencias significativas respecto a la EG y PN. El n.º total de días de estancia en el año 2002 (1.612) fue casi 9 veces superior al del año 1992 (183) lo que supuso un gasto total de 515.220 euros frente a los 32.892 euros de 1992.
Conclusiones:A la vista de este estudio cabe destacar el progresivo incremento de la carga asistencial y gasto sanitario generado por la población inmigrante, con mayor tasa de natalidad y menor control gestacional. La posible mezcla racial y la carga poblacional de determinadas patologías puede agravar este problema en un futuro no muy lejano.
P158NEUMONITIS CRONICA DE LA INFANCIA EN UN SDE DE EEC
Juan José Agüera Arenas, Arancha Escribano Muñoz, Ángel B. Brea Lamas, Manuel Sánchez Solis de Querol, Juan José Quesada López, Carlos Pérez Cánovas y Vicente Bosch Jiménez
Hospital Universitario Virgen de la Arrixaca, El Palmar.
Introducción:La neumonitis crónica de la infancia (NCI) es una rara enfermedad diagnosticada en lactantes pequeños. Fue descrita por primera vez en 1995. El diagnóstico es histopatológico y se caracteriza por ensanchamiento difuso e hipercelularidad del septo alveolar, hiperplasia de neumocitos tipo II y acumulación de macrófagos intraalveolares. El sdr de EEC es un cuadro poco frecuente que incluye ectrodactilia, displasia ectodérmica y fisura labio-palatina. Presentamos un caso clínico de Sdr. de EEC diagnosticado con biopsia de (NCI).
Caso clínico:RN, segundo hijo de padres sanos, no consanguíneos. No antecedentes de malformaciones congénitas. Embarazo controlado sin incidencias. Parto a término, cesárea por no progresión. APGAR 6/8 precisando reanimación tipo II. PN: 2450. Talla 47. PC 33. Ingresa inicialmente por distrés respiratorio y síndrome polimalformativo, destacando en su exploración: ectrodactilia, alteraciones ectodémicas (ausencia de cejas y pestañas, pelo fino y escaso, coloración albina, blefarofimosis, orificios preauriculares...) y fisura palatina. Todo compatible con sde de EEC. A las pocas horas del nacimiento precisó ventilación mecánica con VAFO y NO, siendo diagnosticado de probable neumonía aspirativa de L.A. Posteriormente se diagnosticó en 2 ocasiones de neumonía bacteriana. En la evolución posterior las imágenes radiológicas fueron compatibles con neumonía intersticial. A los 2 1/2 meses seguía precisando V.M con altas prestaciones. Una vez descartada causas secundarias de neumopatía intersticial (VRS, CMV, clamidias...) con LBA no concluyente, se decide realizar biopsia pulmonar detectando hallazgos compatibles con (NCI). Se inició tratamiento con corticoides y cloroquina y tras 5 meses sigue precisando ventilación mecánica.
Discusión:1. La neumonía intersticial idiopática incluye un grupo infrecuente y heterogéneo de enfermedades entre las que se encuentra la (NCI). Para su diagnóstico es necesario realizar una biopsia pulmonar y su tratamiento, muy controvertido, incluye corticoides, inmunosupresores, antipalúdicos... con resultados variables y pronóstico incierto. 2.El EEC es un síndrome que puede incluir infecciones respiratorias recurrentes y crónicas, pudiendo ser éste el mecanismo fisiopatológico de la (NCI) según los autores que la describieron.3. No hemos encontrado en la literatura médica la asociación entre el síndrome EEC y la (NCI).
P159ESTUDIO DESCRIPTIVO DE LAS HERNIAS DIAFRAGMATICAS CONGÉNITAS EN NUESTRO HOSPITAL EN LOS ULTIMOS 15 AÑOS
Pedro Mondéjar López, Juan José Agüera Arenas, Ángel B. Brea Lamas, Antonio Vicente Santos y Vicente Bosch Jiménez
Hospital Universitario Virgen de la Arrixaca, El Palmar.
Introducción:La hernia diafragmática congénita (HDC) se define como la herniación de contenido abdominal hacia la cavidad torácica a través de un canal pleuroperitoneal permeable. La más frecuente es la hernia de Bochdalek. Su incidencia varía entre 1/2000-5000 RN, siendo la presentación más frecuente la de un RN con distrés respiratorio y abdomen excavado. Su mortalidad oscila entre el 30-70 % dependiendo del grado de hipoplasia pulmonar subyacente.
Objetivo:Realizar un estudio descriptivo de las HDCs diagnosticadas en nuestro hospital en los últimos 15 años.
Material y métodos:Hemos revisado las HDCs ingresadas en nuestro Hospital desde el 1 de Enero de 1988 hasta el 31 de Diciembre de 2002, recogiendo los siguientes datos: sexo, duración del embarazo, asfixia perinatal, PN, existencia de dx. Prenatal, tipo de hernia, momento de dx. Postnatal y de cirugía si la hubo, malformaciones asociadas, mortalidad y confirmación diagnóstica con necropsia.
Resultados:Fueron diagnosticados de HDC 61 pacientes, 34 varones (55 %) y 27 mujeres. La incidencia fue de 1/3300 RN vivos. Fueron prematuros (< 37 semanas) un 13 % y PEG un 11,5 %. Tuvieron asfixia perinatal moderada-grave un 36 %. Estaban diagnosticados prenatalmente el 39,5 % y las hernias fueron izquierdas en un 70,5 % de los casos. Se diagnosticaron postnatalmente dentro de las primeras 24 horas el 62,5 % de los casos. Antes de 1995 se realizó cirugía dentro de las primeras 24 horas el 78 % frente la 35 % en años posteriores. Se encontró algún tipo de malformación asociada en un 41 % de los casos (más frecuente la malrotación intestinal). La mortalidad global fue de un 46 % (50 % pre95 y 41 % post95) obtuviendo confirmación diagnóstica (necropsia) en un 32 % de los casos.
Conclusiones:1. La HDC no parece aumentar la incidencia de prematuridad o de bajo peso aunque sí la existencia de asfixia perinatal.2. La incidencia de HDC izquierda es inferior a la de otras series.3. La incidencia de MA es similar a otras series así como el tipo de malformación más frecuente.4. El índice de mortalidad tiende a descender probablemente por la demora de la cirugía tras estabilización, y las nuevas técnicas de ventilación, siendo similar a la de otros hospitales salvo aquellos que disponen de ECMO.
P160IMPACTO DE LA APLICACION DE PRESION DE DISTENSION CONTINUA NASAL (NEEP) EN UNA UNIDAD DE CUIDADOS INTENSIVOS NEONATALES
Ángela Ruiz Extremera, Esther Ocete Hita, Alejandro de Goicoechea Vera, Francisco Contreras Chova, M. Pilar Rodríguez Santano, Victoria Ramos Ramos, Ana Garach Gómez y Rogelio Bayes García
Hospital Clínico Universitario San Cecilio, Granada y Universidad de Granada.
Antecedentes y objetivo:El surfactante, los corticoides prenatales y los modernos respiradores han mejorado el pronóstico de los recién nacidos (RN) con distres respiratorio. Los nuevos generadores de NEEP y sobre todo los nuevos sistemas de aplicación a la nariz del RN, en los que se permite el escape de flujo, ha puesto de manifiesto su eficacia. El objetivo fue conocer si la disponibilidad de este método de terapia respiratoria disminuye la frecuencia y el tiempo de ventilación mecánica (VM) y el tiempo de estancia en UCIN.
Pacientes y métodos:450 RN encamados en UCIN que fueron dados de alta a su domicilio entre el año 98, 99, 00 y 01divididos en dos grupos, Grupo1: 154 RN atendidos antes de junio del año 1999 y 296 atendidos con posterioridad (fecha de adquisición de 3 generadores de NEEP: junio 1999). Se han excluido los exitus. La recogida de datos incluyó edad gestacional (EG), peso (P), ventilación mecánica (días), NEEP (días), tiempo de estancia en la UCIN y diagnósticos al ingreso y evolutivos.
Resultados:El 21 % fueron prematuros menores de 1.500 g, 41 % con peso entre 1.500-2.500g y 38 % mayores de 2.500g. La incidencia de VM antes de la NEEP y después fue de 85/154 (55 %) vs 104/296 (35 %) (p < 0,001). El tiempo de VM (12,9 ± 2,1 vs 11 ± 2,2 días ± ES, p < 0,001). En el grupo 2 recibieron NEEP 122 (51 %) RN; 71 (24 %) RN de forma aislada y 51 (17 %) previa o post-VM. La estancia media ± ES en UCIN de los RN del grupo 1 fue de 20 ± 2,15 días y del grupo 2 de 16,2 ± 1 días (p < 0,001). La estancia media de los niños ventilados fue en el Grupo 1: 30,4 ± 3,4 y en el II de 26,1 ± 2,2 (p < 0,001), mientras que la estancia media de los niños sin VM y sin NEEP fue de 7,5 ± 9 días vs 8 ± 9 días respectivamente (NS). Los días totales de VM y/o NEEP (Grupo 1 = 85 RN vs Grupo 2 = 175RN) fue de 12,9 ± 2,1 vs 8,7 ± 1,4 días ± ES.
Conclusiones:El uso CPAP nasal en UCIN constituye un gran avance para los pacientes que requieren apoyo respiratorio, permitiendo disminuir la incidencia de ventilación mecánica, el tiempo de la misma, el tiempo de apoyo respiratorio global (VM y/o NEEP) y la estancia media de estos pacientes en UCIN.
P161 EVOLUCION DE LA MORTALIDAD NEONATAL PRECOZ DEL HOSPITAL CLINICO S. CECILIO DE GRANADA, DURANTE CATORCE AÑOS. SITUACION EN ESPAÑA Y ANDALUCIA
Ángela Ruiz Extremera, P. Carretero, M. Cruz Martínez, A. Caño Aguilar, L. Delgado, L. Garrigosa, Esther Ocete Hita y Alejandro de Goicoechea Vera
Hospital Clínico Universitario San Cecilio, Granada.
Antecedentes y objetivos:Aunque el peso al nacer ha sido un determinante de la mortalidad neonatal precoz, los mejores cuidados perinatales están consiguiendo unas tasas de mortalidad muy bajas. El objetivo fue conocer la evolución de la mortalidad neonatal precoz de nuestro hospital en los últimos catorce años en relación con el peso al nacimiento y con España y nuestra Comunidad Autónoma.
Pacientes y métodos:Entre 1989 al 2002, nacieron en el hospital 38.859 recién nacidos vivos y de ellos fallecieron de forma precoz 123 niños. Se analizo la tasa de mortalidad neonatal precoz (TMNp) para cada grupo de peso y la distribución en el total de las muertes precoces. Los pacientes se clasificaron en grupos básicos de peso siguiendo la clasificación de la OMS: G1: < 1.000 g, G2: 1.000-1499 g, G3: 1.500-2.499 g y G4: > de 2.499 g. Se calcularon las tasas de Andalucía, datos publicados por el Instituto de Estadística Andaluz (IEA) hasta el año 1998 y para las nacionales de la Sección de Medicina Perinatal de la SEGO hasta el 2000. Para el análisis de las variables se utilizó el SPSS.10.
Resultados:El descenso observado es de 16 pacientes en 1999 (G1 = 7, G2 = 1, G3 = 1, G4 = 7), a uno en el año 2002 (G2 = 1). Esta disminución ha sido progresiva por años y en los grupos 1 y 4 fundamentalmente. La evolución de la TMNp fue del 5,17 ‰ a 0,39 ‰ con un valor de p < 0,05. En los grupo menores de 2500g (1-2-3) la disminución de la mortalidad fue muy significativa (p < 0,01) y en el G4 se observo una disminución pero sin diferencias significativas. La TMNp en Andalucía fue en 1989: 4,86 ‰ a 1989: 2,41 ‰) y en España (1989: 4,56 ‰ a 2000: 2,26 ‰).
Conclusiones:La mayor supervivencia en los primeros siete días de vida observada en nuestro hospital es un reflejo de la conseguida en el resto de nuestro país y es un indicador de calidad en la atención perinatal pero aconseja un control exhaustivo de la morbilidad.
P162CUTIS MARMORATA TELANGIECTASIA CONGÉNITA E TROMBOSE VENOSA PROFUNDA NO RECÉM-NASCIDO
Alexandra Emílio, Ana Bettencourt, Luís Gago Leal, Teresa Tomé y Teresa Costa
Maternidade Alfredo da Costa, Lisboa (Portugal).
Os autores apresentam um caso clínico de um recém-nascido do sexo masculino, raça negra, 2.º filho de pais jovens, saudáveis, não consanguíneos; irmão de 2 anos saudável; outros antecedentes familiares desconhecidos. Gravidez vigiada desde o 1.º trimestre, sem intercorrências. Parto eutócico na MAC, às 37 semanas de gestação, Apgar 9/10, PN 3460 g. AIG. Observação do fenótipo compatível comCutis Marmorata Telangiectasia Congenita: hemangioma reticulado gigante disseminado por todo o corpo, leucocória bilateral, máculas hipomelanóticas no tronco, sindactilia do 2.º e 3.º dedos do pé esquerdo, sem outras alterações. Ecografia transfontanelar realizada ao 2.º dia de vida sem alterações. Alta para domicílio ao 3.º dia, acompanhado a mãe. RMN-CE ao 17.º dia de vida mostrando trombose extensa do Seio Longitudinal Superior e hemorragia dos Plexos Coroideus, o que motivou reinternamento, na Unidade de Cuidados Intermédios Neonatais da MAC, para vigilância clínica, estudo de trombofilia congénita e eventual terapêutica. Clinicamente bem. Primeiro estudo etiológico de trombofilia (mãe, pai e RN) sem alterações. Ao 36.º dia de vida surge trombose venosa profunda na Veia Femural Esquerda. Iniciou Ácido Acetilsalicílico e Enoxaparina, com resolução das alterações trombóticas ao 6.º dia de terapêutica. Manteve terapêutica com antiagregante plaquetar e heparina de baixo peso molecular durante 4 meses, com melhoria clínica significativa. Do estudo etiológico de trombofilia foi detectada mutação do 4G/5G na posição 675 do gene do Inibidor do Activador do Plasminogénio 1 (PAI-1), situação clínica extremamente rara como causa de trombofilia congénita. Controlos posteriores em consulta de Pediatria, Cirurgia Vascular, Oftalmologia e Cardiologia Pediátrica. Actualmente com 14 meses e tem desenvolvimento psicomotor adequado, edema linfático do membro inferior esquerdo, atraso do desenvolvimento estaturo-ponderal e anemia ferropénica devido a erros alimentares sistemáticos e contexto social carenciado. A Síndrome de van Lohuizen ou Cutis Marmorata Telangiectasia Congenita é uma síndrome extremamente rara, de etiologia desconhecida, diagnosticada pela clínica. Existe um caso descrito de Trombose Venosa Profunda associado ao Factor V de Leiden mas nenhum associado à mutação do 4G/5G na posição 675 do gene do PAI-1.
P163TRATAMIENTO DEL QUILOTORAX NEONATAL CON SOMATOSTATINA
Mónica Belda Anaya, Lorea Ruiz Pérez, José Luis Tarazona Fargueta, Fernando Alonso Ortega, Jesús Mira Navarro y Bartolomé Jiménez Cobo
Hospital General Universitario, Alicante.
Introducción:El Quilotórax neonatal es una patología poco frecuente. En la mayoría de los casos es una complicación post-quirúrgica. Puede provocar insuficiencia respiratoria, pérdida proteica e inmunodeficiencia. Inicialmente se realiza tratamiento médico, reservándose el quirúrgico para casos refractarios. Se han comunicado recientemente algunos casos tratados con éxito con somatostatina o análogos. Presentamos dos casos de quilotórax neonatal tratados con somatostatina.
Caso clínico 1:Neonato pretérmino de 26 semanas gestación, 920 gr, con EMH grado II y ductus que precisó cierre quirúrgico. Presenta a los 34 días de vida, en el contexto de sepsis por cándida y síndrome de vena cava superior con catéter venoso central, quilotórax izquierdo. Tratado inicialmente con nutrición parenteral total y drenaje pleural, presenta múltiples recidivas que requieren punciones pleurales evacuadoras. Se inicia tratamiento con somatostatina (2 μg/kg/12 horas) durante 3 días, con lo que se resuelve sin recidivas.
Caso clínico 2:RN de 37 semanas y 3.000 g, ingresa por quilotórax congénito (diagnóstico prenatal del derrame pleural). Tras tratamiento inicial conservador habitual no presenta mejoría y se inicia tratamiento con somatostatina (2 mg/kg/12 horas) durante 7 días. Presenta resolución clínica y radiológica a las 48 horas de inicio del tratamiento.
Discusión:El tratamiento conservador basado en una intervención nutricional y drenaje pleural es la primera opción terapeútica ante un quilotórax neonatal.
En los casos prolongados, derrame de gran flujo o cuando aparecen complicaciones secundarias, la somatostatina parece ser un tratamiento efectivo, no invasivo, con pocos efectos secundarios y que reduce la necesidad de intervención quirúrgica. El mecanismo de acción es desconocido pero parece disminuir el flujo sanguíneo en territorio esplácnico y producir vasoconstricción de los vasos linfáticos, reduciendo secundariamente la producción de quilo. En nuestros dos casos, utilizando dosis bajas de somatostatina, conseguimos una resolución rápida y eficaz del quilotórax. Son cada vez más los casos publicados tratados con éxito con somatostatina.
Conclusión:Aunque se necesitarían más estudios prospectivos para establecer las dosis adecuadas, la forma de administración (infusión continua o varias dosis al día) y la duración del tratamiento, la somatostatina parece una alternativa a considerar en el tratamiento del quilotórax antes de la cirugía.
P164ICTERICIA NEONATAL. ESTUDIO OBSERVACIONAL PROSPECTIVO
María Bengoa Caamaño, M. José Hernández Bejarano, Carmen Pedraz García, Laura San Feliciano Martín, M. Ángel Arias Consuegra y M.ª Candelas Martínez Hernández
Hospital Universitario de Salamanca. Hosp. Clín. y Hosp. Virgen de la Vega.
Objetivos:Observar la incidencia y factores asociados a la aparición de ictericia neonatal en recién nacidos a término asistidos en el H. Clínico Universitario de Salamanca en un periodo de 6 m.
Material y métodos:Estudio prospectivo observacional de todos los neonatos a término, nacidos en HCUS entre 1 marzo del 2002 y el 30 de septiembre de dicho año. Se diseñó un protocolo previo al estudio para recogida de datos perinatales y socioculturales de estos niños.
Resultados:De los 484 niños estudiados 124 (25,6 %) desarrollaron ictericia neonatal; precisando ingreso, por este motivo, en la unidad de neonatología 38 (30,6 %). Dentro de este grupo se encontraron 11 casos (28,9 %) de incompatibilidad AB0 y 2 (5 %) de Rh. Así mismo 12 neonatos ingresados por otro motivo desarrollaron ictericia apareciendo en 2 (16 %) incompatibilidad AB0. En todos los casos el tratamiento recibido fue fototerapia continua, no necesitando en ninguno exanguinotransfusion. No se presentaron complicaciones secundarias a la hiperbilirrubinemia.
Entre los recién nacidos con ictericia que no precisaron ingreso (86 casos) se encontraron 9 casos de incompatibilidad AB0 (66 % antiA). Otros factores asociados al desarrollo de ictericia han sido: partos instrumentales, traumatismos obstétricos, pérdidas ponderales superiores al 10 %, enfermedades metabólicas maternas. Ninguno de estos niños requirió tratamiento.
De los niños que no desarrollaron ictericia 11 presentan incompatibilidad AB0 (81 % antiA) y un caso de incompatibilidad Rh (antiD).
Conclusiones:1. La ictericia neonatal es una entidad muy frecuente en nuestro medio.2. La situación más frecuente es la presencia de ictericia por sumación de factores (75 %), seguida de la incompatibilidad AB0, siendo la antiA la más frecuente.3. No todos los niños con isoinmunización AB0 desarrollan ictericia.4. La mayor parte de los niños con ictericia no requieren ingreso ni tratamiento.
P165ESTUDIO DE FACTORES PRONOSTICOS DE MORTALIDAD DE LAS HERNIAS DIAFRAGMATICAS CONGÉNITAS
Antonio Vicente Santos, Juan José Agüera Arenas, Ángel B. Brea Lamas, Pedro Mondéjar López y Vicente Bosch Jiménez
Hospital Universitario Virgen de la Arrixaca, El Palmar.
Introducción:La mortalidad de la hernia diafragmática congénita (HDC) se ha logrado reducir en la última década en parte a la introducción de la oxigenación de membrana extracorporea (ECMO). El conocimiento de factores pronósticos de mortalidad junto a los criterios ya existentes, nos pueden servir de ayuda para valorar la necesidad de traslado a centros hospitalarios que dispongan de ECMO.
Objetivo:Estudio de factores pronósticos que puedan tener relación con la mortalidad de los pacientes diagnosticados de HDC en nuestro Hospital.
Material y método:Se han incluido en el estudio 54 casos de HDC diagnosticadas prenatalmente o en de las primeras 72 horas de vida. Se han recogido como probables factores pronósticos de mortalidad: traslado hospitalario, prematuridad, peso al nacimiento, dx prenatal, localización de la hernia, momento del dx postnatal y de cirugía (< 24 horas o 24-72 horas de vida), asfixia perinatal, y la primera determinación arterial de ph, PCO2, IO e IV a su ingreso.
Resultados:La muestra se compone de 54 casos, 28 (53 %) varones, 4 (7,8 %) de bajo peso y 5 (9,2 %) prematuros. Se mostraron factores pronósticos de mortalidad con significación estadística:1. De los que tenían undx prenatal fallecieron un 72,7 % (16) (X2 = 5,5, p = 0,019),2. Con IO > 30 fallecieron 92,3 % (12) (X2 = 15, p < 0,005).3. Con IV > 500 fallecieron 78,6 % (11) (X2 = 5,43, p = 0,038).4. De los nacidosprematuros fallecieron el 100 % (5) (X2 = 4,73, p = 0,038).5. Si eldx postnatal se realizó en < 24 h. de vida fallecieron el 60 % (27) (X2 = 4,015, p = 0,045).6. Si lacirugía se realizó en < 24 h. de vida fallecienron 93,8 % (15) (X2 = 17,37, p < 0,005).7. De aquellos que presentaronasfixia fallecieron el 90,5 % (19) (X2 = 25,38, p < 0,005).8. Con ph < 7,2 fallecieron el 86,4 % (19) (X2 = 21,4, p < 0,005).9. Con PCO2 > 60 mmHg fallecieron el 80 % (16) (X2 = 12,02, p < 0,005).
Conclusiones: 1.La dificultad de adaptación postnatal inmediata, y de compromiso respiratorio grave en las primeras 24 horas de vida, fundamentalmente en niños prematuros, se han mostrado como los factores pronósticos de mortalidad con mayor significación en niños con dx de HDC.2. No se han mostrado como factores pronósticos de mortalidad la necesidad de traslado, el bajo peso al nacimiento, y la localización de la hernia.
P166ESTUDIO COMPARATIVO DE LAS VARIABLES PESO Y TALLA EN NIÑOS DE DIFERENTE ORIGEN ÉTNICO AL NACER Y AL AÑO DE VIDA
José Luis Fillat Claver y María R. Gutiérrez Grau
Instituto Catalán de la Salud, Barcelona.
Antecedentes y objetivo:Dado que el fenómeno migratorio ha ido adquiriendo cada vez más relevancia en nuestro pais desde 1995, planteamos en este estudio, como una aproximación a esta realidad, la posibilidad de encontrar diferencias significativas en el peso y/o talla entre niños españoles y de diferente origen étnico tanto al nacer y/o al año de vida.
Métodos:Estudio analítico, transversal y retrospectivo utilizando los registros antropométricos procedentes de las revisiones pediátricas de una muestra aleatoria de niños de una Area Básica de Salud, nacidos en nuestro pais, entre los años 1995 y 2001, tomados al nacer y a los 12 meses.
Se analizan un total de 118 registros de los cuales 52 pertenecen a niños españoles (27 varones/25 hembras), 30 a niños magrebies (21 hembras/9 varones) y 36 a niños subsaharianos (11 varones/15 hembras).
Se incluyen tan solo niños nacidos a término y de peso adecuado para su edad gestacional.
Los datos registrados fueron informatizados mediante el programa Excel, calculándose para cada uno de los 6 grupos la media y desviación estándar.
Resultados:

Conclusiones:No existen diferencias significativas entre el peso y la talla del niños españoles, magrebies y subsaharianos al nacer o al año de vida. En niños y en niñas megrabies se observan una talla y peso al nacer discretamente superior al de los otros colectivos que se estabiliza al año de vida. Posteriores estudios con una muestra más amplia de población serian necesarios para confirmar esta observación.
P167
SEGUIMIENTO NEUROLOGICO DE RECIÉN NACIDOS MENORES DE 1.500 GRAMOS A LOS 2 AÑOS DE EDAD. PERIODO 1998-99
Amalia Arce Casas, Martí Irondo Sanz, Javier Krauel Vidal, Rafael Jiménez González, Jaume Campistol Plana, M. Pilar Poo Argüelles y Margarita Ibáñez
Hospital San Joan de Deu, Barcelona.
Antecedentes:Existe un interés creciente en el seguimiento de recién nacidos de muy bajo peso al nacimiento con la mejoría de la asistencia obstétrica y neonatal.
Objetivos:Evaluar las cifras de supervivencia de los recién nacidos de muy bajo peso, junto con la proporción y los tipos de secuelas que se observan a los dos años de edad.
Material y método:Estudio observacional de seguimiento de los recién nacidos de menos de 1500 gramos nacidos entre los años 1998 y 1999. Se evalúan los datos obtenidos en el programa de seguimiento compuesto por visitas a Pediatría, Maduración, Neurología, Psicología y Oftalmología; y la realización de Potenciales Evocados Visuales y Auditivos. Se clasifican las secuelas obtenidas en leves, moderadas y graves.
Resultados:Ingresan en el periodo de estudio un total de 136 recién nacidos de muy bajo peso. La supervivencia es del 77,9 %. Completan el seguimiento hasta los dos años de edad un 83,9 % de los supervivientes. La aparición de secuelas se observa en un 20,2 % de los niños, siendo un 9 % graves, un 1,1 % moderadas y un 10,1 % leves. Las secuelas con más frecuencia observadas afectan a la función neuromotora y a los órganos sensoriales (visión y audición). Los pacientes perdidos en el seguimiento tienen mayor peso al nacimiento, mayor edad gestacional y mayor normalidad ecográfica que los pacientes seguidos.
Conclusión:Se obtiene un incremento en la supervivencia de los recién nacidos de muy bajo peso con la mejoría de los cuidados neonatales. La presencia de secuelas es similar a la obtenida en otros estudios de seguimiento. Se considera relevante la muestra de pacientes perdidos y su influencia en los porcentajes de secuelas obtenidas.
P168ÉPULIS CONGÉNITO. UNA RARA ENTIDAD
Estefanía Romero Castillo, M. Ester Guerrero Vega, Patricia Martín Cendón, M. José Añón Requena, Simón Pedro Lubián López, Manuel Matías Vega y José M. Angulo Madero
Hospital Puerta del Mar, Cádiz.
El épulis congénito del recién nacido es un tumor benigno que nace generalmente del reborde alveolar del maxilar superior. Se suele presentar como masa única pediculada, de consistencia blanda y de la misma coloración de la mucosa, que protuye de la cavidad oral. Su frecuencia es muy baja. Marcada predisposición por el sexo femenino (8:1). Posee una inmunohistoquímica característica.
Caso clínico:Recién nacido a término, de sexo femenino, que nace de parto eutócico. Apgar 9/10. A la exploración presenta masa polipoide de 5,5 x 4,5 x 2 cm que nace del reborde gingival del maxilar superior y protuye hacia el exterior de la cavidad oral, no provocando obstrucción de la vía aérea. Se procede a exéresis quirúrgica con evolución postoperatoria favorable.
Estudio anatomopatológico:Celulas poligonales con citoplasma eosinófilo y granular, rodeadas de numerosos vasos sanguíneos.
Inmunohistoquímica:Los gránulos citoplasmáticos se ponen de manifiesto con tinción de PAS. La población celular tumoral mostraba intensa positividad para vimentina y negatividad para la proteína S-100 y la desmina. Aspecto granular de las células proliferantes.
Conclusiones:1. El épulis congénito continúa siendo una entidad rara para los pediatras.2. Es obligado realizar diagnóstico histopatológico para diferenciarlo de otros tumores.3. Destacar la benignidad de la tumoración. No se han descrito casos de recidiva, malignidad ni daño futuro en la dentición.4. A pesar de las regresiones espontáneas publicadas, el tratamiento de elección es el quirúrgico de forma precoz.5. Las indicaciones quirúrgicas surgen de problemas potenciales como obstrucción de la vía aérea, imposibilidad de alimentación y del aspecto grotesco de la tumoración que crea gran inquietud familiar.
P169QUILOTORAX NEONATAL. DIEZ AÑOS DE EXPERIENCIA
Julio Álvarez Pitti, María Macho Díaz, M. Concepción Centeno Monterrubio, Lourdes Román Echevarría y Lucila Martín Vargas
Hospital de Cruces, Baracaldo.
Objetivos:Determinar la incidencia y las causas del quilotórax (QT) neonatal en nuestro Hospital, valorando su evolución clínica y tratamiento.
Métodos:Se realiza una revisión retrospectiva de las historias de los 8 pacientes registrados en nuestra base de datos con diagnóstico de quilotórax neonatal desde 1992 a 2003. Se describe la población recogiendo las causas, métodos diagnósticos, tratamientos (Tto), duración y complicaciones. Definimos QT cuando el líquido pleural (LP) tiene más de 1000 cels/ml y al menos 70 % de linfocitos.
Resultados:Se diagnostican 8 QT de los que el 62 % (5) son varones. Seis de los casos (75 %) son posquirúrgicos (PCx), 5 tras cirugía cardíaca (2,5 % de 199 cirugías cardíacas) y 1 atresia de esófago; dos (25 %) congénitos (QTc). Los 2 QTc se diagnostican en el 1.º y 4.º día de vida, y los PCx en una media de 12,2 (2-33) días tras la cirugía. Todos debutan con distres respiratorio y derrame pleural en Rx. de tórax. Sólo en 2 de los casos PCx el LP es lechoso, aunque todos reciben alimentación enteral tras la cirugía durante 8,2 días (2-17). Requieren ventilación mecánica los QTc y 2 de los PCx. El tratamiento inicial en los congénitos es nutrición parenteral (NP) durante 26 y 90 días respectivamente, con drenaje de 28 y 70 días de duración; tras resolverse el derrame, se continúa con alimentación enteral con fórmula (Monogén®) con triglicéridos de cadena media (TCM) durante su estancia (13 y 60 días) y tras el alta. Uno precisa cirugía a los 34 días de evolución. De los PCx, 4 reciben TCM durante 24,2 días (13-37). En 1 caso fracasa, requiriendo NP. El volumen total medio (VTM) de drenaje de los QTc es de 2421 mL/kg y de 72 mL/kg en el 7.º día de Tto. En los PCx el VTM es de 396,2 mL/kg y a los 7 días de 14,4 mL/kg Todos los casos presentan hiponatremia, hipoproteinemia y pérdida de peso respecto del previo al diagnóstico (28 % de media en los QTc y 5,4 % en los PCx). Reciben antibióticos 7/8 (47,5 días de media en los QTc y 12,8 en los PCx). Todos sobreviven. No hay recidivas.
Conclusiones:El aspecto del LP tiene relativa utilidad diagnóstica. Este se establece mediante la clínica, la Rx de Tx y el análisis del LP. Los QTc son menos frecuentes, presentan VTM mayores, requiriendo NP, presentando más complicaciones, por lo que requieren un planteamiento diferente. En los PCx el Tto con TCM resulta eficaz aunque su uso requiere control.
P170DISTROFIA MIOTONICA DE STEINERT NEONATAL
Javier Mallo Castaño, Leticia Castañón López, Fernando Fernández Calvo, M. Teresa Palau Benavides, Ana Díaz Moro, Carmen Villafañe Landeira y M. del Carmen Fuentes Acebes
Hospital de Léon.
Introducción:La Distrofia Miotónica de Steinert en su forma neonatal grave presenta una herencia AD con penetrancia y expresividad variables. El defecto genético se localiza en el cromosoma 19q 13,3 y consiste en la expansión de repetición de tripletes (CTG) asociando el fenómeno de anticipación. La mayor parte de los casos son transmitidos a través de la línea materna. Presenta una incidencia de 1/30.000 RN, afectando a múltiples sistemas con mal pronóstico y careciendo de tratamiento específico.
Caso 1:RN varón pretérmino de BPEG. AF: madre, tíos y abuelo en rama materna con D. Miotónica Steinert. AP: 1.º/1, embarazo normal, parto cesárea, EG: 33 sem, PRN: 1,269. EF: hipotonía generalizada marcada, llanto débil, respiración espontánea con aceptable movilidad torácica. EC: hemograma con anemia, bioquímica con ictericia colostática inespecífica de predominio directo (BT 19,4 BD 7,9 GOT 343 GPT 129 GGT 195) y CK 103, Rx tórax con atelectasias por tapones mucosos, EKG y Ecocardiograma normales, Exploración oftalmológica normal, Eco y TAC cerebral con hidrocefalia, quiste porencefálico y atrofia cerebral marcada, EMG con patrón miopático distal y actividad denervativa espontánea sin descargas miotónicas, genotipo de RN: heterozigoto con alelo normal y otro expandido en rango patológico heredado de la madre que corresponde con proceso de anticipación, estudio genético de RN: 1700 repeticiones (Steinert congénito).
Caso 2:RN mujer pretérmino de PAEG. AF: madre y hermana con D. Miotónica Steinert. AP: 2.º/2, embarazo normal, parto cesárea, EG: 32 sem, PRN: 1,350. EF: hipotonía generalizada marcada, insuficiencia respiratoria severa con bradicardia. EC: hemograma, bioquímica y Rx tórax similares al caso previo (ictericia colostática y parálisis diafragmática derecha), Ecocardiograma con DAP sin HTP y pequeña CIA, Eco cerebral con hidrocefalia y atrofia cerebral severa, EMG y genotipo de RN similares al caso previo, estudio genético de RN: 2000 repeticiones (Steinert congénito).
Evolución:Desfavorable con éxitus en el segundo caso antes del año de vida
Comentarios:Presentamos estos dos casos de Síndrome de Steinert grave neonatal por su baja incidencia y la proximidad de aparición de ambos entre sí (intervalo de diez días). El estudio genético permitió confirmar el diagnóstico
P171SINDROME DE JARCHO-LEVIN
M. Isabel López Conde, Ana García González, Esther Vázquez López, Carmen García-Plata González, Carlos Somoza Rubio y Ramón Morales Redondo
Complexo Hospitalario Xeral-Calde, Lugo.
El síndrome de Jarcho-Levin o displasia espondilotorácica es una enfermedad genética rara caracterizada únicamente por malformaciones de las vértebras y de las costillas.
Presentamos el siguiente caso clínico: RN hembra, fruto de la primera gestación de una madre sana de 34 años. Embarazo que cursó con amenaza de parto pretérmino y placenta previa oclusiva recibiendo tratamiento con tocolíticos y corticoides para maduración pulmonar fetal. Parto por cesárea a las 35 semanas de gestación Apgar 9/10. Exploración física del RN: peso 2.325 g. Talla 41 cm. PC 33 cm. Cuello corto, tórax muy corto, abdomen prominenete. Resto de la exploración física por órganos y aparatos dentro de la normalidad. Radiología: pérdida de alineación de cuerpos veretebrales, columna dorsal compuesta por hemivértebras con normalidad de columna lumbar y cervical, nueve pares de costillas, con costillas gráciles. Ecografía abdominal, exploración ocular, estudio metabólico: normales.
Conclusión:Los trastornos en el desarrollo y crecimiento del esqueleto pueden presentarse en el contexto de multitud de cuadros clínicos y etiológicos y en muchos casos se acompañan de afectación de estructuras extraesqueléticas, pero dado que en el caso presentado las alteraciones costovertebrales no se asociaban a otras anomalías congénitas el disgnóstico fue de Síndrome de Jarcho-Levin o Displasia espondilotorácica.
P172DISPLASIA ALVÉOLO-CAPILAR
Ester Sanz López, Elena Mederuelo, Manuel Sánchez Luna, Belén Bernardo Atienza, M. Luisa Franco, Rocío Chacón Aguilar, Gema Arriola Pereda y Cristina Menéndez Hernando
Hospital General Universitario Gregorio Marañón, Madrid.
Introducción:La displasia alvéolo-capilar (DAC) es una enfermedad pulmonar letal poco referida en la literatura, de difícil diagnóstico y cuya incidencia probablemente sea mayor de la conocida. El diagnóstico es histológico y su sospecha clínica se realiza en distrés respiratorio neonatal severo con hipertensión pulmonar (HTP) con respuesta parcial o transitoria a óxido nítrico inhalado e hipoxemia progresivamente refractaria a tratamiento médico.
Objetivo:Se expone la experiencia en el diagnóstico y manejo de la DAC en una unidad de referencia de ECMO.
Se presentan dos recién nacidos a término remitidos a nuestra unidad de ECMO por distrés respiratorio con hipoxemia severa refractaria a tratamiento convencional y con respuesta parcial a óxido nítrico inhalado. En el primer caso se indicó entrada en ECMO venovenoso a las 24 horas de vida y en el segundo entrada en ECMO venoarterial a los siete días de vida. En ambos casos se realizó biopsia pulmonar abierta (a los 25 y a los 17 días de vida respectivamente), confirmándose en ambos el diagnóstico de DAC, con malalineamiento de las venas pulmonares en el primer caso.
Conclusión:La DAC es una entidad de difícil diagnóstico que ha de sospecharse en aquellos recién nacidos con hipoxemia refractaria al tratamiento convencional y respuesta parcial o transitoria a óxido nítrico inhalado.
P173INDICE DE LOBULARIDAD LEUCOCITARIA (IL) Y CONCENTRACION DE MIELOPEROXIDASA (MPX) COMO PREDICTORES DE INFECCION NEONATAL
Fco. Javier Aguirre Rodríguez, José Luis Gómez Llorente, Antonio Bonillo Perales, José Espín Gálvez, Moisés Leyva Carmona, M. Ángeles Llamas Guisado, Juan López Muñoz, Pedro Cortés Mora y Raúl Sánchez Pérez
Hospital Torrecárdenas del SAS, Almería.
Justificación y objetivos: En el presente estudio nos propusimos analizar los factores relacionados con riesgo de infección neonatal y conocer la capacidad predictiva del Il y la MPX en relación a los habitualmente utilizados en la práctica clínica.
Material y métodos:Analizamos los últimos 28 recién nacidos con sepsis neonatal de nuestro Hospital que comparamos prospectivamente con 30 recién nacidos con riesgo infeccioso que no cursaron sepsis neonatal (en los que además analizamos sangre venosa de cordón umbilical). En el estudio estadístico se utilizó: "r" de Pearson, "t" student para muestras independientes y análisis de regresión logística múltiple.
Resultados:a) En el estudio de correlación encontramos las siguientes relaciones estadísticamente significativas: Horas bolsa rota vs PCR en cordón umbilical; r = 0,446, p < 0,01. Edad gestacional vs IL cordón; r = 0,334, p = 0,03. PesoRN vs MPX de cordón umbilical; r = 0,264, p < 0,043. VSG cordón vs MPX RN; r = 0,733, p = 0,025. PCR cordón vs horas de bolsa rota; r = 0,452, p < 0,001.MPX RN vs leucocitos RN; r = 0,311, p = 0,045.b) En el estudio comparativo entre grupos:

c) En el análisis de regresión logística múltiple observamos que la única variable que predice significativamente la aparición de sepsis neonatal es el % de neutrófilos del recién nacido (OR: 0,91 IC95 %: 0,85-0,97, p = 0,03).
Conclusiones:El índice de lobularidad leucocitaria no es un factor relacionado con mayor probabilidad de sepsis neonatal. Aunque la MPX es diferente en recién nacidos con y sin infección neonatal, la única variable predictora de sepsis neonatal es un porcentaje de neutrófilos inferior a un 50 %.
P174CAUSA INFRECUENTE DE MENINGITIS NEONATAL
Gerardo García Matas, Rocío Marín Ravina, Carlos Sáenz Reguera, Antonia López Sanz, Lucas Eduardo Durán de Vargas y Antonio González-Meneses
Hospital Universitario Virgen Macarena, Sevilla.
Antecedentes y objetivo:La sepsis neonatal tardía con afectación meníngea por Neisseria meningitidis B es infrecuente, siendo pocos los casos descritos en la literatura. Presentamos un recién nacido de 15 días de vida con meningitis por N. meningitidis B.
Caso clínico:Recién nacido varón a término de peso adecuado que ingresa con 15 días de vida en UCI Neonatal por presentar aspecto de enfermedad grave, fiebre con rechazo de las tomas de 24 horas de evolución. Antecedentes familiares y personales: madre de 20 años, primípara. Embarazo controlado. Parto eutócico y test de Apgar de 9 y 10 al 1 y 5 minutos de vida respectivamente. Ingreso previo en neonatología por madre portadora de estreptococo del grupo B con profilaxis incompleta, siendo dado de alta a las 72 horas de observación sin datos clínicos ni analíticos de infección. Exploración física:Peso 3400 gramos. Temperatura 39 °C. Mal estado general, color terroso con palidez de piel y mucosas. Decaído, quejido y polipnea. Mala perfusión periférica. Tendencia a la hipertonía con opistótonos. Taquicardia con hipotensión arterial. Fontanela normotensa. Resto sin hallazgos. Pruebas complementarias: leucocitos 5000/mm3. PCR 241 mg/l. Bioquímica del LCR con pleocitosis, hipoglucorráquia e hiperproteinorráquia. Cultivo de LCR donde se aisla Neisseria meningitidis serotipo B. Hemocultivo negativo.
Con estos datos clínicos y analíticos se llega al diagnóstico de sepsis neonatal tardía con meningitis por N. meningitidis serotipo B.
Evolución favorable tras tratamiento con drogas inotrópicas, Antibioterápia i.v., expansores de volumen, sedación y nutrición parenteral.
Conclusiones:La sepsis neonatal tardía es la infección bacteriana generalizada con clínica multisistémica y cultivo positivo de sangre y/o de LCR entre la 2.ª y la 4.ª semana de vida. La incidencia de meningitis en RN es de 0,2 a 0,4 por 1000 RN vivos. Entre los agentes etiológicos más frecuentes se encuentran estreptococo del grupo B, E. coli K1, siendo escasa la incidencia de N. meningitidis B como causante de sepsis y/o meningitis en el periodo neonatal. Se puede presentar con una sintomatología poco específica, siendo la irritabilidad y la fontanela hipertensa los hallazgos más frecuentes. La tasa de mortalidad oscila entre el 20 y el 50 % con complicaciones tardías en el 40-50 % de los casos.
P175 ANALISIS DE MORTALIDAD NEONATAL EN EL AÑO 2002
Gerardo García Matas, Rocío Marín Ravina, Andrés Rodríguez Sacristán, Julia Pérez Losada, Carlos Sáenz Reguera, Lucas Eduardo Durán de Vargas y Antonio González-Meneses González-Meneses
Hospital Universitario Virgen Macarena, Sevilla.
Objetivo:Estudio de la mortalidad neonatal en nuestra sección de Neonatología durante el año 2002, analizando las causas generales de mortalidad en los distintos periodos neonatales.
Material y método:Realizamos un estudio retrospectivo revisando historias clínicas y archivos de mortalidad de la sección de Neonatología y del servicio de Obstetricia de nuestro hospital correspondiente al año 2002. El número total de RN fue de 3560, incluyendo 29 muertes fetales. Se utilizan fórmulas estadísticas correspondientes a las tasas de mortalidad específica por edad.
Hicieron exitus 15 RN, 9 varones y 6 mujeres. 3 procedían de partos multiples, 2 presentanción en pelvianas, 9 nacieron tras cesárea y 2 partos instrumentales.
Por distribución por grupos de peso, de un total de 20 RN vivos con peso inferior a 1.500 g 8 fueron exitus. Si tomamos los RN con peso entre 500 y 1.000 g de peso (8) 5 fallecieron. Menores de 2.500 g de peso fallecieron 12, 10 en la primera semana de vida. 5 RN eran de peso bajo para la edad gestacional.
Entre los diagnósticos principales, la prematuridad (5 casos) junto con las malformaciones (5 casos) fueron las dos causas más frecuentes. Una muerte fue postneonatal, 2 fueron en el periodo neonatal tardío por malformación y por encefalopatía hipóxico-isquémica en un prematuro, el resto fallecio en el periodo neonatal precoz.
Entre los factores de riesgo durante la gestación 3 casos presentaron líquido meconial, 2 diabetes gestacional, 2 gestaciones con dispositivo intrauterino y 2 casos de ruptura precoz de membranas.
Resultados:La tasa de mortalidad neonatal en nuestra unidad fue en el año 2002 de 3,9 por mil RN, similar a la encontrada en otros hospitales de nuestro país, atribuyendose el 33 % de la misma a la mortalidad neonatal inmediata, con una tasa de 1,40 por mil RN. El resto se distribuye entre la prímera semana de vida con una mortalidad neonatal precoz del 3,37 por mil RN, una mortalidad en el periodo neonatal tardío de 0,56 por mil RN.
La mortalidad perinatal se situa en el 7,8 por mil RN con una tasa de mortalidad perinatal ampliada del 11,8 por mil RN.
P176 HIPERTERMIA NEONATAL. REVISION DE CASOS EN PERIODO 2001-2002
Rocío Marín Ravina, Gerardo García Matas, Pedro Terol Barrera, M. Rosario Santano Gallinato, Antonia López Sanz, Carlos Sáenz Reguera, Francisco Camacho González, Lucas Eduardo Durán de Vargas y Antonio González-Meneses González-Meneses
Hospital Universitario Virgen Macarena, Sevilla.
Introducción:La hipertermia neonatal puede observarse en el segundo y tercer día de vida en Neonatos clínicamente sanos. Esta situación es relativamente frecuente en niños alimentados al pecho o expuestos a temperaturas elevadas.
Objetivo:Revisar la incidencia de hipertermia en los Neonatos ingresados en nuestra unidad durante el periodo 2001-2002, con especial referencia a la "fiebre de sed".
Material y métodos:De los 6.000 recién nacidos en dicho bienio, revisamos los 15 ingresos por hipertermia. Todos habían nacido en nuestro hospital, 9 procedían de maternidad y 6 del servicio de urgencias. Edad media de ingreso 3 días.
Resultados:La mayoría sin antecedentes obstétricos y perinatales de interés. Cesárea: 4 casos. Peso medio: 3.480 g. Sexo: mayor n.º de varones (9). Alimentación: 12 con lactancia materna y 3 con lactancia mixta; 6 presentaron rechazo con poco aporte dietético. Temperatura corporal: 38-38,8. Valoración hemodinámica: estables. Pérdida de peso: superior al 10 %: 4 casos.
Diagnósticos:1. Fiebre de sed, 8 casos (53,3 %) La mitad con sequedad de piel y mucosas y 7 de ellos con sodio mayor de 145. Urea elevada, proteinuria, y, acidosis metabólica leve: 2 casos. Pérdida de peso: 9,7 %. Tratamiento: soluciones de rehidratación endovenosa en 5 neonatos. Antibióticos: se inició tratamiento en 4 casos. Se retiran tras confirmar "fiebre de sed". Estancia media hospitalaria: 5 días. Alimentación al alta: lactancia materna y artificial si lo requieren.2. Síndrome febril, 5 casos (33,3 %). En todos ellos la exploración fue negativa, desapareciendo la fiebre a las pocas horas de su ingreso. Bioquímica urea y sodio: normales. Hemocultivo: negativo.3. Sepsis, 2 casos (13,3 %). 1 caso de sepsis neonatal precoz por EGB. Tratamiento antibiótico 14 días. El otro caso, sepsis neonatal tardía por E. coli. Tratamiento antibiótico: 14 días. Alta a los 20 días.
Conclusiones:De los 648 recién nacidos a término ingresados en nuestra unidad durante el bienio 2001-2002, un 2,3 % ingresaron por hipertermia. En nuestro estudio la causa más frecuente de hipertermia fue la fiebre de sed, cuya evolución fue favorable en la mayoría de los casos con un buen aporte lácteo. En un porcentaje de recién nacidos que debutaron por hipertermia, no se ha objetivado la causa de la misma catalogándose de síndrome febril no filiado (33,3 %). De todas las sepsis ingresadas en el bienio 2001-2002, sólo dos casos debutaron con fiebre lo que reafirma la escasa presentación de fiebre en las sepsis.
P177BLUEBERRY MUFFIN BABY COMO MANIFESTACION INICIAL DE INFECCION CONGÉNITA POR CITOMEGALOVIRUS
Laura Castells Vilella, Martín Morales Sánchez, M. Rosa Martorell Albareda, Paloma Araujo Salinas, Josep Quilis Esquerra, M. Consuelo Sánchez Garre, Ramón Vidal Sanahuja y Carlota Rovira Zurriaga
Hospital Mutua de Terrassa.
Introducción:El Citomegalovirus (CMV) sigue siendo la causa más frecuente de infección vírica congénita en todo el mundo. A pesar de que la mayoría de los neonatos infectados están asintomáticos, aproximadamente un 10 % presentan bajo peso al nacer, microcefalia, coriorretinitis, ictericia, hepatoesplenomegalia, rash cutáneo y ocasionalmente eritropoyesis dérmica o lesiones en "blueberry muffin".
Aportamos el caso de un neonato que presenta lesiones en "blueberry muffin" como manifestación inicial de una infección congénita sintomática por CMV.
Caso clínico:Recién nacido varón, de pocos minutos de vida, que presenta exantema petequial con máculo-pápulas violáceas. Fruto de una 1.ª gestación de 37 2/6 semanas, que cursó sin incidencias. Serologías maternas a lúes, HIV y HBsAg negativas, e inmune para Rubeola y Toxoplasma. Amniorrexis 26 horas antes del parto con líquido teñido de meconio. Fórceps con Apgar 6-8-8, pérdida del bienestar fetal y un peso de 2.570 g. A la exploración destaca un neonato hipotrófico e hipotónico, palidez cutánea con exantema petequial generalizado y lesiones maculares violáceas, distrés respiratorio leve e importante hepatoesplenomegalia. La analítica muestra neutropenia, trombopenia, hiperbilirrubinemia e hipertransaminasemia. En la biopsia de las lesiones cutáneas se observan focos de hematopoyesis extramedular. Ante la sospecha de infección congénita por CMV se practica estudio virológico que confirma la presencia del virus en sangre y orina. Se realiza ecografía cerebral que es normal, y abdominal que evidencia esplenomegalia de 7,5 cm. Actualmente presenta retraso psicomotor leve, sordera neurosensorial y hepatitis ictérica colestásica.
Comentarios:El "blueberry muffin baby" es un síndrome neonatal caracterizado por múltiples máculo-pápulas y nódulos violáceos cutáneos. Estas lesiones se asocian a infecciones prenatales, anemias crónicas severas y procesos neoplásicos. En la infección congénita por CMV, así como en la infección por rubéola, toxoplasma, y parvovirus B19, éstas representan focos de hematopoyesis extramedular que involucionan espontáneamente. Aunque no es una manifestación frecuente, su aparición debe hacernos pensar en estos diagnósticos y obliga a su confirmación por biopsia cutánea.
NEUROLOGIA
P178 LA CEFALEA COMO MOTIVO DE CONSULTA EN UN HOSPITAL DE AREA
Sonia Fernández Fernández, M.ª Llanos Carrasco Marina, Cristina Calvo Rey, María Vázquez López, Verónica Nebreda Pérez y Rafael Díaz Delgado
Hospital Severo Ochoa, Leganés.
Introducción:La cefalea es un motivo frecuente de consulta en pediatría debido a la gran preocupación que suscita en los padres, al considerarla como un síntoma de patología subyacente grave. Sin embargo, es sabido que la mayoría de las veces la causa es banal, y una meticulosa historia clínica y exploración física puede llevarnos al diagnóstico.
Objetivos:Establecemos un estudio descriptivo prospectivo de 42 casos de cefaleas con el objetivo de analizar el diagnóstico, tratamiento y evolución de las mismas.
Material y método:Revisión prospectiva de 42 pacientes con cefalea remitidos desde atención primaria para valoración en nuestro hospital.
Resultados:Revisamos prospectivamente 42 pacientes con edades comprendidas entre los 4 y los 13 años, siendo el 69 % mujeres y el 31 % varones; del total, el 85 % presentaba antecedentes familiares de cefalea y sólo el 7 % antecedentes personales de interés (convulsiones febriles o no, infecciones del sistema nervioso central, trumatismo craneoencefálico severo...
En más del 95 % de los casos, una cuidadosa historia clínica y exploración física permitieron llegar al diagnóstico. En la anamnesis se recogió un tiempo de evolución de los síntomas de más de 6 meses en un 59,5 %, con una frecuencia predominantemente semanal en el 52 % de los casos (diaria en el 40 %, mensual en el 7 % y progresiva en el 4,7 %). Se recogió también las características propias de las cefaleas (localización, duración, síntomas acompañantes, factores desencadenantes...), sólo el 2,3 % presentaba signos sugerentes de organicidad.
Se realizó pruebas de neuroimagen sólo en un 14 % de los casos, siendo todas normales, excepto un hallazgo casual de mega cisterna magna. Se clasificaron como jaqueca en un 40,4 %, tensional 35,7 %, mixta 23,8 % y otro tipo en un 5 %. El 14,2 % se trató con profilaxis antimigrañosa, psicoterapia en un 11,9 %, 2,3 % tratamiento antibiótico y el resto tratamiento analgésico intermitente.
Conclusión:Las cefaleas en su gran mayoría son posibles de diagnosticar y filiar por la historia clínica sin necesidad de más pruebas complementarias, por tanto, en muchos casos no será necesario remitir estos pacientes al hospital.
P179ACIDOSIS LACTICA CONGÉNITA COMO FORMA DE PRESENTACION DE ENFERMEDAD MITOCONDRIAL POR DÉFICIT DEL COMPLEJO III DE LA CADENA RESPIRATORIA
Enrique Villalobos Pinto, Juan José García Peñas, Gabriel Ángel Martos Moreno, M. Luz Ruiz-Falco de Rojas y Luis Gutiérrez Solana
Hospital del Niño Jesús, Madrid.
Introducción:Las acidosis lácticas congénitas (ALC) son un grupo heterogéneo de procesos que agrupan patologías con clínica y etiología variables.
Objetivos:Presentación de un caso de ALC por déficit del complejo III de la cadena respiratoria que debutó constatus convulsivo neonatal, analizando la evolución clínica y los hallazgos bioquímicos, neurofisiológicos y neurorradiológicos.
Caso clínico:Paciente de 23 días de vida que requiere ingreso en UCIP por un cuadro destatus convulsivo. Debut clínico con llanto intenso, rechazo de tomas y episodio de rigidez tónica generalizada, con revulsión ocular, con evolución posterior astatus refractario a la terapia habitual, requiriendo tratamiento con midazolán en perfusión a altas dosis (1,2 mg/kg/hora) y pentobarbital a 6 mg/kg/hora No existían antecedentes familiares de epilepsia. No hubo incidencias durante la gestación y no presentó evidencia de sufrimiento fetal agudo. El periodo neonatal inicial y las pruebas metabólicas fueron normales.
EEG:Patrón paroxismo-supresión persistente independiente del tratamiento anticomicial. TAC Craneal:normal. ECO Cerebral:Normal. Bioquímica:Acidosis metabólica severa con cetonuria y gran aumento de ácido láctico y pirúvico. Hipoglucemias ocasionales sin cetonemia.LCR: Discreta pleocitosis con proteínas en límites normales.Serología TORCH: Negativa.Estudio de metabolopatías congénitas:inicialmente descarta hiperglicinemia no cetósica, déficit de piruvato-carboxilasa, acidurias orgánicas y aminoacidopatías. Evolutivamente confirma patrón de acidosis láctica congénita.Complejos de cadena respiratoria en biopsia muscular:déficit del complejo III de la cadena respiratoria. Se inicia tratamiento con tiamina, piridoxina, carnitina, biotina y cianocobalamina desde el periodo agudo convulsivo. En su evolución presenta un desarrollo psicomotor adecuado y está libre de crisis tras dos años de seguimiento.
Conclusiones/comentarios: 1. Ante todostatus convulsivo de debut neonatal tardío se debe investigar una posible metabolopatía congénita e instaurar tratamiento empírico a la espera de los resultados.2. Las enfermedades mitocondriales por déficit de complejos de la cadena respiratoria son una patología heterogénea en su presentación clínica y evolución.
P180A PROPOSITO DE UN CASO DE SINDROME DE FREY
Santiago Gallego Vela, Jesús M.ª Garagorri Otero, M. Teresa Urgel Gómez, Pilar Arnauda Espatolero, Elena Lucas Sáez, M. Dolores Yecora Navarro y Jorge Pons García
Hospital Clínico Universitario Lozano Blesa, Zaragoza.
Introducción:El síndrome de Frey es una entidad clínica rara, originada por la lesión que se produce en la rama auriculotemporal del nervio trigémino, relacionada anatómicamente con la glándula parótida. En los niños, dicha rama se lesiona en el momento del parto mediante el uso de fórceps. La clínica viene definida por una mácula eritematosa recidivante en la zona malar, como consecuencia de una reacción parasimpática de la rama aberrante, desencadenada tras estímulos gustatorios. En la mayoría de los casos no requiere tratamiento.
Caso clínico:Niña de 6 meses que acude a la consulta de alergología infantil para estudio, por posible alergia alimentaria. Como antecedentes personales de interés: parto cefálico mediante fórceps. La niña presenta una mácula eritematosa de forma triangular, que va desde la comisura labial, hasta la región malar y pabellón auricular, que aparece súbitamente tras la ingesta de frutas (naranja, pera, manzana...), con unos 20 minutos de duración. La paciente no presenta otra sintomatología acompañante, ni rechazo alimentario. Las pruebas de reacción cutánea y anticuerpos específicos resultaron negativas. Se realizó una prueba de provocación con frutas y resultó positiva, evidenciándose la lesión. La niña acudió al tercer mes a la consulta y continuaba con la sintomatología. No recibió ningún tratamiento ni restricciones alimentarias.
Conclusiones:Se presenta un Síndrome de Frey, que si bien es una patología infrecuente, hay que tener en cuenta ante niños con sospecha de alergia alimentaria, que presenten un eritema facial recidivante y parto mediante fórceps. Su consideración evita la realización de numerosas y costosas pruebas complementarias.
P181SINDROME DE PRADER-WILLI POR DISOMIA UNIPARENTAL
Josefina Márquez Fernández, M. Inmaculada Correa Ruiz, M. Ángeles Carrasco Azcona, M. del Carmen Medina Gil, M. Ángeles Aguilera Llovet y Gabriel Cruz Guerrero
Hospital Universitario de Valme, Sevilla.
Introducción:El Síndrome de Prader-Willi (SPW) es un desorden multisistémico de origen genético que en el 28 % de los casos corresponde a disomía uniparental materna del cromosoma 15. Su incidencia es de 1/15.000 RN vivos. Asocia una grave hipotonía al nacimiento, retraso mental, baja estatura, obesidad, hipogonadismo hipotalámico y una apariencia característica.
Descripción de los casos:Presentamos dos niñas que al nacimiento tienen hopotonía, trastornos de succión y rasgos dismórficos. En la exploración destaca en los dos casos, talla baja con perímetro craneal en percentil mayor a la talla. Se practican exámenes radiológicos para descartar malformaciones cerebrales, abdominales y cromosomopatías siendo normales, salvo en el 2.º caso donde se aprecian riñones con menor tamaño del correspondiente. Asímismo, se realiza despistaje de errores innatos del metabolismo que resultan negativos.
Vistas en consulta de Neurología a los 8 y 6 meses respectivamente por persistir la hipotonía (aunque con evolución favorable), mostraban rasgos dismórficos inespecíficos y continuaban con talla en percentil bajo e hipotonía tipo suprasegmentaria. Se realizan estudios de enzimas musculares, AGCML, ecografía abdominal en el 2.º caso (normal) y estudio genético para SPW objetivándose alteración compatible con dicho síndrome por disomía uniparental en ambos casos.
Evolución:Ambas niñas están en la actualidad en tratamiento con hormona de crecimiento.
Conclusiones:1. Aunque su incidencia es baja, el SPW es una entidad a tener en cuenta, ya que el diagnóstico precoz (siguiendo los criterios establecidos) permite poner en marcha tratamientos que suponen un gran beneficio en la evolución y desarrollo de estos niños. Por lo tanto, en la actualidad, un RN con hipotonía suprasegmentaria y trastornos de succión debería ser indicación de estudio genético de Prader-Willi.2. Debemos tener en cuenta que en los casos de disomía uniparental las características fenotípicas se presentan de forma más tardía que en los casos de delección, por lo que la sospecha del SPW se hace más dificultosa.
P182ESCLEROSIS MULTIPLE SECUNDARIAMENTE PROGRESIVA DE PRESENTACION TEMPRANA
Óscar Rubio Cabezas, L. Ayala Bernaldo de Quirós, M. Luz Ruiz-Falco de Rojas, Juan José García Peñas y Luis González Gutiérrez-Solana
Hospital del Niño Jesús, Madrid.
Introducción:La esclerosis múltiple es una enfermedad rara en la infancia, que suele aparecer en niños mayores o adolescentes. Se presenta un caso de aparición temprana y evolución fatal.
Caso clínico:Niña de 2 años y 6 meses, previamente sana, que consultó en su hospital de origen por un cuadro de 2 semanas de evolución de pérdida de fuerza en miembro inferior izquierdo con afectación posterior de miembro inferior derecho. En el momento del ingreso presentaba diplejia fláccida y arrefléxica sin otros signos de afectación neurológica. Tras realizar estudios electrofisiológicos y análisis de LCR fue diagnosticada de síndrome de Guillain-Barré y tratada con gammaglobulina i.v., con mejoría inicial. Posteriormente presentó afectación de miembros superiores, evolucionando a tetraparesia fláccida, por lo que fue trasladada a la UCIP de nuestro hospital. La exploración neurológica en ese momento mostraba paresia facial central derecha, arreflexia en miembros superiores y miembro inferior derecho e hiperreflexia en miembro inferior izquierdo, sin afectación sensitiva aparente y con retención fecal y sudoración profusa, por lo que se descartó el diagnóstico inicial. En los días siguientes se añadieron al cuadro inicial datos de neuritis óptica y afectación troncoencefálica. Las pruebas de neuroimagen demostraron afectación desmielinizante a nivel encefálico y medular. Los estudios neurofisiológicos evidenciaron un patrón neurógeno con signos de afectación mieloradiculoneurítica. Fue tratada con inmunoglobulinas y corticoides i.v., con mejoría parcial. La paciente presentó numerosas reagudizaciones sin remisión completa entre ellas. El análisis del LCR mostró elevación de los niveles de IgG, albúmina y proteína básica de la mielina, con beta2-microglobulina normal. Tras el diagnóstico de esclerosis múltiple secundariamente progresiva, se realizaron varios ciclos de plasmaféresis con escasa mejoría. La afectación neurológica intercrítica fue progresivamente mayor y la paciente falleció 30 meses después del comienzo de los síntomas.
Conclusiones: 1.Debe considerarse el diagnóstico de esclerosis múltiple en toda enfermedad neurológica progresiva que asocie signos encefálicos y medulares.2. La edad no debe ser un criterio excluyente para el diagnóstico de las formas secundariamente progresivas de esclerosis múltiple.
P183TUMOR CEREBELOSO EN UNA NIÑA CON TRASTORNO GENERALIZADO DEL DESARROLLO
L. Ayala Bernaldo de Quirós, Óscar Rubio Cabezas, M. Luz Ruiz-Falco de Rojas, Juan José García Peñas y Luis González Gutiérrez-Solana
Hospital del Niño Jesús, Madrid.
Introducción:Se han descrito diversas alteraciones del cerebelo y fosa posterior en niños con trastorno generalizado del desarrollo (TGD). Presentamos un caso de TGD asociado a astrocitoma quístico de cerebelo, con mejoría significativa tras la extirpación del tumor.
Caso clínico:Niña de 21 meses, sin antecedentes familiares de interés, que consultó por falta de progresión en el desarrollo del lenguaje, alteración de la interacción social recíproca y ausencia de juego simbólico, más evidentes en los 3 meses previos. Embarazo, parto y período neonatal normales; desarrollo psicomotor normal durante el primer año. Tras realizar exploración psicológica y neuropediátrica (incluyendo EEG y cariotipo normales) fue diagnosticada de trastorno generalizado del desarrollo con rasgos autistas. A los 28 meses de vida, ante la progresión del TGD hacia un síndrome autista, se realizó RM craneal que puso de manifiesto un proceso expansivo en hemisferio cerebeloso izquierdo con afectación de la amígdala y el vermis y distorsión de la porción inferior del IV ventrículo. La lesión fue extirpada a los 30 meses de edad y el examen histológico mostró alteraciones características de astrocitoma fibrilar.
A partir de la intervención quirúrgica, se observó una mejoría progresiva de los rasgos autistas, aunque la paciente desarrolló posteriormente un TDAH, que está siendo tratado con risperidona debido a falta de respuesta a metilfenidato.
Conclusión:Las pruebas de neuroimagen, especialmente la RM craneal, deberían realizarse de forma sistemática en el estudio inicial de los niños con sospecha de trastorno generalizado del desarrollo.
P184NEUROCISTICERCOSIS COMO CAUSA DE EPILEPSIA EN PACIENTES INMIGRANTES
Cristina Ruiz Serrano, M.ª Verísima Barajas Sánchez, Miriam Blanco Rodríguez, M. Elena Fernández Villalba, M. Elena Zamora Flores, Ana Leal Orozco y Mercedes Bernacer Borja
Fundación Jiménez Díaz, Madrid.
Introducción:Una de las características diferenciales de nuestro servicio de Pediatría y de las áreas a las que atiende es la alta tasa de inmigrantes que acuden al servicio de Urgencias y que presentan patología hasta ahora poco frecuente en nuestro medio.
Caso 1:Paciente dominicano de 11 años que ingresa para estudio tras presentar episodio de desconexión del medio, con visión borrosa y disartria. Posteriormente, aparición de crisis tónico-clónicas de miembro superior derecho.
Exploración física:Glasgow 15/15. No rigidez de nuca. Signos meníngeos negativos. No focalidad neurológica. Resto exploración normal
Exploraciones complementarias: Hemograma:11.210 leucocitos (86 % N, 9,5 % L). EEG: actividad focal lenta de elevado voltaje en cuadrante posterior izquierdo. RM cerebral: lesión en surco parietal medio izquierdo con captación anular de contraste. IgG cisticercosis en suero débilmente positivo. Ag de cisticercosis en suero y LCR, IgG cisticercosis en LCR, AcEchinococcus granulosus en suero y LCR: negativos.
Caso 2:Paciente dominicano de 12 años que ingresa por presentar acúfenos y disartria, con posterior pérdida de consciencia y movimientos generalizados tónico-clónicos de segundos de duración. Episodios similares previos.
Exploración física:Glasgow 15/15. No focalidad neurológica. No rigidez de nuca. Signos meníngeos negativos. Resto normal.
Exploraciones complementarias: Hemograma: 7.000 leucocitos (60 % N, 32 % L). Serología cisticercosis en suero y LCR negativa. Mantoux negativo. EEG: asimetría interhemisférica con actividad lenta irregular difusa derecha. RM cerebral: lesión parietal derecha con captación anular de contraste y calcificaciones.
Ambos pacientes recibieron tratamiento con Albendazol, corticoides y fenitoína, con buena evolución clínico-radiológica y desaparición de las crisis convulsivas.
Conclusiones: 1.La causa más frecuente de epilepsia en los niños de origen hispano es la cisticercosis. 2. La enfermedad parasitaria cerebral más prevalente es la producida por la larva de la Tenia Solium.
P185SINDROME DE MILLER FISHER PEDIATRICO
Alfonso Rodríguez Albarrán, Olga González Calderón, Ana Gloria Andrés Andrés, Ricardo Torres Peral, Antonio Grande Benito, Emilio Nava García y Javier López Ávila
Hospital Universitario de Salamanca. Hosp. Clín. y Hosp. Virgen de la Vega.
Introducción:El Síndrome de Miller Fisher, también denominado síndrome de oftalmoplejía, ataxia y arreflexia, es un cuadro excepcional en la infancia. La mayor parte de los autores creen que es una variante del Síndrome de Guillain Barré. Suele presentar una disociación albúmina-citológica y la determinación de anticuerpos antigangliósidos GQ1b confirman el diagnóstico. El tratamiento se realiza mediante medidas de soporte y tratamiento farmacológico en función de la repercusión clínica. Produciéndose la recuperación clínica en unas 10 semanas.
Caso clínico:Niño de 5 años de edad, residente habitual en Alemania, que como antecedentes personales refiere una picadura por garrapata hace 6 meses, y una infección de vías respiratorias altas sin fiebre hace 8 días, tratada con un producto naturista (derivado del Pelargonium). Comienza con un cuadro de somnolencia, cefalea, inestabilidad y diplopía de unas 12-24 horas de evolución. A la exploración presenta: Glasgow 14, leve ptosis palpebral izquierda, midriasis bilateral normoreactiva, parálisis del III y VI par craneal, nistagmus moderado, hiporreflexia tendinosa de extremidades inferiores y una marcha atáxica con Romberg negativo.
Durante la evolución clínica se observa una persistencia de la diplopía y del estado de somnolencia. Además de un aumento de la ataxia, una pérdida del reflejo fotomotor, una ptosis palpebral bilateral y una arreflexia tendinosa de extremidades inferiores con flexión plantar normal.
Ante la clínica y la exploración física se inicia el estudio para descartar las distintas posibilidades diagnosticas: infeccioso, toxico, tumoral, vascular... objetivándose una disociación albúmina-citológica a las dos semanas de inicio del cuadro y un índice de Anti GQ1b por enzimoinmunoanálisis de 4,58 (normal < 1) que junto con la clínica nos confirma el diagnóstico de Síndrome de Miller-Fisher.
A partir de la primera semana comienza a mejorar desde el punto de vista neurológico. Se valoró la administración de inmunoglobulinas intravenosas pero debido a la necesidad de volver a su país de origen no se pudo llevar a cabo.
Conclusiones:Es importante tener presente este síndrome ante una cínica compatible y valorar la determinación de los anticuerpos Anti GQ1b al ser positivos en más del 90 % de los casos cuando se realizan en las primeras 4 semanas.
P186 DISECCION DE LA ARTERIA VERTEBRAL EN LA INFANCIA
Olga González Calderón, Alfonso Rodríguez Albarrán, Ana Gloria Andrés Andrés, Raquel Páez González, Antonio Grande Benito y Emilio Nava García
Hospital Universitario de Salamanca. Hosp. Clín. y Hosp. Virgen de la Vega.
Introducción:La disección de las arterias del territorio vertebrobasilar es una patología poco frecuente en pediatría. Pueden ser traumáticas o espontáneas y a su vez ser del trayecto intracraneal de la arteria, o ser extracraneales. El dolor es un síntoma prodrómico que aparece en más del 70 % de los casos. Clínicamente suele presentarse como un infarto cerebral.
Caso clínico:Niña de 14 años de edad con cefalea frontoccipital de 5 días de evolución, con tendencia al sueño progresiva y febrícula en las últimas 24 horas. A la exploración destaca, Glasgow 14 (O3, V5, M6), desorientación témporo-espacial, discreta hipotonía y una bradicardia de 48 l.p.m. Se inicia estudio, solicitándose las siguientes pruebas complementarias: analítica sanguinea, de LCR, de orina, fondo de ojo, EEG, serologías y estudios de imagen (TAC y RMN) que sugieren encefalitis de origen herpético.
Evolución:Se inicia tratamiento con Aciclovir y Dexametasona con aparente mejoría transitoria. Al tercer día se constata hemiparesia derecha con pérdida de sensibilidad ipsilateral, Babinsky positivo, disartria, esterotipias verbales y amnesia lacunar. Se realizan nuevas pruebas de imagen apreciándose lesiones sugerentes de isquemia multifocal dependiente de territorio vertebro-basilar izquierdo. Ante la sospecha de infarto cerebral se inicia tratamiento antiagregante con evolución favorable. A la semana reingresa con hemiparesia izquierda y disartria, constatándose disección selectiva de la arteria vertebral izquierda comenzándose tratamiento con dicumarínicos. Se produce una remisión clínica, descartándose coagulopatía de base, y a los tres meses se objetiva una repermeabilización de la arteria vertebral.
Conclusiones:La importancia radica en que un gran número de casos pueden no ser reconocidos ya que los síntomas prodrómicos son inespecíficos, máxime si no existe antecedente traumático previo. El pronóstico es bueno, aunque no es excepcional el fallecimiento. Estudios seriados arteriográficos, han demostrado que al menos el 85 % se resuelven o tienen una mejoría sustancial en 2 a 3 meses, siendo la recurrencia sintomática infrecuente.
P187IDEBENONA: LA ESPERANZA EN LA ATAXIA DE FRIEDREICH
Esther Álvarez García, M. Teresa de Benito Guerra, M. Ester Guerrero Vega, Myrian Macarena Ley Martos y Rocío Montiel Crespo
Hospital Puerta del Mar, Cádiz.
Enfermedad hereditaria AR producida por la mutación del gen codificador de la frataxina. Su ausencia provoca un aumento del hierro intramitocondrial originando la muerte de esta por la aparición de radicales libres. El sistema nervioso, corazón y páncreas son especialmente susceptibles a estos radicales. Es la ataxia hereditaria más frecuente (50 %) afectando a 1:50.000 personas siendo degenerativa mixta con afectación de cordones posteriores y cerebelo. Se manifiesta con alteraciones del movimiento intencional, del equilibrio y de la coordinación. Aparece entre los 5-15 años y la muerte sobreviene en los 1.º años de vida adulta generalmente por fallo cardíaco. Se está intentando la terapia con idebenona,antioxidante que frenaría la progresión de la enfermedad fundamentalmente a nivel cardíaco.Caso clínico: niña de 11 años enviada por temblor postural que aumenta con los movimientos voluntarios. Se inicia a los 5 años y progresa hasta hacerse más evidente en los últimos 2 meses. El temblor no fluctúa a lo largo del día y aparece en todas las actividades cotidianas. AF y AP sin interés.Exploración: Cifoescoliosis. Pies cavosbilaterales. Fuerza, pares craneales y pupilas normales. Dismetría de miembros.
Superiores e inferiores. Temblor postural que aumenta con los movimientos voluntarios incluyendo cuello y cabeza. Marcha ataxica que aumenta al cerrar los ojos. RMP abolidos en miembros inferiores. Sensibilidad protopática alterada. Ataxia de la mirada.Ex. complementarios: glucemias normales. EEG y RMN craneal: normal. RMN columna:pérdida de volumen del segmento extendido desde bulbo caudal a C2. Rx raquis: escoliosis de convexidad izquierda. Ecocardio normal. EMG y ENG: polineuropatia sensitiva. Estudio genético compatible.
Evolución:A los 4 meses del diagnóstico y del inicio de rehabilitación,realizamos escala de valoración de ataxia (International Cooperative ataxia rating scale: World Federation of Neurology) previo al tratamiento con Idebenona. Se revisa a los 6 meses encontrándose estabilizada y sin efectos secundarios.
Conclusiones:Dado el pronóstico de la enfermedad, aunque no existan suficientes estudios que demuestren la eficacia del tratamiento, hay que intentarlo ya que no parece perjudicial y no existe alternativa mejor. No hay que olvidar las alteraciones cardíacas, ya que aunque tardías, son la principal causa de muerte, encontrandose mejorías a este nivel en casos de inicio precoz del tratamiento.
P188HIPEREKPLEXIA NEONATAL PRECOZ
Marta Ruiz Jiménez, M. Teresa Cortés Coto, Javier Martínez Sarries, Beatriz Martínez Menéndez, Ana Pilar Galera Peinado, María Fernández Ibieta, Lucía Cabanillas Villaplana, Marta Muro Brussi, Esther Iglesias González-Nicolás y Ernesto Antonio Sáez Pérez
Hospital Universitario de Getafe.
Introducción:La hiperekplexia o enfermedad del sobresalto, es una enfermedad rara, caracterizada por reacciones de sobresalto exagerados frente a estímulos mínimos auditivos, visuales y táctiles. Puede cursar con hipertonía, y mioclonías durante el sueño. Puede manifestarse ya en el periodo fetal como movimientos intrauterinos anormales o más tarde desde el periodo neonatal al adulto. Los espasmos tónicos producidos por estímulos pueden conducir a apnea y muerte súbita durante la época de lactante. Suele ser familiar, generalmente autosómica dominante, aunque existen casos esporádicos.
Caso clínico:Varón de 2 días que ingresa por presentar desde las 24 h de vida, tremulaciones en miembros superiores y barbilla, en relación con estímulos, presentando a las 48 h de vida un episodio aislado de mioclonías de miembros superiores. No antecedentes perinatales de interés. Antecedentes familiares: Madre sobresaltos frecuentes. Exploración física: Mioclonías segmentarías, focales y generalizadas desencadenadas por estímulos auditivos, y en menor grado por estímulos táctiles; no disminuyen e incluso aumentan con estímulos repetidos. Reflejos y moro simétrico hiperactivo. Tono muscular normal. Pruebas complementarias: normales.
Evolución:Tras la sospecha clínica de hiperekplexia se inicia tratamiento con clonacepam, observándose buena respuesta clínica. Monitorización domiciliaria. En la actualidad el paciente tiene 14 meses, sigue tratamiento con clonacepam en pauta descendente. Evolución favorable con sobresaltos mínimos ante estímulos intensos y sin repercusión en la marcha. Desarrollo psicomotor normal.
Conclusiones:1. La hiperekplexia, aunque es una enfermedad rara, debe tenerse en presente en el diagnóstico diferencial de tremulaciones, hipertonía y trastornos epilépticos del neonato.2. El diagnóstico de la enfermedad es clínico, siendo las pruebas complementarias normales. El espasmo flexor generalizado en respuesta a estímulos en el puente nasal se considera marcador clínico de la enfermedad. 3. El tratamiento de elección es el clonacepan.4. Estos pacientes deben ser monitorizados durante el 1.º año de vida por la posibilidad de pausas de apnea provocadas por la hipertonía. La flexión de cabeza y piernas hacia el tronco logra muchas veces que ceda la crisis de apnea al producir la relajación del enfermo.
P189NEUROPATIA BRAQUIAL IZQUIERDA POSTINFECCIOSA. SINDROME DE PARSONAGE TURNER
Raquel Amo Rodríguez, Sonia Vilanova Fernández, M. de los Ángeles Ruiz Gómez, Gustavo Pico Fuster, M. Antonia Grimalt Calatayud y Juana M. Román Piñana
Hospital Son Dureta, Palma de Mallorca.
Introducción:La plexopatía braquial aguda en la infancia es más rara que en el adulto. Puede estar precedida por una infección (más frecuentemente respiratoria) o una inmunización. El inicio de la clínica es agudo, se acompaña de dolor del miembro afecto, seguido de la impotencia funcional y debilidad de éste.
Objetivos:Presentación de un caso clínico de paresia braquial izquierda de origen postinfeccioso. Se comentan los hallazgos clínicos y neuroradiológicos asociados, diagnóstico diferencial, tratamiento y evolución.
Caso clínico:Lactante de 6 meses que tras 5 días de presentar un proceso infeccioso febril con catarro de vías altas, gastroenteritis aguda, presenta de forma súbita, al despertarse, parálisis braquial izquierda, con dolor a la movilización (sin antecedente traumático). A la exploración se evidencia postura asimétrica con brazo izquierdo en extensión y pronación interna, déficit de supinación de la mano, brazo-antebrazo y ausencia de flexión palmar, reflejos osteotendinosos del brazo afecto débiles. Resto de la exploración clínica y neurológica normal. Antecedentes sin interés. Pruebas complementarias: electromiograma: lesión postganglionar de predominio en hombro izquierdo, RMN medular: imagen hidromiélica lateral en C3-C6 en asta anterior de médula, punción lumbar: normal, serología: coxackie B6 y CMV IgM positivos, coprocultivo: positivo a campylobacter jejuni, estudio metabólico: normal, estudio inmunológico: ligera disminución de L T CD4.
Diagnóstico diferencial: traumatismo, procesos expansivos de columna cervical, trastornos inmunológicos,... Tratamiento: corticoides, antibióticos, antivíricos y rehabilitación. Evolución clínica y neuroradiológica favorable.
Comentarios: 1.Hallazgo en nuestro caso concomitante de lesión hidromiélica medular cervical sin aparente relación con la afectación neurológica.2. Ante una paresia braquial aguda en un lactante hay que descartar una etiología post o parainfecciosa.3. La RMN es una prueba esencial para el estudio y el diagnóstico de las posibles entidades etiológicas de una paresia braquial aguda. 4. El EMG es necesario para evaluar la extensión de la lesión.
NUTRICION
P190 EVOLUÇÃO DA CONDIÇÃO NUTRICIONAL DE CRIANÇAS OBESAS EM PROGRAMA DE ATUAÇÃO DE EQUIPE INTERPROFISSIONAL
Ana L.G. Demarchi, Arnaldo V. Silva, Christianne F.L. Nascimento, Custódia V. de N. Mäder, Daniella M. Mazzaferro, Kazue Sato, Lilian C.Q. Mattoso, Vanda M. Falcone, Roseli S. Sarni y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil), Programa Einstein de Nutrição na Comunidade de Paraisópolis, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Introdução:A obesidade apresenta elevada prevalência na população mundial e tem ascendido rapidamente em populações carentes. Esta doença de múltiplas repercussões é hoje problema de saúde pública, com necessidade de diagnóstico e intervenção precoces. O Programa Einstein de Nutrição na Comunidade (PENC), desenvolve semanalmente, com equipe interprofissional, atendimentos grupais e individuais, com crianças obesas e suas famílias. Objetivo: avaliar a evolução da condição nutricional de crianças obesas submetidas ao programa com atuação de equipe interprofissional. Casuística: 106 crianças com excesso de peso na faixa etária de 0 a 10 anos (40 % do sexo masculino) receberam intervenção interprofissional: reeducação alimentar, na cozinha de aprimoramento, com participação das mães, orientadas por nutricionistas e atuação psicopedagógica, sob supervisão de psicóloga e pedagoga (de janeiro de 2001 a novembro de 2002).
Método:A avaliação foi realizada na admissão ao programa e 9 meses após a intervenção. Definiu-se obesidade segundo a relação P/E > a 120 % e sobrepeso com relação P/E entre 110 e 120 %. Intervenção: reeducação alimentar na cozinha de aprimoramento, com importante participação das mães orientadas por nutricionistas, atividade física 2 vezes por semana, orientada por professor de educação física. Atuação psicopedagógica na dinâmica familiar com supervisão de psicóloga e pedagoga. Acompanhamento médico mensal.
Resultados:A evolução foi satisfatória em 49 crianças (46,2 %): 19 (38,8 %) passaram de formas graves de obesidade para formas moderadas e leves; 19 (38,8 %) foram de obesidade para sobrepeso; 7 (14,3 %) de obesidade para eutrofia e 4 (8,1 %) de sobrepeso para eutrofia. As 57 crianças restantes não conseguiram acompanhar o programa.
Conclusão:A intervenção interprofissional propiciou expressiva melhora da condição nutricional, em função principalmente da reeducação alimentar, com efetiva participação das mães na cozinha de aprimoramento, e da atuação junto à família; daí o melhor resultado quando comparado à literatura.
P191AVALIAÇÃO E EVOLUÇÃO DA ANEMIA FERROPRIVA EM CRIANÇAS OBESAS
Ana L.G. Demarchi, Christianne F.L. Nascimento, Custódia V. de N. Mäder, Daniella M. Mazzaferro, Fernanda Oliveira, Kazue Sato, Lilian C.Q. Mattoso, Vanda M. Falcone y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil), Programa Einstein de Nutrição na Comunidade de Paraisópolis, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Introdução:A obesidade infantil presente nas populações de nível sócio econômico baixo é frequentemente acompanhada de carência de micronutrientes. O Programa Einstein de Nutrição na Comunidade (PENC), desenvolve semanalmente, com equipe interprofissional, atividades grupais e individuais com crianças obesas e suas famílias. Objetivo: avaliar os níveis de hemoglobina e depósitos de ferro em crianças obesas na admissão ao programa e ao final do tratamento. Casuística: foram avaliadas 86 crianças de 0 a 10 anos, atendidas pelo PENC, no período de janeiro de 2001 a novembro de 2002. Metodologia: hemograma (contagem automática), ferritina (quimioluminescência) e ferro sérico (química seca). Foi diagnosticada anemia ferropriva em crianças com hemoglobina abaixo dos limites estabelecidos pela OMS, ferritina abaixo de 12 ng/dl; RDW maior que 14,5 % e ferro sérico abaixo de 40 μg/dl. Intervenção medicamentosa: as crianças com anemia ferropriva receberam 4 mg/kg/dia de ferro elementar sobre a forma de sulfato ferroso com período médio de 3 meses; e as crianças com ferropenia receberam 2 mg/kg/dia de ferro elementar por período médio de 2 meses. Intervenção educativa: orientação teórica e prática na alimentação visando a utilização de alimentos ricos em ferro, com boa disponibilidade.
Resultados:21 crianças (24,4 %) com anemia, sendo 85,7 % menores de dois anos e 4 (4,6 %) com diminuição dos depósitos de ferro. Antes da intervenção constatou-se que todas as crianças recebiam alimentação com excesso de alimentos lácteos não enriquecidos e pobres em ferro. Após 3 meses de tratamento, todas as crianças normalizaram os parâmetros laboratoriais.
Conclusão:A reeducação alimentar, com efetivas mudanças no hábito alimentar das famílias, teve importância fundamental nos resultados obtidos. Destaque-se que as atividades das mães na cozinha de aprimoramento, foram um dos grandes diferenciais na obtenção dos bons resultados conseguidos, além da adesão ao tratamento medicamentoso.
P192 TRATAMENTO DE CRIANÇAS OBESAS: EVOLUÇÃO DO PERFIL LIPIDICO E DE REPERCUSSÕES CLINICO LABORATORIAIS
Ana L.G. Demarchi, Arnaldo V. Vicente, Christianne F.L. Nascimento, Custódia V. de N. Mäder, Daniella M. Mazzaferro, Kazue Sato, Lilian C.Q. Mattoso, María Arlete Escrivão, Vanda M. Falcone y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil), Programa Einstein de Nutrição na Comunidade de Paraisópolis, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Introdução:O Programa Einstein na Comunidade de Paraisópolis (PECP) atua na segunda maior favela de São Paulo/Brasil. O Programa Einstein de Nutrição na Comunidade (PENC), atende crianças obesas por meio de equipe interprofissional, atuando junto à criança e sua família com atividades grupais e individuais.
Objetivos:Avaliar a evolução do perfil lipídico e de algumas repercussões clínico laboratoriais.
Casuística:Foram avaliadas 93 crianças de 0 a 10 anos atendidas no PENC no período de janeiro de 2001 a novembro de 2002.
Método:Considerou-se obesidade a relação P/E ≥ 120 % e dislipidemia de acordo com a American Heart Association (1986).Análises laboratoriais: lipidograma por eletroforese; glicemia por química seca e insulina por enzima imuno ensaio.Intervenção: reeducação alimentar com redução da ingestão das gorduras saturadas e de alimentos ricos em colesterol; atividade física regular, no mínimo duas vezes por semana e atividades psicopedagógicas com os pacientes e seus familiares.
Resultados:Na avaliação inicial do perfil lipídico: dislipidemia em 60 (64,5 %), predominando na faixa de 2 a 3 anos, assim distribuídas: aumento de colesterol total em 20 crianças (21,5 %), LDL em 13 (13,9 %); triglicérides em 36 (38,7 %); e redução do HDL em 27 (27,9 %) De 86 crianças que fizeram insulina basal e curva de resistência à insulina 7 (8,15 %) apresentaram alterações, levando ao diagnóstico clínico presuntivo de hiperinsulinemia. Em 10 crianças (13,3 %) foi diagnosticada acantose nigricans, que se manteve durante o acompanhamento. Hipertensão arterial foi encontrada em 8 crianças (10,6 %). Com o tratamento 47 crianças (78,3 %) normalizaram os lipídios séricos. Em todos os casos de hipertensão ocorreu normalização dos níveis pressóricos, sendo que terapia medicamentosa foi necessária em 1 paciente. Houve normalização da insulina e da curva de resistência em 6 crianças (85,7 %).
Conclusão:Os resultados obtidos indicam que a abordagem da obesidade por equipe interprofissional abrangendo a criança e sua família, com atividades psicopedagógicas, semanais grupais, aliadas às atividades práticas de reeducação alimentar e promoção de hábitos alimentares saudáveis promovidas na cozinha de aprimoramento para as mães e crianças, parece ser o melhor método para o tratamento desta patologia complexa de etiologia multifatorial.
P193 CARACTERISTICAS PSICOLOGICAS DE MÃES DE CRIANÇAS DESNUTRIDAS E A RELAÇÃO COM O VINCULO MÃE/FILHO
Christianne F.L. Nascimento, Custódia V. de N. Mäder, María Arlete Escrivão, Patricia V. Spada, Vanda M. Falcone y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil), Programa Einstein de Nutrição na Comunidade de Paraisópolis, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
A desnutrição é considerada distúrbio multifatorial, relacionado não só com a escassez de recursos econômicos, mas com alimentação inadequada, condições socioculturais e psicológicas e, que costumam acompanhar a pobreza. A literatura ressalta que os aspectos psicológicos maternos e o vínculo mãe/filho são importantes fatores de risco para a desnutrição. O Programa Einstein de Nutrição na Comunidade de Paraisópolis (segunda maior favela de São Paulo, Brasil, com 50.000 habitantes), faz parte do Programa Einstein na Comunidade e atende crianças desnutridas de baixo nível sócio-econômico. O objetivo deste estudo foi avaliar as características psicológicas de mães de desnutridos com idade entre 0 e 3 anos e sua relação com o vínculo mãe/filho. As mães foram avaliadas segundo a presença de transtornos mentais (Self Reporting Questionnaire, SRQ), a presença de depressão (Beck Depression Inventory) e o nível intelectual (Escala Geral de Raven). Quanto ao vínculo, utilizou-se protocolo específico construído por nossa equipe. Estes instrumentos foram aplicados ao ingresso do programa. Para a análise estatística foi utilizado o teste Qui-quadrado de Mantel-Haenszel e teste Exato de Fisher, aceitando-se como significante p < 0,05. Foram avaliadas 59 mães, verificando-se presença de distúrbio mental em 39,0 % (23), presença de depressão em 22,0 % (13) e nível intelectual rebaixado em 50,9 % (30). O vínculo mãe/filho mostrou-se ruim em 69,5 % (41). Houve associação significante entre nível intelectual rebaixado e baixa escolaridade materna (p = 0,0367). O vínculo mãe/filho ruim mostrou diferença estatisticamente significante em mães com menos de 30 anos (p = 0,0057). A presença de depressão mostrou-se associada à idade materna maior ou igual a 30 anos (p = 0,0259). Verificou-se associação significante entre depressão e distúrbios mentais (p = 0,0120), bem como entre depressão e vínculo mãe/filho ruim (p = 0,0044). Não houve associação significante entre as demais variáveis analisadas: escolaridade e idade maternas, número de filhos com as características psicológicas e vínculo mãe/filho.
Conclusão:A presença de depressão associada a vínculo mãe/filho ruim, que prejudica a capacidade materna para cuidar da criança desnutrida, justifica a necessidade de intervenção na área psíquica no tratamento da desnutrição.
P194AVALIAÇÃO DA SAUDE MENTAL DE GESTANTES
Custódia V. de N. Mäder, Christianne F.L. Nascimento, María Arlete Escrivão, Patricia V. Spada, Vanda M. Falcone y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Estudos sugerem que ansiedade, estresse e depressão, durante a gestação, podem provocar complicações neste período, no parto e pós-parto. Podem também prejudicar a relação mãe-bebê e futuramente o desenvolvimento da criança. Objetivo: identificar e intervir com equipe interdisciplinar nos transtornos mentais presentes em gestantes.
Métodos:Estudamos 80 gestantes de baixo nível sócio-econômico (49 adultas e 31 adolescentes) atendidas no Programa Einstein na Comunidade de Paraisópolis, segunda maior favela de São Paulo, Brasil, com 50.000 habitantes. Ao ingressar no programa, as gestantes foram atendidas individualmente por psicóloga ou pedagoga treinadas para avaliação da saúde mental, que se repetia no momento da saída. Foram utilizados os instrumentos: Self Reporting Questionnaire SRQ (presença de transtornos mentais) e Beck Depression Inventory (presença de depressão). Foram realizados dez encontros semanais, com duas horas de duração, divididas da seguinte forma: 40 minutos com abordagem no vínculo mãe/feto, com propostas de: relaxamento, auto-massagem, cantigas de ninar, massagem em bebê, dinâmicas para trabalhar sentimentos e dúvidas que emergem na gestação/parto e relação entre mãe/concepto e mãe/concepto/pai, sob a coordenação de psicóloga e pedagoga. A seguir, 60 minutos para abordar os temas: mudanças no corpo, cidadania, alimentação da gestante, aleitamento materno, parto e cuidados no pós parto, maternidade e paternidade, cuidados com o recém-nascido, sob a coordenação dos membros da equipe interdisciplinar e 40 minutos para esclarecimentos de dúvidas com a enfermagem. Para comparação da saúde mental antes e após intervenção usou-se o teste do x2, aceitando-se como significante p < 0,05.
Resultados:1. Avaliação do SRQ antes da intervenção: sem distúrbios 46 e com distúrbios 62 e após: 34 sem distúrbios e com distúrbios 18; x2 = 7,29 e p = 0,007;2. Avaliação do Beck antes da intervenção: sem depressão 63 e com depressão 17 e após: 71 sem depressão e com depressão 9; x2 = 2,94 e p = 0,086.
Conclusão:A freqüência de transtornos mentais e depressão foi expressiva nessa população. O SRQ se mostrou efetivo na identificação de melhora dos transtornos mentais pós-intervenção interdisciplinar em gestantes de baixo nível sócio-econômico (p = 0,007). Com relação a depressão o resultado mostra tendência a melhoria, sem entretanto ser significante (p = 0,086).
P195AVALIAÇÃO DO VINCULO MATERNO-FETAL
Custódia V. de N. Mäder, Christianne F.L. Nascimento, María Arlete Escrivão, Patricia V. Spada, Vanda M. Falcone y Fernando José de Nóbrega
Centro de Promoção e Atenção à Saúde, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
A relação da mãe com seu filho começa na gestação, baseada em elementos de realidade e de fantasias da mulher, ocorridos antes e durante o processo de desenvolvimento fetal. Este relacionamento, descrito como apego/vínculo materno ao feto, tem sido considerado atributo para o sucesso do desenvolvimento da criança.
Objetivos:Conhecer e fortalecer o Vínculo materno/fetal (VMF).
Métodos:Elaborou-se instrumento para VMF que consiste em 5 itens:1. Toma os cuidados necessários com alimentação, vitaminas, pré-natal.2. Sente-se feliz com a gravidez.3. Demonstra afetividade ao feto.4. Consegue perceber os movimentos fetais.5. Sente-se muito angustiada e com dúvidas em relação a gravidez. Estabeleceu-se escore em função do conhecimento das questões acima, sendo que cada sim dos itens 1 a 4 vale um ponto, no item 5, 1 ponto a resposta negativa. Escore, até 3 pontos é considerado ruim e acima de 3, bom vínculo mãe/feto. Participaram do atendimento, 80 gestantes (adultas e adolescentes), atendidas no Programa Einstein na Comunidade, da favela de Paraisópolis (50 mil habitantes), São Paulo, Brasil. Foram realizados dez encontros semanais, com as seguintes propostas: relaxamento/auto-massagem/cantigas de ninar/massagem em bebê/dinâmicas para trabalhar sentimentos e dúvidas que emergem na gestação e relação entre mãe/concepto e mãe/concepto/pai, além de exposição de temas técnicos: alimentação na gestação/aspectos psicológicos presentes no parto e pós parto/aleitamento materno/cuidados básicos com o bebê/gravidez e modificações no organismo materno. As gestantes eram avaliadas na matrícula e após o tratamento. Para a comparação dos resultados à entrada e à saída, usou-se o teste do x2, aceitando-se como significante p < 0,05.
Resultados:Avaliação do vínculo materno-fetal Adultas à entrada: bom 26, ruim 23 e na alta: bom 46, ruim 3; (x2 = 20,94; p = 0,0000047). Adolescentes à entrada: bom 10, ruim 21, na alta: bom 31, ruim 0; (x2 = 31,76; p = 0,0000000). Total de gestantes adultas e adolescentes: à entrada: bom 36, ruim 44, na alta: bom 77, ruim 3; (x2 = 50,64; p = 0,0000000).
Conclusão:A atuação desenvolvida pela equipe interprofissional, conseguiu de forma altamente significativa, bom vínculo materno/fetal.
P196EFEITOS DA DESNUTRIÇÃO NO DESENVOLVIMENTO NEUROPSICOLOGICO
Mônica C. Miranda, Elaine G. Sinnes, Kazue Sato, Marcel Taga, Orlando F.A. Bueno y Fernando José de Nóbrega
Universidad Federal de São Paulo, Brasil, Centro de Promoção e Atenção à Saúde, São Paulo (Brasil) y Instituto de Ensino e Pesquisa do Hospital Albert Einstein, São Paulo (Brasil).
Introdução e objetivos:O presente estudo investigou os efeitos da desnutrição no desenvolvimento de funções cognitivas que ainda não estão bem estabelecidas em diversos estudos, como as funções relacionadas ao cór

