


La porfiria aguda intermitente ([PAI] AIP por sus siglas en inglés; MIM# 176000) es una enfermedad genética autosómica dominante, con penetrancia incompleta (10-15%) y expresividad variable. Se debe a la deficiencia parcial de la enzima hidroximetilbilano sintetasa (HMBS) por mutaciones en el gen HMBS. La prevalencia reportada recientemente en España es de 6,3 casos/millón de habitantes1, pero en la Región de Murcia se estima superior debido al efecto fundador de la mutación c.669_698del2.
Se manifiesta clásicamente como crisis intermitentes de dolor abdominal acompañado de otros síntomas gastrointestinales, cardiovasculares, neurológicos y/o psiquiátricos, y se conocen múltiples factores desencadenantes como ayuno prolongado, ingesta de alcohol o consumo de determinados fármacos (tabla 1).
Fármacos precipitantes de crisis
| Aminoglutetimida | Barbitúricos |
| Carbamacepina | Cloranfenicol |
| Clemastina | Clonidina |
| Cotrimoxazol | Danazol |
| Dapsona | Dihidralazina |
| Dimenhidrinato | Dipirona |
| Derivados ergóticos | Eritromicina |
| Etamsilato | Etosuximida |
| Etomidato | Griseofulvina |
| Ketoconazol | Meprobamato |
| Metildopa | Sulfonamidas |
| Metisergida | Ácido nalidíxico |
| Orfenadrina | Oxcarbazepina |
| Tolbutamida | Fenilbutazona |
| Fenitoína | Pirimidona |
| Pirazolona | Pirazinamida |
La prevalencia de la PAI en la infancia se desconoce, los casos sintomáticos son excepcionales, y la sintomatología descrita suele ser muy variable. Solo hay un estudio prospectivo en población pediátrica sueca (Hultdin et al., 20033) que incluye 61 pacientes menores de 18 años, de los cuales el 10% presentó crisis (todos, dolor abdominal leve).
Aunque el diagnóstico en el niño es todo un reto, se debe sospechar en casos con sintomatología inespecífica gastrointestinal, neurológica y/o psiquiátrica, y debería incluirse en el diagnóstico diferencial del dolor abdominal de causa desconocida. Ante su sospecha es de elección como cribado el test de Hoesch (detección cualitativa de PBG en orina), cada vez más utilizado en España, gracias a la puesta en marcha del proyecto PAGORA (Protocolo de cribado de porfiria aguda en pacientes con dolor abdominal no filiado en urgencias) y, posteriormente, el estudio enzimático y/o molecular específico. El tratamiento se basa en evitar factores desencadenantes, tratar los síntomas y aumentar la ingesta de hidratos de carbono en casos leves-moderados, en los graves la administración de hemina en forma de arginato ha demostrado ser eficaz4, y recientemente se ha publicado una nota (Andreeva et al., 20145) sobre un proyecto de investigación acerca del uso de terapia génica en este grupo de pacientes.
Se realiza una revisión retrospectiva de historias clínicas de pacientes menores de 18 años con diagnóstico molecular de la PAI tras el análisis por antecedentes familiares positivos (progenitor afectado) en seguimiento por la sección de genética médica. Análisis de prevalencia, fenotipo y genotipo de estos pacientes.
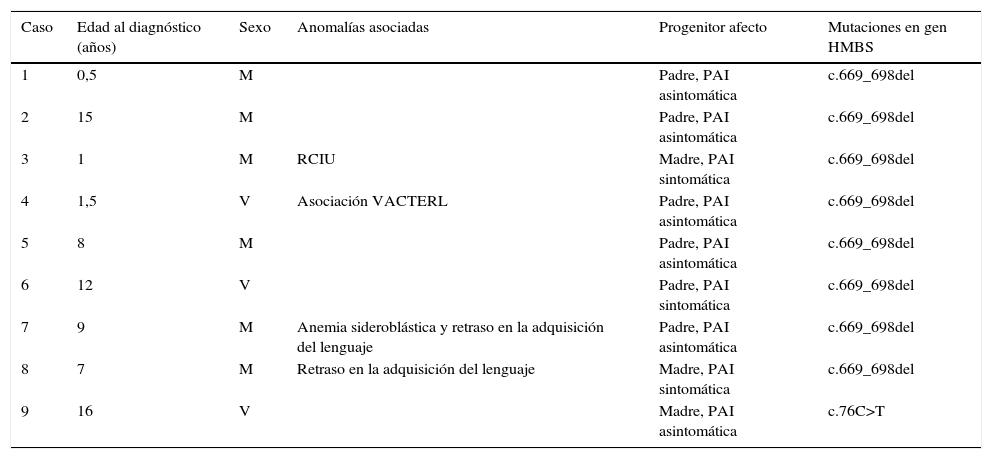
Describimos 9 casos, con edad media al diagnóstico de 7 años (tabla 2). La prevalencia es de 28 casos/millón de habitantes (población total ≤18 años en Murcia, 320.698). El periodo medio de seguimiento tras el diagnóstico fue de 23 meses (rango: 3-40 meses), y durante el mismo no se registraron crisis documentadas en ningún paciente. El caso 9 a los 2 años de edad, previo diagnóstico molecular de PAI, presentó episodio de dolor abdominal moderado por lo que fue ingresado y se diagnosticó de etiología desconocida (no se determinó PBG urinario para valorar una posible crisis).
Resumen de datos epidemiológicos, fenotipo y genotipo de los casos
| Caso | Edad al diagnóstico (años) | Sexo | Anomalías asociadas | Progenitor afecto | Mutaciones en gen HMBS |
|---|---|---|---|---|---|
| 1 | 0,5 | M | Padre, PAI asintomática | c.669_698del | |
| 2 | 15 | M | Padre, PAI asintomática | c.669_698del | |
| 3 | 1 | M | RCIU | Madre, PAI sintomática | c.669_698del |
| 4 | 1,5 | V | Asociación VACTERL | Padre, PAI asintomática | c.669_698del |
| 5 | 8 | M | Padre, PAI asintomática | c.669_698del | |
| 6 | 12 | V | Padre, PAI sintomática | c.669_698del | |
| 7 | 9 | M | Anemia sideroblástica y retraso en la adquisición del lenguaje | Padre, PAI asintomática | c.669_698del |
| 8 | 7 | M | Retraso en la adquisición del lenguaje | Madre, PAI sintomática | c.669_698del |
| 9 | 16 | V | Madre, PAI asintomática | c.76C>T |
En 4 casos (45%) se detectaron anomalías asociadas. El caso 7 y 8 presentaron retraso en la adquisición del lenguaje. El caso 7 padece además, anemia sideroblástica debida a mutación c.683G>T en gen SLC25A38 en homocigosis. El caso 5 anomalías congénitas múltiples compatibles con asociación VACTERL (atresia anorrectal con fístula rectovesical, hemivértebra lumbar, vena cava superior que drena a seno coronario, riñón derecho multiquístico y reflujo vesicoureteral izquierdo grado V).
El caso 4 (antecedente de CIR, peso al nacimiento 1.630g; p<1) presenta retraso pondoestatural significativo a los 3 años (peso y talla en percentil [p] <1).
El diagnóstico de la madre del caso 4 se realizó durante la gestación. Presentó crisis aguda durante la misma y precisó administración de hematina intravenosa. El parto fue mediante cesárea urgente por crecimiento intrauterino retardado (CIR) y preeclampsia materna en semana 36 de gestación.
Como conclusiones, los resultados de nuestro estudio apoyan la hipótesis de que en nuestra región, debido al efecto fundador de la mutación c.669_698del, la prevalencia de PAI en la población pediátrica es alta: 28 casos/millón de habitantes. Todos los casos han permanecido asintomáticos al diagnóstico y durante su seguimiento (crisis dudosa en el caso 9), lo que puede explicarse por la corta edad media de la población estudiada (7 años). Describimos una alta tasa de anomalías asociadas (retraso del lenguaje, anomalías congénitas múltiples, etc.), no descrita previamente, por lo que sería necesario realizar estudios de seguimiento en muestras más amplias. La aparición de crisis durante la gestación podría ser factor riesgo de CIR en el RN y de hipertensión materna, pero existe controversia al respecto6.
El diagnóstico precoz es fundamental al permitir adoptar medidas para evitar los factores desencadenantes de crisis, abordar los problemas crónicos que asocia la enfermedad y realizar un adecuado asesoramiento familiar.
A los pacientes y familiares por su colaboración. Este estudio ha sido financiado en parte por la UCAM-Universidad Católica de Murcia (PMAFI/09/14) y M. Barreda-Sánchez es una estudiante predoctoral becada por esta misma universidad.

