

La poliquistosis renal autosómica recesiva (PQRAR) tiene gran relevancia en pediatría dada su gravedad y morbimortalidad precoz. La variabilidad fenotípica de esta enfermedad es amplia, la mayor parte de los pacientes se trata de casos de novo, y la correlación exacta entre genotipo y fenotipo aún no ha sido establecida1. Todo esto genera en las familias gran angustia acerca del pronóstico de sus hijos y una demanda de consejo genético.
El objetivo de nuestro trabajo consiste en describir retrospectivamente una cohorte de pacientes pediátricos con PQRAR tratados en un único centro a lo largo de 25 años. Se pretende además establecer asociaciones entre las distintas manifestaciones y el pronóstico a largo plazo.
Se identificaron retrospectivamente los pacientes con diagnóstico de PQRAR seguidos por el servicio de nefrología pediátrica entre 01/1991-12/2016. Se incluyeron 16 pacientes (12 varones y 4 mujeres) con una mediana de edad en el momento del estudio de 16,5 años. El 62% fueron diagnosticados de forma prenatal mediante ecografía. Entre los criterios ecográficos de la enfermedad, el aumento del tamaño renal fue la característica más frecuentemente descrita, en un 75% de casos, seguido de la presencia de quistes renales visibles que se observó en el 72%.
Se realizó estudio genético en 2 pacientes, uno de ellos presentó la mutación R1804fs del gen PKHD1 y otro una mutación en doble heterocigosis en PKHD1: NM_138694.3: c.3350_3351delTA NP_619639: p.I1117Kfs*7 / NM_138694.3: c.3765_3766delinsG NP_619639: p.Q1256Rfs*47.
Dos pacientes fallecieron en periodo neonatal por insuficiencia respiratoria secundaria a hipoplasia pulmonar. Los 14 restantes sobreviven en la actualidad. El 57% ha desarrollado enfermedad renal crónica (ERC), la mediana de edad de este subgrupo en el momento del estudio son 22,9 años (tabla 1).
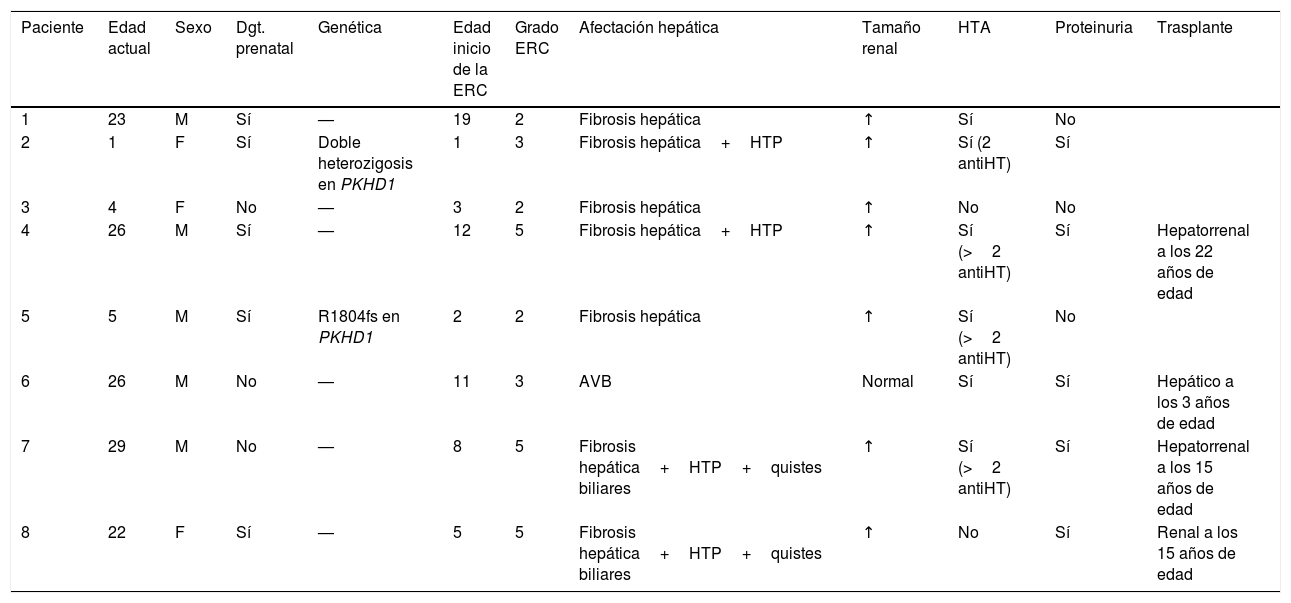
Resumen de las principales variables recogidas en el grupo de pacientes con ERC
| Paciente | Edad actual | Sexo | Dgt. prenatal | Genética | Edad inicio de la ERC | Grado ERC | Afectación hepática | Tamaño renal | HTA | Proteinuria | Trasplante |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 23 | M | Sí | — | 19 | 2 | Fibrosis hepática | ↑ | Sí | No | |
| 2 | 1 | F | Sí | Doble heterozigosis en PKHD1 | 1 | 3 | Fibrosis hepática+HTP | ↑ | Sí (2 antiHT) | Sí | |
| 3 | 4 | F | No | — | 3 | 2 | Fibrosis hepática | ↑ | No | No | |
| 4 | 26 | M | Sí | — | 12 | 5 | Fibrosis hepática+HTP | ↑ | Sí (>2 antiHT) | Sí | Hepatorrenal a los 22 años de edad |
| 5 | 5 | M | Sí | R1804fs en PKHD1 | 2 | 2 | Fibrosis hepática | ↑ | Sí (>2 antiHT) | No | |
| 6 | 26 | M | No | — | 11 | 3 | AVB | Normal | Sí | Sí | Hepático a los 3 años de edad |
| 7 | 29 | M | No | — | 8 | 5 | Fibrosis hepática+HTP+quistes biliares | ↑ | Sí (>2 antiHT) | Sí | Hepatorrenal a los 15 años de edad |
| 8 | 22 | F | Sí | — | 5 | 5 | Fibrosis hepática+HTP+quistes biliares | ↑ | No | Sí | Renal a los 15 años de edad |
antiHT: fármaco antihipertensivo; AVB: atresia vías biliares; Dgt. prenatal: diagnóstico prenatal; Edad actual: años de edad en el momento en el que se desarrolla el estudio; Edad inicio de la ERC: años de edad en el momento del inicio de la ERC; ERC: enfermedad renal crónica; F: femenino; HTA: hipertensión arterial; HTP: hipertensión portal; M: masculino.
Llama la atención la rápida evolución clínica en el paciente con diagnóstico genético de doble heterocigosis, en consonancia con publicaciones2 que relacionan la presencia de dos mutaciones truncadas en el gen PKHD1 con un fenotipo más grave.
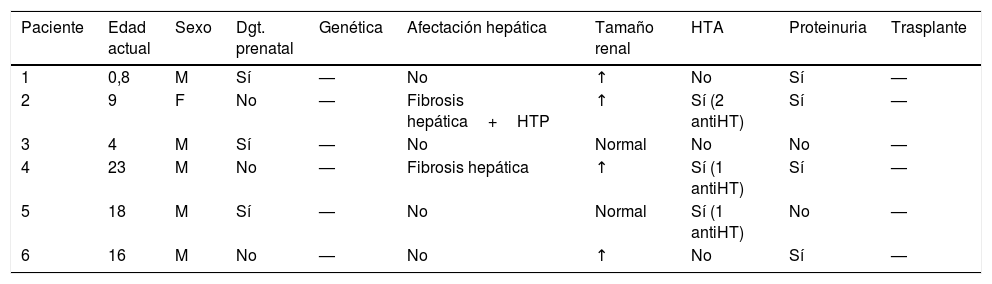
Por otro lado, el 43% de la muestra no ha desarrollado por el momento ERC, la mediana de edad de este subgrupo son 12,3 años (tabla 2).
Resumen de las principales variables recogidas en el grupo de pacientes sin ERC
| Paciente | Edad actual | Sexo | Dgt. prenatal | Genética | Afectación hepática | Tamaño renal | HTA | Proteinuria | Trasplante |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0,8 | M | Sí | — | No | ↑ | No | Sí | — |
| 2 | 9 | F | No | — | Fibrosis hepática+HTP | ↑ | Sí (2 antiHT) | Sí | — |
| 3 | 4 | M | Sí | — | No | Normal | No | No | — |
| 4 | 23 | M | No | — | Fibrosis hepática | ↑ | Sí (1 antiHT) | Sí | — |
| 5 | 18 | M | Sí | — | No | Normal | Sí (1 antiHT) | No | — |
| 6 | 16 | M | No | — | No | ↑ | No | Sí | — |
antiHT: fármaco antihipertensivo; Dgt. prenatal: diagnóstico prenatal; Edad actual: años de edad en el momento en el que se desarrolla el estudio; ERC: enfermedad renal crónica; F: femenino
HTA: hipertensión arterial; HTP: hipertensión portal; M: masculino.
No se encontró asociación entre el tamaño renal y la afectación hepática o la hipertensión arterial (HTA) y/o proteinuria. Tampoco entre el diagnóstico prenatal y el desarrollo de ERC.
Sí que se observó asociación entre la presencia de nefromegalia al diagnóstico y el desarrollo posterior de la ERC: OR: 3,5; IC 95%: 0,24-51,9, aunque sin significación estadística: test exacto de Fisher p 0,53.
En nuestro estudio la mortalidad de la PQRAR viene determinada por la hipoplasia pulmonar neonatal. Superado el periodo neonatal la supervivencia es alta. Datos similares han sido descritos en la literatura previa3, aunque con cifras de mortalidad superiores a las de nuestra muestra. La mejoría en las cifras de supervivencia probablemente se debe a que las series publicadas4 tienen varios años de antigüedad y no reflejan los avances médicos en cuidados neonatales y en el seguimiento de los pacientes en unidades de referencia. El diagnóstico prenatal, ahora mayoritario, y la posibilidad de interrupción del embarazo, es otro factor a tener en cuenta en la epidemiología de la enfermedad.
La morbilidad de los supervivientes viene dada por la HTA de difícil control, el desarrollo de la ERC y la hepatopatía, mientras que la función pulmonar progresa correctamente. En sintonía con la literatura, más del 50% de los individuos progresan a ERC. En las series publicadas5, el deterioro renal se establece a lo largo de la primera década de vida, sin embargo, en nuestra muestra el 43% de los pacientes presenta una función renal normal superados los 10 años de vida.
Varios estudios6 han señalado que el tamaño renal es un potencial parámetro para evaluar la gravedad de la enfermedad. En nuestra muestra identificamos una tendencia entre la presencia de nefromegalia y la ERC, aunque dicha asociación no alcanza significación estadística, lo cual puede deberse al reducido número de pacientes.
Este trabajo muestra que la PQRAR es una enfermedad crónica tratable, con supervivencia prolongada una vez superado el periodo neonatal, con HTA significativa y precoz, y raramente con proteinuria grave.
Como limitaciones de nuestro estudio destaca el escaso número de pacientes de la muestra, que restringe la extrapolarización de datos a la población y disminuye la capacidad estadística de los análisis. Además, el estudio genético fue realizado únicamente en 2 pacientes lo cual reduce la capacidad para extraer conclusiones acerca del papel del estudio genético.

