


Hypophosphatasia is a very rare bone metabolism disorder caused by a deficiency in alkaline phosphatase activity, due to mutations in the ALPL gene. Its clinical hallmark is the impairment of skeletal and tooth mineralization, although extra-skeletal manifestations are frequent. Its phenotypic spectrum is widely variable from a subtype with exclusive odontological impairment (odontohypophosphatasia) to five subtypes with systemic involvement, classified according to the age at the onset of the first symptoms (four of them in the paediatric age range: perinatal lethal, perinatal benign, infant and childhood hypophosphatasia). Those subtypes of hypophosphatasia with an earliest onset usually involve a worse prognosis, due to the risk of developing potentially lethal complications, such as seizures or severe respiratory insufficiency, secondary to rib cage malformations. Due to the extremely low prevalence of the severe forms of hypophosphatasia, its clinical variability and overlapping phenotypic features with several more prevalent conditions, the diagnosis of hypophosphatasia in the clinical setting is challenging. However, its potential lethality and impact on the patient's quality of life, along with the recent availability of an enzyme replacement therapy, increases the relevance of the early and accurate identification of patients affected with hypophosphatasia. On the basis of published evidence and clinical experience, this article suggests an algorithm with practical recommendations for the differential diagnosis of childhood hypophosphatasia, as well as an updated review of current therapeutic options.
La hipofosfatasia es una enfermedad ultra-rara del metabolismo mineral óseo causada por un déficit de actividad de la fosfatasa alcalina, debido a la existencia de mutaciones en el gen ALPL. Clínicamente, se caracteriza por el desarrollo de hipomineralización esquelética y dental, junto con la frecuente aparición de manifestaciones extraesqueléticas. Su espectro fenotípico es muy variable y engloba una forma de afectación exclusivamente odontológica (odontohipofosfatasia) y 5 subtipos de afectación sistémica diferenciados según el momento de inicio de los síntomas (4 de los cuales se desarrollan en la edad pediátrica: formas perinatal letal, perinatal benigna, del lactante e infanto-juvenil). Las formas de inicio más precoz presentan, generalmente, peor pronóstico, debido a la posibilidad de desarrollar complicaciones potencialmente letales, como la dificultad respiratoria grave por malformaciones torácicas o la presencia de convulsiones. Debido a la baja prevalencia de las formas graves de la enfermedad, y a su variabilidad y solapamiento fenotípico con otras patologías más prevalentes, el diagnóstico de la hipofosfastasia en la práctica clínica constituye un reto. No obstante, su potencial gravedad e impacto sobre la calidad de vida de los pacientes, así como la reciente disponibilidad de un tratamiento de reemplazo enzimático específico, confieren particular relevancia a la correcta identificación de los pacientes afectos de hipofosfatasia. A partir de la evidencia publicada y la experiencia clínica, en el presente artículo se propone un algoritmo con recomendaciones prácticas para el diagnóstico diferencial de la hipofosfatasia en niños, así como una revisión actualizada de las opciones de tratamiento existentes.
Hypophosphatasia (HPP) is an extremely rare disease of calcium and phosphate metabolism with systemic involvement and a progressive course, characterized by defective bone mineralization due to reduced activity of the tissue-nonspecific isoenzyme of alkaline phosphatase (TNSALP).1,2 Alkaline phosphatase (ALP) is a key enzyme in bone and tooth mineralization of which there are at least 4 isoenzymes: TNSALP (which accounts for 95% of the total serum ALP activity) and 3 tissue-specific isoenzymes (intestinal, placental and germ cell).1,2 The TNSALP isoenzyme is encoded by the ALPL gene (1p36.12; OMIM 171760), which is mainly expressed in liver, bone, kidney, muscle and sensory areas of the cerebral cortex.3 The presence of variants in ALPL associated with changes in TNSALP function determines the development of HPP, and more than 330 mutations have been described to date.4
The main substrates of TNSALP are inorganic pyrophosphate (PPi), pyridoxal 5′-phosphate (PLP) and phosphoethanolamine (PEA).3 In patients with HPP, low TNSALP activity leads to accumulation of these substrates and the manifestations associated with the disease. The high concentration of PPi in the extracellular osseous matrix prevents the nucleation of calcium and phosphate crystals, interfering with the growth of hydroxyapatite crystals and leading to skeletal abnormalities.3 On the other hand, the dephosphorylation of PLP (an active metabolite of vitamin B6 or pyridoxine) to pyridoxal (PL) by TNSALP is necessary for pyridoxine to cross the cell membrane or the blood-brain barrier. Pyridoxal 5′-phosphate is an essential cofactor of inhibitory neurotransmitters such as gamma-aminobutyric acid. Symptomatic neonatal and infantile forms of HPP characteristically manifest with seizures secondary to a central deficiency of vitamin B6, despite the accumulation of PLP in plasma.1–3 In addition, the frequent presence of inflammatory damage in the joints in patients with HPP suggests that TNSALP plays a role in inflammatory processes activated by the deposition of calcium pyrophosphate dihydrate crystals.
Historically, hypophosphatasia has been classified into a form with exclusive involvement of tooth (odontohypophosphatasia) and 5 subtypes with systemic involvement classified according to age at onset (4 paediatric forms and 1 adult form) (Tables 1–3). At present, it is also important to differentiate between mild and severe forms, at least in the infantile and childhood subtypes.1 Furthermore, there have been descriptions in the literature of another disease with manifestations that are similar to those of HPP but with a normal or even mildly elevated serum TNSALP activity (pseudohypophosphatasia),1 although in these cases, activity in the normal range might be due to intercurrent conditions.1
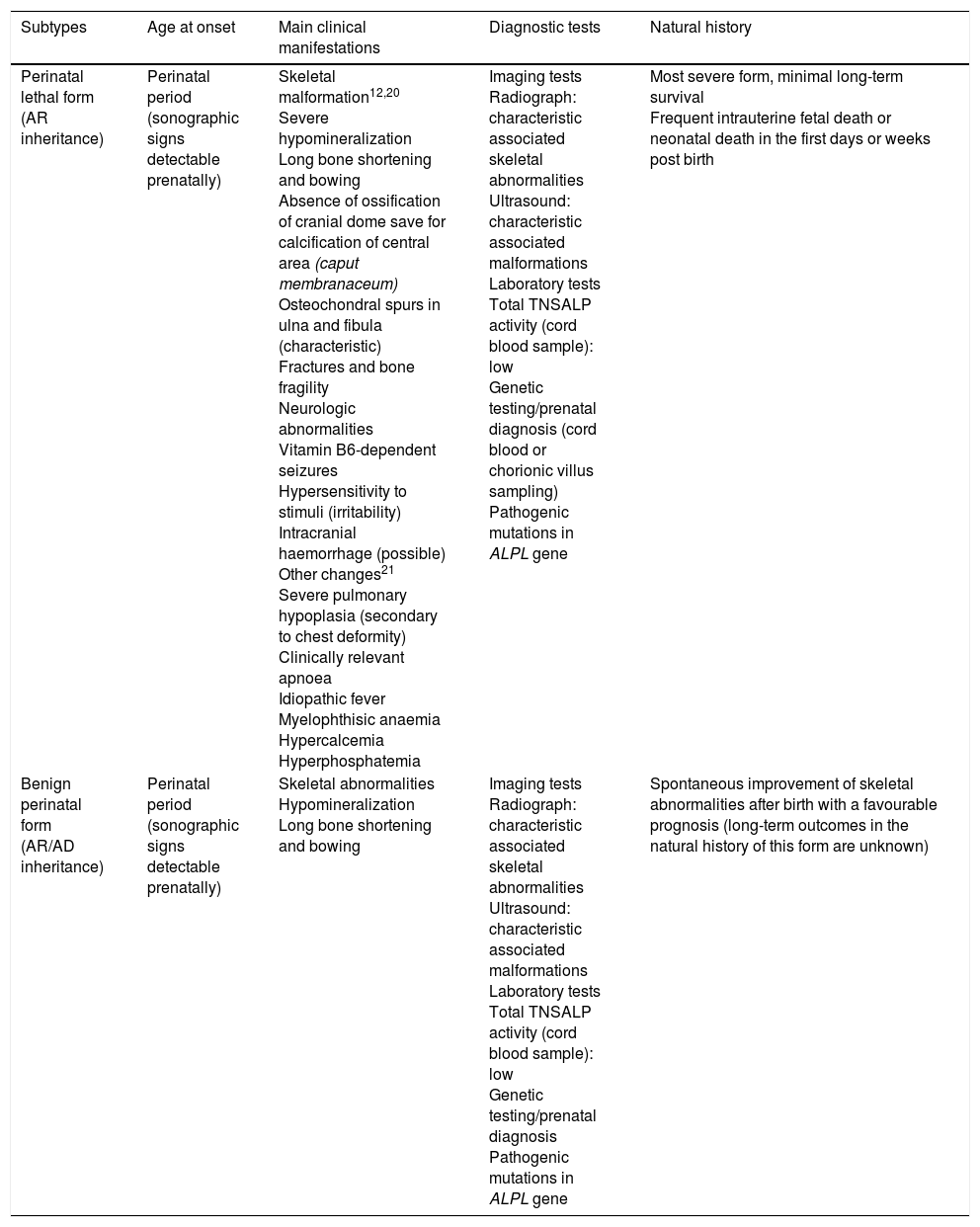
Clinical features of perinatal forms of hypophosphatasia.
| Subtypes | Age at onset | Main clinical manifestations | Diagnostic tests | Natural history |
|---|---|---|---|---|
| Perinatal lethal form (AR inheritance) | Perinatal period (sonographic signs detectable prenatally) | Skeletal malformation12,20 Severe hypomineralization Long bone shortening and bowing Absence of ossification of cranial dome save for calcification of central area (caput membranaceum) Osteochondral spurs in ulna and fibula (characteristic) Fractures and bone fragility Neurologic abnormalities Vitamin B6-dependent seizures Hypersensitivity to stimuli (irritability) Intracranial haemorrhage (possible) Other changes21 Severe pulmonary hypoplasia (secondary to chest deformity) Clinically relevant apnoea Idiopathic fever Myelophthisic anaemia Hypercalcemia Hyperphosphatemia | Imaging tests Radiograph: characteristic associated skeletal abnormalities Ultrasound: characteristic associated malformations Laboratory tests Total TNSALP activity (cord blood sample): low Genetic testing/prenatal diagnosis (cord blood or chorionic villus sampling) Pathogenic mutations in ALPL gene | Most severe form, minimal long-term survival Frequent intrauterine fetal death or neonatal death in the first days or weeks post birth |
| Benign perinatal form (AR/AD inheritance) | Perinatal period (sonographic signs detectable prenatally) | Skeletal abnormalities Hypomineralization Long bone shortening and bowing | Imaging tests Radiograph: characteristic associated skeletal abnormalities Ultrasound: characteristic associated malformations Laboratory tests Total TNSALP activity (cord blood sample): low Genetic testing/prenatal diagnosis Pathogenic mutations in ALPL gene | Spontaneous improvement of skeletal abnormalities after birth with a favourable prognosis (long-term outcomes in the natural history of this form are unknown) |
AD, autosomal dominant; ALPL, tissue-nonspecific alkaline phosphatase gene; TNSALP, tissue non-specific alkaline phosphatase; AR, autosomal recessive.
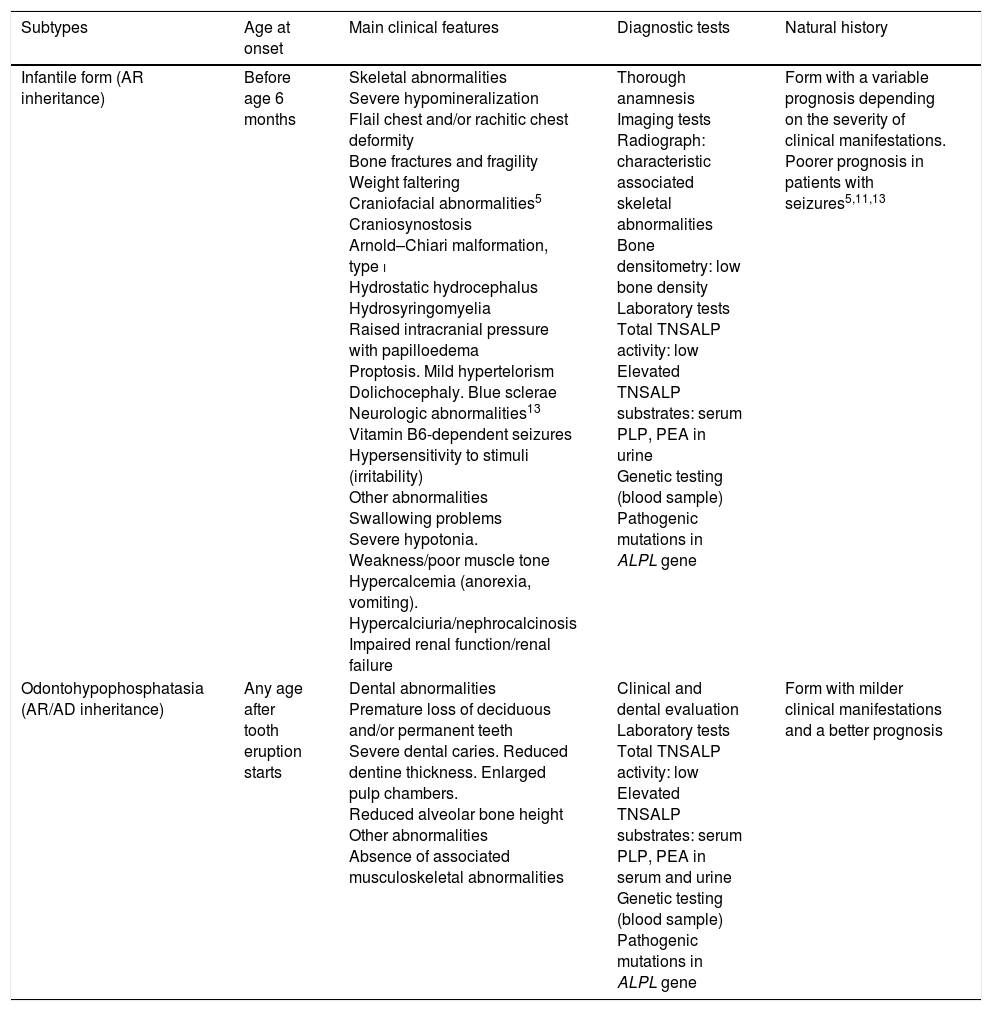
Clinical features of infantile hypophosphatasia and odontohypophosphatasia.
| Subtypes | Age at onset | Main clinical features | Diagnostic tests | Natural history |
|---|---|---|---|---|
| Infantile form (AR inheritance) | Before age 6 months | Skeletal abnormalities Severe hypomineralization Flail chest and/or rachitic chest deformity Bone fractures and fragility Weight faltering Craniofacial abnormalities5 Craniosynostosis Arnold–Chiari malformation, type i Hydrostatic hydrocephalus Hydrosyringomyelia Raised intracranial pressure with papilloedema Proptosis. Mild hypertelorism Dolichocephaly. Blue sclerae Neurologic abnormalities13 Vitamin B6-dependent seizures Hypersensitivity to stimuli (irritability) Other abnormalities Swallowing problems Severe hypotonia. Weakness/poor muscle tone Hypercalcemia (anorexia, vomiting). Hypercalciuria/nephrocalcinosis Impaired renal function/renal failure | Thorough anamnesis Imaging tests Radiograph: characteristic associated skeletal abnormalities Bone densitometry: low bone density Laboratory tests Total TNSALP activity: low Elevated TNSALP substrates: serum PLP, PEA in urine Genetic testing (blood sample) Pathogenic mutations in ALPL gene | Form with a variable prognosis depending on the severity of clinical manifestations. Poorer prognosis in patients with seizures5,11,13 |
| Odontohypophosphatasia (AR/AD inheritance) | Any age after tooth eruption starts | Dental abnormalities Premature loss of deciduous and/or permanent teeth Severe dental caries. Reduced dentine thickness. Enlarged pulp chambers. Reduced alveolar bone height Other abnormalities Absence of associated musculoskeletal abnormalities | Clinical and dental evaluation Laboratory tests Total TNSALP activity: low Elevated TNSALP substrates: serum PLP, PEA in serum and urine Genetic testing (blood sample) Pathogenic mutations in ALPL gene | Form with milder clinical manifestations and a better prognosis |
AD, autosomal dominant; ALPL, tissue-nonspecific alkaline phosphatase gene; TNSALP, tissue non-specific alkaline phosphatase; AR, autosomal recessive; PEA, phosphoethanolamine; PLP, pyridoxal 5′-phosphate.
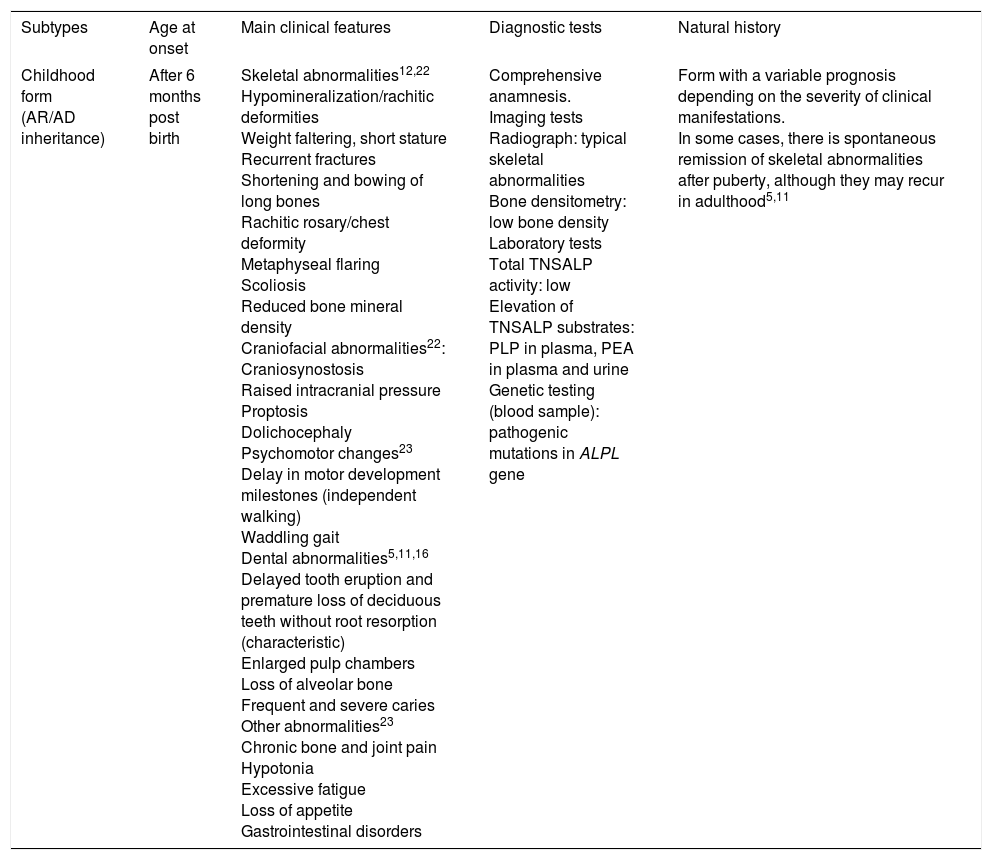
Clinical features of childhood hypophosphatasia.
| Subtypes | Age at onset | Main clinical features | Diagnostic tests | Natural history |
|---|---|---|---|---|
| Childhood form (AR/AD inheritance) | After 6 months post birth | Skeletal abnormalities12,22 Hypomineralization/rachitic deformities Weight faltering, short stature Recurrent fractures Shortening and bowing of long bones Rachitic rosary/chest deformity Metaphyseal flaring Scoliosis Reduced bone mineral density Craniofacial abnormalities22: Craniosynostosis Raised intracranial pressure Proptosis Dolichocephaly Psychomotor changes23 Delay in motor development milestones (independent walking) Waddling gait Dental abnormalities5,11,16 Delayed tooth eruption and premature loss of deciduous teeth without root resorption (characteristic) Enlarged pulp chambers Loss of alveolar bone Frequent and severe caries Other abnormalities23 Chronic bone and joint pain Hypotonia Excessive fatigue Loss of appetite Gastrointestinal disorders | Comprehensive anamnesis. Imaging tests Radiograph: typical skeletal abnormalities Bone densitometry: low bone density Laboratory tests Total TNSALP activity: low Elevation of TNSALP substrates: PLP in plasma, PEA in plasma and urine Genetic testing (blood sample): pathogenic mutations in ALPL gene | Form with a variable prognosis depending on the severity of clinical manifestations. In some cases, there is spontaneous remission of skeletal abnormalities after puberty, although they may recur in adulthood5,11 |
AD, autosomal dominant; ALPL, tissue-nonspecific alkaline phosphatase gene; TNSALP, tissue non-specific ALP; AR, autosomal recessive; PEA, phosphoethanolamine; PLP, pyridoxal 5′-phosphate.
Generally, forms with earlier onset have a poorer prognosis1,2,5 due to the potentially fatal complications of skeletal abnormalities (severe respiratory difficulties due to chest deformities) or extraskeletal manifestations (presence of seizures).6
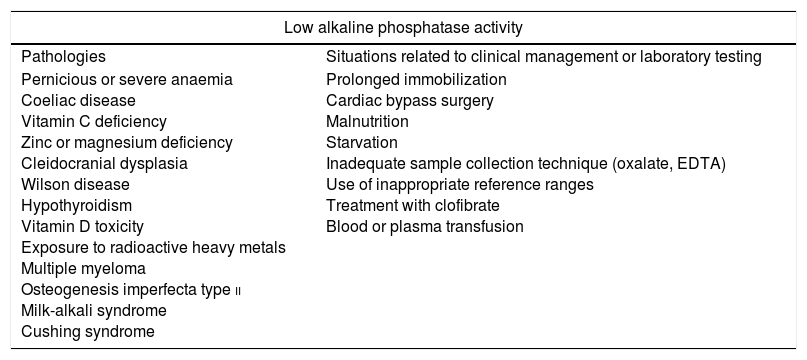
The prevalence of severe forms of HPP is estimated at 1 case per 300,000 births in Europe.5 It varies between populations, with severe forms being most frequent in Canadian Mennonites (1:2500)2 and perinatal lethal forms in Japan.7 The prevalence of mild forms with a dominant pattern of inheritance is estimated at 1 case in 6370 births in Europe.8 In contrast, the prevalence of hypophosphatasaemia (low serum ALP activity) is much higher, as this state may be due to multiple causes in the absence of HPP (Table 4).1
Situations that may be associated with low alkaline phosphatase activity.
| Low alkaline phosphatase activity | |
|---|---|
| Pathologies | Situations related to clinical management or laboratory testing |
| Pernicious or severe anaemia Coeliac disease Vitamin C deficiency Zinc or magnesium deficiency Cleidocranial dysplasia Wilson disease Hypothyroidism Vitamin D toxicity Exposure to radioactive heavy metals Multiple myeloma Osteogenesis imperfecta type ii Milk-alkali syndrome Cushing syndrome | Prolonged immobilization Cardiac bypass surgery Malnutrition Starvation Inadequate sample collection technique (oxalate, EDTA) Use of inappropriate reference ranges Treatment with clofibrate Blood or plasma transfusion |
EDTA, ethylenediaminetetraacetic acid.
The diagnosis of HPP in clinical practice poses challenges due to the low prevalence of the disease and the considerable phenotypic overlap with other diseases that are more common. Furthermore, the little importance that is normally given to low serum ALP activity (compared to the usual assessment that follows detection of hyperphosphatasaemia) is a limiting factor in the diagnosis of HPP. Yet, diagnosis is particularly important given the potential severity of the disease, the considerable impact it has in the quality of life of many of those affected, or the iatrogenic problems that could result from misdiagnosis. Furthermore, a targeted enzyme replacement therapy for HPP has recently become available,9,10 so correct diagnosis is now even more crucial to allow early and appropriate treatment.
The main aim of this article is to provide general recommendations to facilitate the identification of paediatric patients with HPP. We recommend that those seeking detailed information on the genetic basis, pathophysiology and clinical manifestations of the disease consult recently published literature reviews on the subject.1,2,6,10
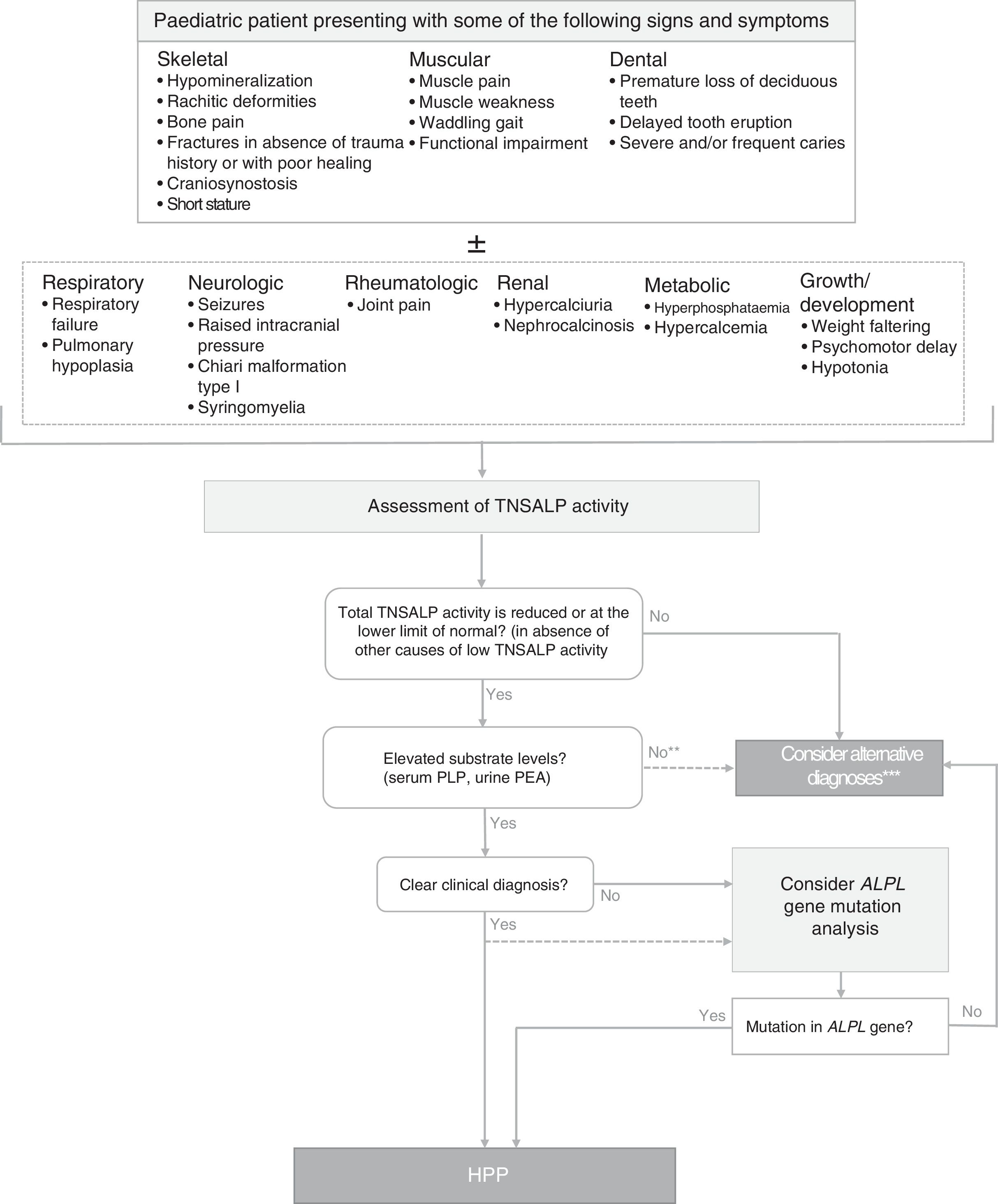
Diagnosis of hypophosphatasiaFig. 1 shows an algorithm for the diagnosis of HPP in paediatric patients based on current evidence and expert recommendations derived from clinical experience. It aims to provide a logical sequence for diagnosis of potential cases of symptomatic HPP, although there are forms of the disease with minimal to non-existent clinical manifestations in childhood and adolescence. In such cases, persistence of low serum TNSALP activity, even in the absence of skeletal manifestations, may suggest the presence of a mild form of HPP (whose severity may increase in adulthood). A diagnosis of HPP should be considered mainly in patients presenting with certain musculoskeletal or dental signs and symptoms (although in severe perinatal and infantile forms, respiratory and neurologic symptoms are strongly suggestive of HPP). Tables 1–3 list the most frequent clinical features of the paediatric forms of HPP.

Algorithm for diagnosis of hypophosphatasia in paediatric patients. ALPL tissue nonspecific alkaline phosphatase gene; TNSALP, tissue non-specific alkaline phosphatase; HPP, hypophosphatasia; PEA, phosphoethanolamine; PLP, pyridoxal 5′-phosphate. *In≥2 measurements, with no history of normal ALP activity levels and in the absence of acute disease. **PEA has a low specificity and is not always elevated in HPP. Patients with HPP often have a total TNSALP activity level at the limit of detection, while in less severe forms of disease the level may be at the lower limit of normal or mildly decreased. A decrease in TNSALP activity results in accumulation of TNSALP substrates. ***A diagnosis of HPP should not be ruled out in patients in whom other causes that may account for the clinical manifestations could not be identified, regardless of the levels of TNSALP substrates.
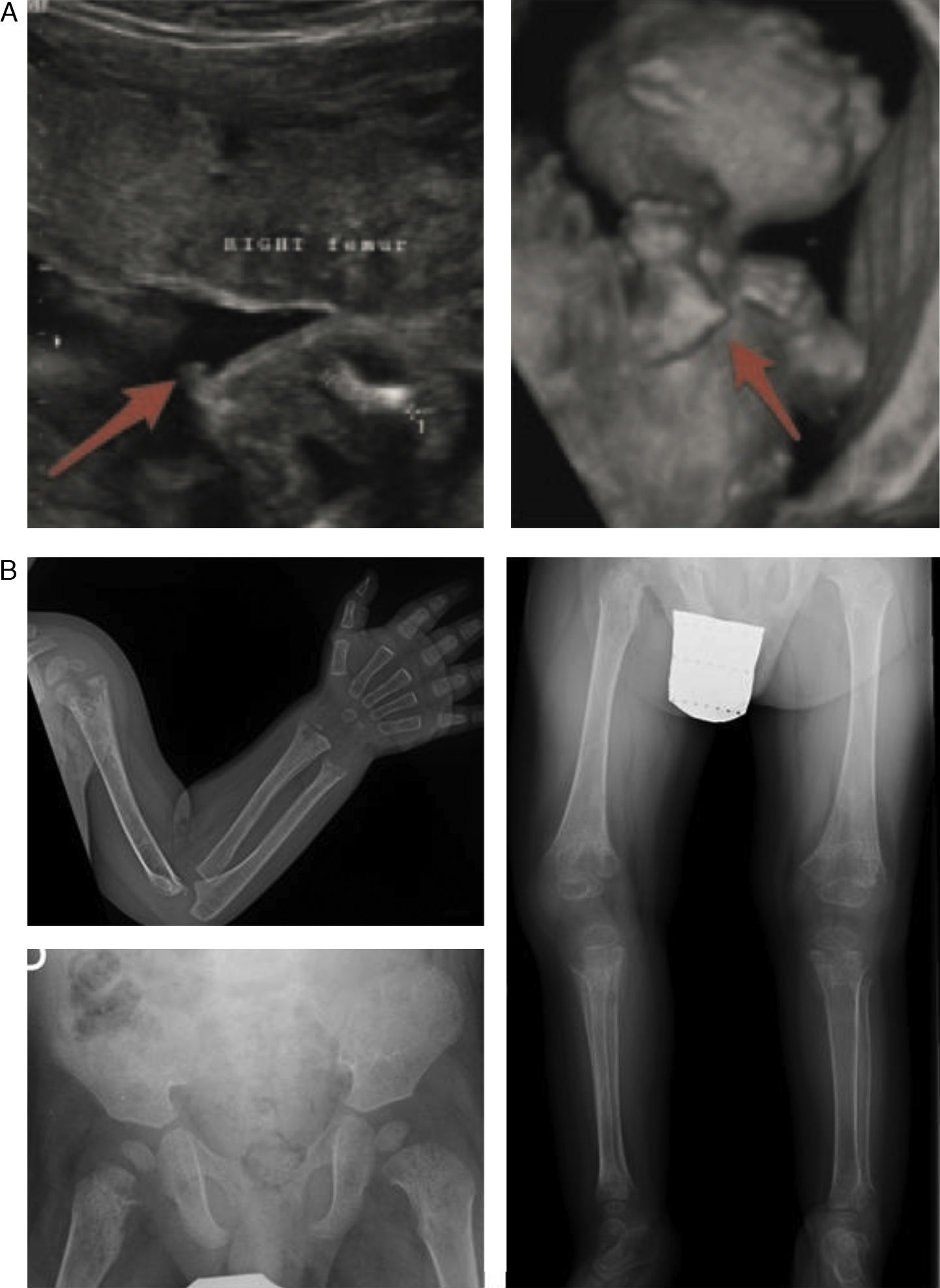
In the lethal perinatal form, the radiologic features are highly indicative of HPP: absence of mineralization in some bones (for which the main differential diagnosis would be osteogenesis imperfecta), caput membranaceum and osteochondral spurs in forearms and legs (a pathognomonic finding) (Fig. 2).2 While hypocalcaemia and hypophosphataemia are associated with different types of rickets, HPP may manifest with hypercalcemia and hyperphosphataemia (prevalence up to 50%), and low serum levels of calcium and phosphate are rare in this disease.11 The cause of hypercalcemia is unknown, although it could be due to impaired calcium uptake in bone. Hyperphosphataemia results from increased tubular reabsorption of inorganic phosphate.1
 and infantile hypophosphatasia (b). (a) Ultrasound at 18 weeks’ gestation: bone spurs in right knew and right elbow (3D). (b) Radiologic evaluation in patient aged 17 months: marked changes in long bone methaphyses (proximal metaphyses of both humeri, distal metaphyses of both radii and ulnae, and proximal metaphyses of both femora, tibiae and fibulae), with metaphyseal flaring, reduced bone density, thickened trabeculae and radiolucent projections that extend from physis to metaphysis. Linear periosteal reaction in the left radius. Images reproduced with the authorization of Zankl A, Mornet E, Wong S. Specific ultrasonography features of perinatal lethal hypophosphatasia. Am J Med Gen Part A. 2008;146A:1200–1204, and Caballero Mora FJ, Martos Moreno GA, García Esparza E, Argente J. Hipofosfatasia infantil. An Pediatr. 2012;76:368–369.")
Characteristic findings of imaging tests in cases of perinatal lethal hypophosphatasia (a) and infantile hypophosphatasia (b). (a) Ultrasound at 18 weeks’ gestation: bone spurs in right knew and right elbow (3D). (b) Radiologic evaluation in patient aged 17 months: marked changes in long bone methaphyses (proximal metaphyses of both humeri, distal metaphyses of both radii and ulnae, and proximal metaphyses of both femora, tibiae and fibulae), with metaphyseal flaring, reduced bone density, thickened trabeculae and radiolucent projections that extend from physis to metaphysis. Linear periosteal reaction in the left radius. Images reproduced with the authorization of Zankl A, Mornet E, Wong S. Specific ultrasonography features of perinatal lethal hypophosphatasia. Am J Med Gen Part A. 2008;146A:1200–1204, and Caballero Mora FJ, Martos Moreno GA, García Esparza E, Argente J. Hipofosfatasia infantil. An Pediatr. 2012;76:368–369.
In the benign perinatal form, prenatal ultrasound examination reveals signs of reduced mineralization in the long bones, but patients start improving spontaneously in the third trimester and may have a normal development.
The most characteristic findings of infantile HPP are bone and muscle pain (with or without fractures), skeletal deformities similar to those observed in patients with rickets, impaired weight gain hypotonia/muscle weakness and delays in achievement of psychomotor development milestones.6 The most frequent skeletal abnormalities are rachitic chest deformity, bowing of the long bones, genu varum (after achieving independent standing), bone fractures and craniosynostosis. This corresponds to detection of metaphyseal widening, fraying and radiolucencies in the long bones in imaging tests5,11,12 (Fig. 2b).
There is often a significant overlap between the presentation of the most severe forms of infantile HPP and the presentation of the perinatal lethal form. Respiratory difficulties resulting from chest deformities predispose infants to pneumonia and respiratory failure and are the leading cause of death in these patients.1,2 Craniosynostosis may cause raised intracranial pressure, which may be associated with Arnold Chiari type I malformation, hydrocephalus and hydrosyringomyelia, among other central nervous system abnormalities.1,2,5,6 Raised intracranial pressure and calcification of the basal ganglia may play a role, along with changes in PLP metabolism, in the development of the seizures characteristic of perinatal and infantile HPP, which are associated with a poor prognosis and a high mortality.5,11,13
Low parathyroid hormone levels are common in infantile HPP. The reduced secretion of this hormone could be due to hypercalcemia and in turn lead to the development of hyperphosphataemia. In addition, hypercalcemia and hypercalciuria could lead to development of nephrocalcinosis, so performance of kidney ultrasound examinations at regular intervals is recommended as part of the follow-up.14
Despite the poor prognosis of this form of HPP, some patients experience a spontaneous improvement in bone mineralization with the corresponding clinical improvement. Patients with infantile HPP may develop neurologic complications associated with craniosynostosis, have a short stature and loose teeth prematurely, but have a more favourable prognosis compared to patients with perinatal HPP.5,11
Childhood HPP follows in the continuum of disease after infantile HPP, with onset usually occurring after age 1 year.1,2 The main radiologic feature is the presence of characteristic projections of non-ossified tissue in the epiphyseal plates (“tongues” of radiolucency), which differentiate HPP from various forms of rickets and metaphyseal dysplasia.12 Presentation with mild hypercalcemia and hypercalciuria is less frequent in the childhood form, and serum levels of parathyroid hormone and vitamin D are usually normal (or slightly reduced in patients with renal compromise).11
Clinicians should take into account the risk of craniosynostosis in every paediatric form of HPP, and patients should undergo neurologic evaluations at regular intervals, including ophthalmoscopy, until they reach adolescence. It is also important to monitor patients that only have dental manifestations, as they may eventually develop other abnormalities. There is evidence that the severity of HPP is associated with the number of teeth lost and inversely correlated to the age at loss of the first tooth.6
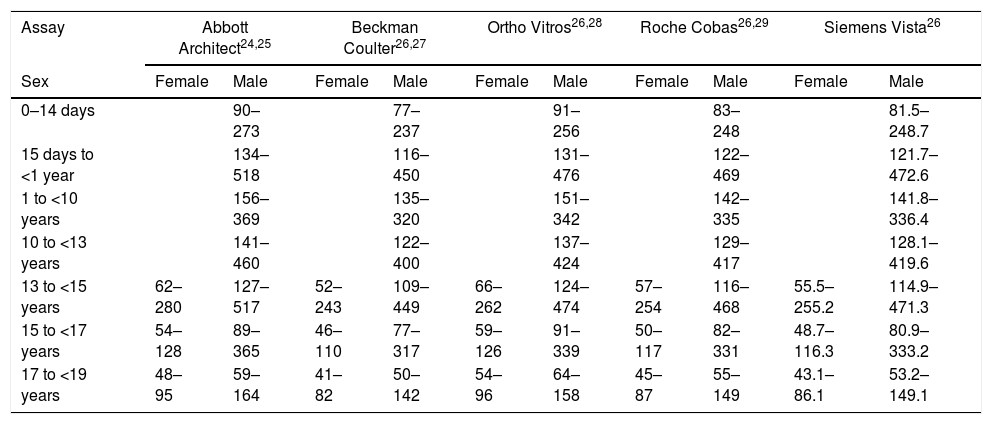
The first step in diagnosis when HPP is suspected is measurement of the total TNSALP activity. Reference values in children vary based on age, sex and the assay used (Table 5). It is important to take into account that mild forms of HPP may manifest with levels of TNSALP activity that are only slightly reduced or even at the lower limit of normal.1 Furthermore, there are multiple conditions that may manifest with a low total serum TNSALP activity (Table 4), so it is also important to assess the potential elevation of certain TNSALP substrates.1 In children with HPP, the degree of PLP accumulation in plasma is associated with the severity of disease, and this is the most sensitive and specific marker for diagnosis of HPP.1 Based on the current international literature, the positive and negative predictive values of PLP for diagnosis in children are very high, although absence of elevation of serum levels of PLP or other substrates of TNSALP (PEA, PPi) is not enough to rule out HPP. Measurement of bone mineral density by dual-energy X-ray absorptiometry may reveal low bone density, although this feature is not required for diagnosis.
Reference intervals (IU/L) for total alkaline phosphatase activity.
| Assay | Abbott Architect24,25 | Beckman Coulter26,27 | Ortho Vitros26,28 | Roche Cobas26,29 | Siemens Vista26 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Female | Male | Female | Male | Female | Male | Female | Male | Female | Male |
| 0–14 days | 90–273 | 77–237 | 91–256 | 83–248 | 81.5–248.7 | |||||
| 15 days to <1 year | 134–518 | 116–450 | 131–476 | 122–469 | 121.7–472.6 | |||||
| 1 to <10 years | 156–369 | 135–320 | 151–342 | 142–335 | 141.8–336.4 | |||||
| 10 to <13 years | 141–460 | 122–400 | 137–424 | 129–417 | 128.1–419.6 | |||||
| 13 to <15 years | 62–280 | 127–517 | 52–243 | 109–449 | 66–262 | 124–474 | 57–254 | 116–468 | 55.5–255.2 | 114.9–471.3 |
| 15 to <17 years | 54–128 | 89–365 | 46–110 | 77–317 | 59–126 | 91–339 | 50–117 | 82–331 | 48.7–116.3 | 80.9–333.2 |
| 17 to <19 years | 48–95 | 59–164 | 41–82 | 50–142 | 54–96 | 64–158 | 45–87 | 55–149 | 43.1–86.1 | 53.2–149.1 |
In patients presenting with clinical manifestations suggestive of HPP and low levels of total serum TNSALP activity (in the absence of another identifiable cause), with or without elevation of TNSALP substrates, other diseases that are more prevalent and whose manifestations overlap with those of HPP need to be ruled out before making the diagnosis (Table 6).
Differential diagnosis of hypophosphatasia in the paediatric age group.
| Differential diagnosis of hypophosphatasia | |
|---|---|
| Perinatal HPP | Osteogenesis imperfecta (severe forms, such as type ii) Congenital dwarfism Campomelic dysplasia Chondrodysplasias with abnormal bone mineralization Severe hyperparathyroidism |
| Infantile and childhood HPP | Inborn errors of energy metabolism Organic aciduria Primary and secondary rickets Osteogenesis imperfecta Achondrodysplasia Periodontitis Cleidocranial dysplasia Cole-Carpenter syndrome Idiopathic juvenile osteoporosis Renal osteodystrophy Chronic nonbacterial osteomyelitis Osteosarcoma Idiopathic hypercalcemia and other causes of nephrocalcinosis Neglect and non-accidental injury |
Detection of mutations in the ALPL gene through molecular analysis can help establish the diagnosis in uncertain cases or serve as confirmation of HPP.15,16 However, some authors consider that in cases with a presentation that is clearly indicative of HPP (clinical manifestations+radiologic features+biochemical profile), mutation analysis is not required for diagnosis.1
Mutation analysis of samples obtained by amniocentesis may be performed in pregnant women in whom fetal abnormalities compatible with HPP have been detected or who have previous children with the disease, although its use is controversial, as it is not possible to differentiate reliably between the lethal and benign perinatal forms until late in the pregnancy.1,2
Management of hypophosphatasiaThe management of HPP should be multidisciplinary and tailored to each patient according to the presentation and severity of the disease. Patients should be followed up in medical centres with specialists in metabolic bone diseases. At present, while there is no cure for HPP, patients can receive enzyme replacement therapy.9,17
Due to the symptoms it produces and its poor prognosis, severe perinatal HPP is managed differently, so we have devoted a separate section to this particular form (the remaining sections refer to the management of all paediatric forms of HPP).
Severe perinatal hypophosphatasiaFirst of all, if hypophosphatasia is suspected before birth, it is recommended to transfer the mother to a hospital with a level III B–C neonatal intensive care unit.
Respiratory support is key in severe perinatal HPP due to the increased risk of developing pulmonary hypoplasia with respiratory failure, which usually requires intubation and mechanical ventilation after birth. The newborn may need high-frequency ventilation, nitric oxide, pharmacological treatment (sildenafil, milrinone) and even extracorporeal life support.
Serum levels of TNSALP activity calcium, phosphate and markers of renal function must be closely monitored, performing measurements every 48–72h in the first week of life and weekly thereafter as required by the patient's condition. A cranial ultrasound examination should be performed in the first 48h post birth, and possibly repeated at a later time based on the condition of the patient.
If prolonged respiratory support is required after the initial acute phase, the patient should be assessed for the presence of tracheobronchomalacia, which requires ventilation with high post-expiratory pressures.
Brain function should be monitored by amplitude-integrated electroencephalography to detect convulsive seizures that may not be apparent based on clinical manifestations, whose presence, if detected, should be confirmed by means of conventional electroencephalography. Seizures are treated with intravenous pyridoxine (loading dose of de 100mg followed by 15–30mg/kg/day administered in 3 doses a day).
Dietary interventions and supportive care (patients of all ages)In case of hypercalcemia, dietary calcium intake should be restricted and care taken to ensure adequate hydration. Treatment with glucocorticoids or diuretic agents may be considered in severe cases, although loop diuretics should be used with caution, as they may exacerbate hypercalciuria. If the serum levels of calcium and phosphate are normal or only mildly elevated, the patient can follow a diet with a normal calcium intake, as calcium supplementation may aggravate hypercalcemia and hypercalciuria. Vitamin D3 supplementation should be avoided unless the patient has a deficiency (although it is not necessary to reduce exposure to sunlight), as calcium and/or vitamin D3 supplementation in these cases has been associated with hypercalcemia, hyperphosphataemia and hypercalciuria.
Dental health should be monitored through regular checkups, promoting adequate oral hygiene.
The degree of functional impairment caused by HPP mainly depends on the musculoskeletal, neurologic and pulmonary abnormalities associated with the disease, so it is important that rehabilitation specialists and physical therapists are involved in the management of these patients.
Medical treatmentThere is no standard medical treatment for HPP, and different approaches have been used with heterogeneous results.
Calcitonin has been used with the aim of decreasing bone resorption, with poor results. Bisphosphonates can be detrimental in HPP.18 They are PPi analogues, so they contribute to the inhibition of hydroxyapatite crystal growth, and they can bind zinc and magnesium cations, thereby compromising any residual TNSALP activity.
Nonsteroidal anti-inflammatories may be useful for pain relief, especially in patients with bone marrow oedema, and for treatment of extra-articular calcifications.18
Enzyme replacement therapy was first tested using intravenous infusions of plasma enriched with TNSALP from patients with Paget bone disease or purified from human placentas. Although there was transient improvement in serum markers, there was no improvement in symptoms or radiologic features, and the results could not be replicated. An isolated increase in the serum level of TNSALP is not sufficient for treatment to be effective, as levels of the enzyme need to be restored within bone tissue. Other approaches for replacement that have been investigated are bone marrow transplantation and transplantation of allogeneic mesenchymal cells committed to the osteoblast phenotype. Although they achieve a degree of clinical improvement, it is important to consider the subsequent need for immunosuppressive therapy and the associated morbidity. Gene therapy with transfection of the ALPL gene through a lentiviral vector has also been tried in ALPL knockout mice.18
Since 2015, asfotase alfa has been authorized by the European Medicine Agency (EMA) for treatment of severe forms of perinatal, infantile and childhood HPP.19 It is a recombinant fusion protein comprising the catalytic domain of the TNSALP linked to the constant region of the IgG1 domain and a terminal decaaspartate motif for bone targeting. The first results of a clinical trial of asfotase alfa for treatment of severe perinatal or infantile HPP were published in 2012. There was significant improvement in skeletal quality, respiratory function, motor and cognitive development and myopathy, and a rapid decline in serum levels of PLP and PPi,9 findings that were corroborated in a later trial with a longer duration of treatment.17 Asfotase alfa has been found to increase survival in patients with HPP (from 27% to 84% at age 5 years), especially in those requiring mechanical ventilation or with B6-dependent seizures (increases in survival from 5% to 76% and 0% to 77%, respectively).10
Asfotase alfa is administered by the subcutaneous route, usually at a total dose of 6mg/kg/week divided in 3 or 6 doses.10,19 The most frequent adverse events are local reactions at the site of injection, occasionally accompanied by local lipodystrophy.17 Although some patients develop antibodies to the protein, this has yet not been proved to have an impact on clinical outcomes.9 Adequate followup is essential to ensure appropriate dose adjustments and to detect potential adverse events. The dose must be adjusted as needed to fit the actual weight of the patient, as a suboptimal dose is associated with rapid clinical and radiographic deterioration.18 Patients must be monitored for the potential development of ectopic mineralization through regular performance of radiography, retinal examination and renal ultrasound. Ectopic mineralization can also occur in the vasculature.18
Surgical treatmentAbnormalities resulting in neurologic pathology require followup, especially craniosynostosis. The need for surgery should be evaluated on a case-to-case basis. Treatment with asfotase alfa does not seem to offer any benefits in this area.9
Surgery may also be required to treat fractures or skeletal deformities such as scoliosis. In case of bone fracture, prolonged loading may increase the risk of fracture healing failure.18
Conclusions- -
Hypophosphatasia is a rare hereditary disease of bone and mineral metabolism characterized by deficient serum TNSALP activity that leads to skeletal and dental hypomineralization and extraskeletal manifestations.
- -
The phenotypic spectrum ranges from potentially lethal early-onset forms to forms with a later onset with fewer and less specific manifestations.
- -
Diagnosis in patients in whom there is a strong suspicion of HPP due to clinical manifestations should be based on the detection of low serum TNSALP activity and radiographic abnormalities, the potential accumulation of TNSALP substrates and the findings of molecular testing, among others.
- -
The recent authorization of asfotase alfa for enzyme replacement therapy in patients with childhood-onset HPP has changed the approach to the management of this disease.
The authors have no conflicts of interest to declare.
We thank Alexion Pharmaceuticals for providing logistic support for the meeting of the working group (although the authors have been solely responsible for the contents produced by the group) and Ogilvy Healthworld for providing editorial support.
Please cite this article as: Martos-Moreno GÁ, Calzada J, Couce ML, Argente J. Hipofosfatasia: manifestaciones clínicas, recomendaciones diagnósticas y opciones terapéuticas. An Pediatr (Barc). 2018;88:356.

