



Conventional cytogenetics diagnoses 3–5% of patients with unexplained developmental delay/intellectual disability and/or multiple congenital anomalies. The Multiplex Ligation-dependent Probe Amplification increases diagnostic rates from between 2.4 and 5.8%. Currently the comparative genomic hybridisation array or aCGH is the highest performing diagnostic tool in patients with developmental delay/intellectual disability, congenital anomalies and autism spectrum disorders. Our aim is to evaluate the efficiency of the use of aCGH as first-line test in these and other indications (epilepsy, short stature).
Patients and methodA total of 1000 patients referred due to one or more of the abovementioned disorders were analysed by aCGH.
ResultsPathogenic genomic imbalances were detected in 14% of the cases, with a variable distribution of diagnosis according to the phenotypes: 18.9% of patients with developmental delay/intellectual disability; 13.7% of multiple congenital anomalies, 9.76% of psychiatric pathologies, 7.02% of patients with epilepsy, and 13.3% of patients with short stature. Within the multiple congenital anomalies, central nervous system abnormalities and congenital heart diseases accounted for 14.9% and 10.6% of diagnoses, respectively. Among the psychiatric disorders, patients with autism spectrum disorders accounted for 8.9% of the diagnoses.
ConclusionsOur results demonstrate the effectiveness and efficiency of the use of aCGH as the first line test in genetic diagnosis of patients suspected of genomic imbalances, supporting its inclusion within the National Health System.
La citogenética convencional detecta un 3-5% de los pacientes con retraso global del desarrollo/discapacidad intelectual y/o malformaciones congénitas. La amplificación de sondas múltiples dependientes de ligación permite incrementar la tasa diagnóstica entre 2,4-5,8%. Actualmente, los arrays de hibridación genómica comparada o aCGH son la herramienta diagnóstica con mayor rendimiento en estos pacientes, en malformaciones congénitas y trastornos del espectro autista. El objetivo del presente trabajo ha sido evaluar la eficiencia del uso del aCGH como técnica de primera línea diagnóstica en estas y otras indicaciones (epilepsia, talla baja).
Pacientes y métodoSe ha estudiado a 1.000 pacientes afectados por las patologías mencionadas mediante la técnica de aCGH.
ResultadosSe detectaron desequilibrios de efecto patogénico en un 14% de los pacientes (140/1.000). Según el fenotipo, se diagnosticaron un 18,9% de los pacientes afectados de retraso global del desarrollo/discapacidad intelectual; un 13,7% de las malformaciones congénitas; un 9,76% de las patologías psiquiátricas, un 7,02% de los casos con epilepsia y un 13,3% de los pacientes con talla baja. Dentro de las malformaciones congénitas destacan las del sistema nervioso central con un 14,9% y las cardiopatías congénitas con un 10,6% de diagnósticos. En las patologías psiquiátricas destacan los pacientes con trastornos del espectro autista, con un 8,9% de diagnósticos.
ConclusionesNuestros resultados demuestran la efectividad y la eficiencia de la utilización del aCGH como test de primera línea en el diagnóstico genético de los pacientes con sospecha de desequilibrios genómicos. Todo ello avala su inclusión dentro del Sistema Nacional de Salud.
In the general population, the prevalence of global developmental delay/intellectual disability (GDD/ID) is estimated at 1–3%,1 the prevalence of autism spectrum disorder (ASD) at 0.7%,2 and the prevalence of congenital anomalies (CAs) at 2–3%,1–3 constituting, overall, a significant health care and social burden. The genetic diagnosis of these patients is very useful in their clinical management, as it allows an accurate prognosis and, above all, is crucial to the prevention of new cases (genetic counselling for family planning). Until recently, these patients were assessed with a set of laboratory techniques, each with a low diagnostic yield in isolation. The most commonly used technique, conventional cytogenetic testing (karyotype analysis), can detect large losses or gains of genetic material and structural rearrangements in 3% to 5% of patients GDD/ID and/or CAs.1,4–6 To improve the diagnostic yield, conventional karyotype analysis has been supplemented with fluorescent in situ hybridisation (FISH) and multiplex ligation-dependent probe amplification (MLPA) techniques. Thanks to their introduction, the detection of abnormalities in patients with unexplained GDD/ID or CAs and normal results of karyotype analysis increased by 2.4–3.7% with subtelomere analysis7,8 and by 5.8% with screening for the most frequent microdeletion and microduplication syndromes.9
The recent development of arrays for the detection of changes in gene dosage (single nucleotide polymorphism arrays [aSNPs] and comparative genomic hybridisation arrays [aCGHs]) has considerably increased the number of diagnoses, allowing molecular diagnosis in 15–20% of patients with GDD/ID, CAs or ASD.5 Single nucleotide polymorphism arrays, originally designed to genotype human DNA at thousands of SNPs across the genome, also allow the assessment of the number of copies of DNA by comparing the intensity of patient DNA hybridisation with the signal intensity of the probes immobilised in the matrix or array.
Comparative genomic hybridisation arrays allow the simultaneous examination of gene dosage at multiple loci of the genome by comparing the relative amount of DNA in 2 genomes (control and patient) marked with different fluorochromes bound to DNA fragments with a known sequence (probes) fixed to a glass slide or support. The fluorescence emitted at each point of the array reflects the relative amount of DNA, allowing the detection of gains or losses of genetic material in specific regions of the genome. A large number of aCGH commercial platforms are currently available, and their diagnostic yield varies significantly. Some platforms are based on bacterial artificial chromosomes (BAC) and others on oligonucleotides. Probes may be distributed at regular intervals to offer whole genome backbone coverage, placed in specific regions that have been associated with a specific disease, or a combination of both.10 The International Standard Cytogenetic Array (ISCA) consortium has proposed standards that have been adopted by most array manufacturers. A multicentre study that compared the main commercial platforms currently available11 concluded that oligonucleotide-based CGH arrays with an ISCA 8×60K design offer the highest benefit–cost ratio. For clinical and economic reasons, scientific guidelines recommend the use of array methods as the first-line diagnostic test in patients with ID/GDD, CA and/or ASD, replacing karyotype analysis, MLPA and FISH.5,12–16
In this article, we present our experience with the use of aCGH as a first-tier diagnostic test in 1000 patients with GDD/ID, CA, psychiatric disorders (ASD, ADHD, schizophrenia, bipolar disorder), epilepsy, short stature and other disorders (dysmorphic features, phenotypic abnormalities, myopathy, overgrowth), replacing techniques such as karyotype analysis, MLPA and FISH. This is the largest case series of these characteristics ever analysed in Spain, as studies published to date have had smaller samples or used aCGH as a third-tier test in patients with normal results of other diagnostic tests, such as karyotype analysis or MLPA.17
Patients and methodsPatientsBetween January 2012 and January 2015, the Array Unit of the Section on Clinical and Molecular Genetics of the Hospital Universitario Vall d’Hebron of Barcelona performed a total of 1222 evaluations using the aCGH method. All these tests were preceded and also followed by a clinical visit. Of all these evaluations, 1000 corresponded to patients with one or more of the following disorders: GDD/ID, CA, psychiatric disorders (ASD, ADHD, schizophrenia, bipolar disorder), epilepsy, short stature, dysmorphic features, phenotypic abnormalities, myopathy or overgrowth syndrome. These patients had not yet undergone molecular testing of any kind and had been referred by the clinical genetics, neurology and paediatric endocrinology departments of the hospital. The remaining evaluations corresponded to 1 of the following 4 categories: (1) family screening (n=167); (2) evaluation of sonographic abnormalities in foetus (n=41); (3) characterisation of anomalies previously diagnosed with other methods, most frequently karyotype analysis (n=4) and (4) patients with positive results in previous genetic tests (n=10).
MethodsTo determine the rate of detection of genetic abnormalities relative to the number of phenotypic abnormalities, we performed an exhaustive review of the health records of all patients to allow the correct interpretation of aCGH results. Through this review, we were able to establish a classification of the disorders present in the patients and count the number of patients whose phenotype fit into each of the categories. Patients included in the study could have 1 or more of the disorders in the classification (Table 1).
Diagnostic detection rate achieved with aCGH for each of the phenotypes under study. Comparison with data from the literature.
| Indication/disease | Total patients tested (N=1000) | Patients with abnormal results (n=140) | Detection rate (%) | Detection rate in the literature (%) |
|---|---|---|---|---|
| GDD/ID | 406 | 77 | 18.9 | 10–1926–28 |
| Malformation | 394 | 54 | 13.7 | 10–125,28 |
| CHD | 188 | 20 | 10.6 | 3.6–2929,30 |
| CNS | 107 | 16 | 14.9 | 14.5–22.531,10 |
| Autism spectrum disorder | 164 | 16 | 9.75 | |
| ASD | 134 | 12 | 8.9 | 732 |
| Epilepsy | 57 | 4 | 7.01 | 7.3–2033–35 |
| Short stature | 105 | 14 | 13.3 | 1019 |
| Other | 298 | 36 | 12.08 |
Psychiatric disorders include: ASD, ADHD, schizophrenia/psychosis, bipolar disorder. Other includes: dysmorphic features, phenotypic abnormalities, myopathy, paresis, overgrowth syndrome. The patients had one or more of the described phenotypes.
ASD, autism spectrum disorder; CHD, congenital heart defect; CNS, central nervous system; GDD/ID, global developmental delay/intellectual disability; ADHD, attention-deficit hyperactivity disorder.
Patient DNA was purified from 3mL peripheral blood samples using the Gentra Puregene Blood kit (Cat. No. 158422, Qiagen, USA) following the directions of the manufacturer. In a few cases, DNA was purified using a variety of methods in other facilities or hospital laboratories.
We obtained the informed consent of the parents before performing the diagnostic tests.
We validated the aCGH method using 2 different oligonucleotide-based array platforms: the CGH Microarray kit 4×180K (Cat. No. G4426B, Agilent Technologies, USA), with ∼180000 evenly spaced backbone probes, and CGH Microarray Kit 8×60K ISCA (Cat. No. G4827A, Agilent Technologies, USA) with fewer probes (∼60000) including 40208 backbone probes and 18851 with denser spacing in ISCA-recommended regions. Tests were performed following the directions of the manufacturer. We decided to perform all aCGH tests using a 8×60K ISCA-design platform on account of its higher yield. The images were scanned at a 3-micron resolution (DNA microarray scanner, Cat. No. G2505C, Agilent Technologies, United States) and analysed with the software Cytogenomics 2.1 (Agilent Technologies, USA). To guide the interpretation of the potential pathogenicity of the copy-number variants (CNVs) detected by aCGH and their gene products, we used EasyArray, a software developed in house using the Python and Visual Basic programming languages. EasyArray compares each of the detected CNVs with the data of control individuals included in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) and an in-house database that includes the main microdeletion/microduplication syndromes, and also checks whether their gene products are included in the UniProt genetic disorder database (http://www.uniprot.org/). Lastly, EasyArray facilitates the consultation of the ISCA Consortium (https://www.iscaconsortium.org), GeneCards (http://www.genecards.org), OMIM (http://www.omim.org) and Decipher (https://decipher.sanger.ac.uk) databases. The programme guides the classification of CNVs as pathogenic, benign or of uncertain significance, and facilitates the writing of reports in adherence to the guidelines of the American College of Medical Genetics.13 To reduce the number of family screenings and achieve a balance between clinical and analytical sensibility, we did not explore CNVs of uncertain significance sized less than 400kb.5 All detected CNVs known to be pathogenic or of uncertain significance were confirmed by FISH, MLPA, karyotype analysis or a second aCGH.
We performed the statistical analysis with the software SPSS version 15.0, using the chi square test to assess the potential association between diagnostic power and the number of phenotypic abnormalities in a single patient.
Finally, in order to compare the diagnostic yield and cost of the different techniques in our population, every abnormality detected by aCGH as the first-tier test was re-evaluated to determine whether it would have been detected with subtelomeric MLPA (P070 probemix, MRC Holland, Netherlands), MLPA assays for the detection of recurrent microdeletions and microduplications (P245 and P297, MRC Holland, Netherlands) and conventional karyotype analysis (aberrations >6Mb)18 with changes in banding patterns considered sufficient by an experienced cytogeneticist. To analyse the cost per test, we followed the guidelines of the Consensus on the Use of Arrays in Clinical Genetics.16
ResultsWhen we compared the results obtained with the 2 different aCGH microarray kits (4×180K and 8×60K ISCA v2, both from Agilent Technologies) we found a clearly higher specificity in the detection of pathogenic CNVs in the 8×60K ISCA v2 microarray. For example, a pathogenic 0.043Mb deletion was detected by only 4 probes using the 4×180K microarray compared to 11 probes using the 8×60K ISCA v2 microarray. In another instance, a pathogenic duplication that affected the entire RAF1 gene was detected by 10 probes using the 4×180K microarray and by 18 probes using the 8×60K ISCA v2 microarray. In light of these results, we decided to use the 8×60K ISCA v2 format, which detected pathogenic genetic imbalances in 14% of the patients (140/1000), and more than one pathogenic CNV in 17 cases. Genetic imbalances of more than 400kb of uncertain significance (variants of uncertain significance [VOUS]) were detected in 6.1% of the patients (61/1.000).
Pathogenic CNVs were detected in 18.9% of patients with GDD/ID (77/406), 13.7% of patients with CAs (54/394), 9.76% of patients with psychiatric disorders (16/164), 7.02% of patients with epilepsy (4/57) and 13.3% of patients referred for evaluation of short stature (14/105). The diagnostic yield was 12.08% (36/298) for the heterogeneous group of disorders grouped under the “other” category (including dysmorphic features, phenotypic abnormalities, myopathy, paresis and overgrowth). The most frequent CAs were abnormalities of the central nervous system, corresponding to 14.9% of the total diagnoses (16/107), and congenital heart defects, corresponding to 10.6% (20/188). In the psychiatric disorders group, the greatest proportion corresponded to patients with ASD, which amounted to 8.9% (12/134) of the total diagnoses (Table 1).
When we grouped patients by number of phenotypic abnormalities, we found a statistically significant increase in diagnostic yield in patients with a greater number of phenotypic abnormalities (P=.0223) (Table 2).
Proportion of patients that received a diagnosis by means of aCGH by number of phenotypic abnormalities presented by the patient.
| Number of phenotypic abnormalities documented in health records | Total patients tested (n=1000) | Patients with abnormal aCGH results (n=140) | Diagnosis (%) |
|---|---|---|---|
| >2 | 68 | 15 | 20 |
| 2 | 288 | 48 | 16.6 |
| 1 | 644 | 77 | 11.9 |
The differences between groups were statistically significant (P=.0223).
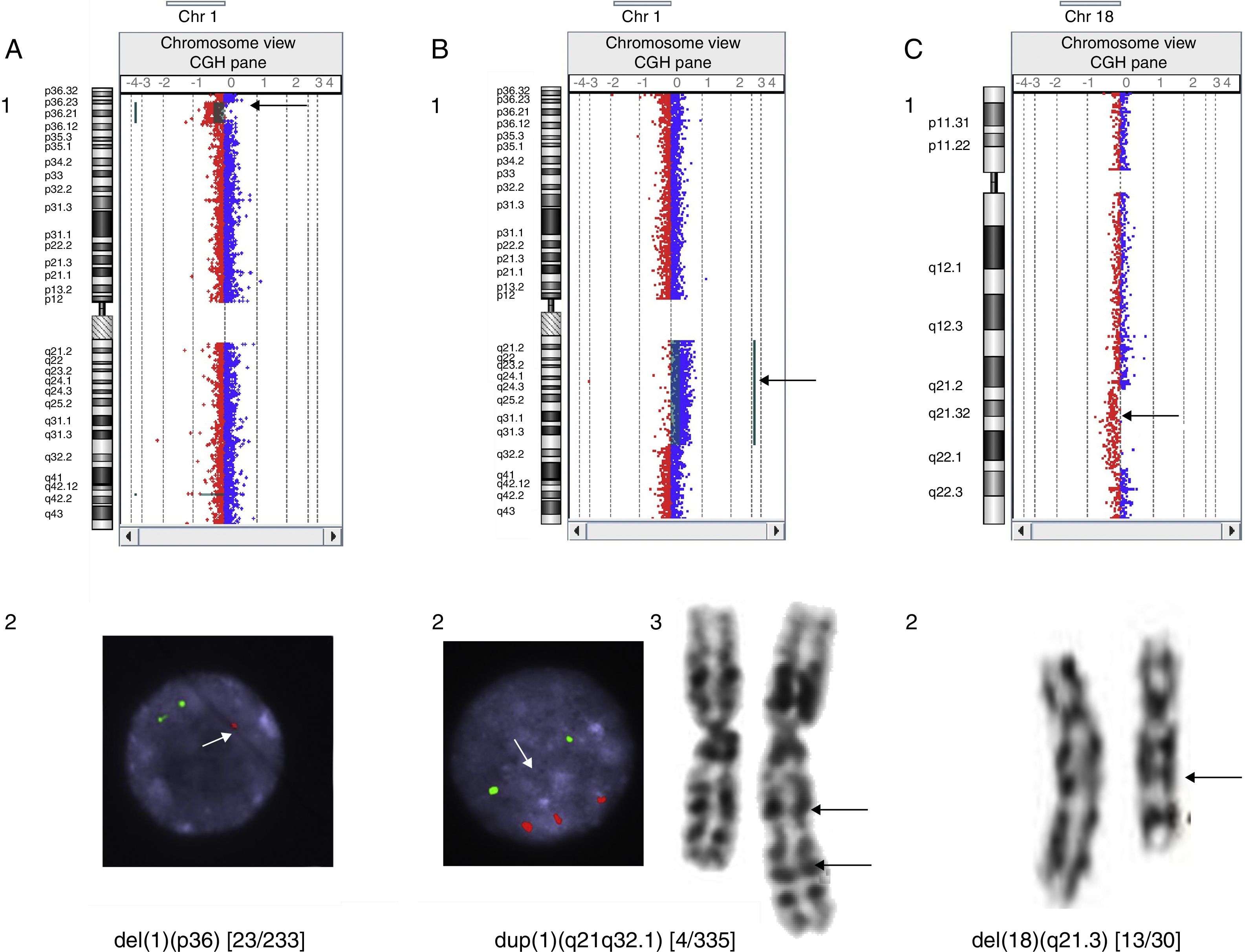
In our series, we detected mosaic microdeletions and microduplications, such as, for example, a 12.6Mb 1p36 deletion in a patient with heart disease that was confirmed by FISH in 9.8% of the cells in a blood culture sample, a 61.38Mb 1q21.1-q32.1 duplication in a patient with epilepsy confirmed by karyotype analysis and FISH in 1.2% of metaphase chromosomes, and a 14.6Mb l 18q21.31-q22.2 interstitial deletion in a patient with GDD that was not detected by the software but visually discernible in the data plot that it generated, with the deletion present in 43.3% of analysed metaphase chromosomes (Fig. 1).
 Mosaic deletion at 1p36 in a patient with heart disease: detected by aCGH (1) and confirmed in 9.8% of cells analysed using FISH. There is only one signal in the nucleus of the image, marked by an arrow, corresponding to 1p36 (2). (B) 1q21.1-q32.1 duplication in a patient with epilepsy: detected by aCGH (1), confirmed in 1.2% of cells analysed with FISH. The three bottom signals in the nucleus of the image, pointed by the arrow, correspond to region 1q21q32.1 (2) and karyotype (3). (C) 18q21.31-q22.2 deletion in a patient with GDD: detected by aCGH (1) and confirmed in 43.3% of cells analysed by karyotyping (2).")
Cases of mosaicism detected by comparative genomic hybridisation. (A) Mosaic deletion at 1p36 in a patient with heart disease: detected by aCGH (1) and confirmed in 9.8% of cells analysed using FISH. There is only one signal in the nucleus of the image, marked by an arrow, corresponding to 1p36 (2). (B) 1q21.1-q32.1 duplication in a patient with epilepsy: detected by aCGH (1), confirmed in 1.2% of cells analysed with FISH. The three bottom signals in the nucleus of the image, pointed by the arrow, correspond to region 1q21q32.1 (2) and karyotype (3). (C) 18q21.31-q22.2 deletion in a patient with GDD: detected by aCGH (1) and confirmed in 43.3% of cells analysed by karyotyping (2).
Last of all, after re-evaluating the anomalies detected by aCGH in the 140 patients, we found that 43 (30.7%) would have been detected by other techniques: 6 (4.2%) with conventional karyotype analysis, 66 (4.3%) with karyotype analysis and MLPA, and 31 (22.1%) with MLPA.
DiscussionThis article presents the largest series of patients with various diseases evaluated by aCGH as the first-tier test in Spain, as the series published to date had either been smaller or used aCGH as a third-tier test in patients that had normal results with other techniques, such as karyotype analysis and MLPA.17
Since abnormalities were detected in 14% of patients (Table 1), our findings confirm that at present, aCGH is the technique with the highest diagnostic yield when used as a single test in patients with GDD/ID, CAs and ASD.5 Its diagnostic yield is not homogeneous and varies widely depending on the type and number of phenotypic abnormalities in the patient (Tables 1 and 2), as observed in our study and those published by other authors (references in Table 1).
The classical indications for evaluation with aCGH include GDD/ID, ASD and CA,5 for which the detection rates with this technique are 18.9%, 14.8% and 13.7%, respectively. Our results and those of other authors19 suggest that short stature should also be considered an indication for evaluation with aCGH, as the test detected abnormalities in 13.3% (14/105) of these patients, of which only 21.42% (3/14) involved the SHOX gene. Our findings also suggest that isolated epilepsy in the absence of malformation of the central nervous system could also be an indication for aCGH screening, as the test allowed molecular diagnosis of 7% of these patients.
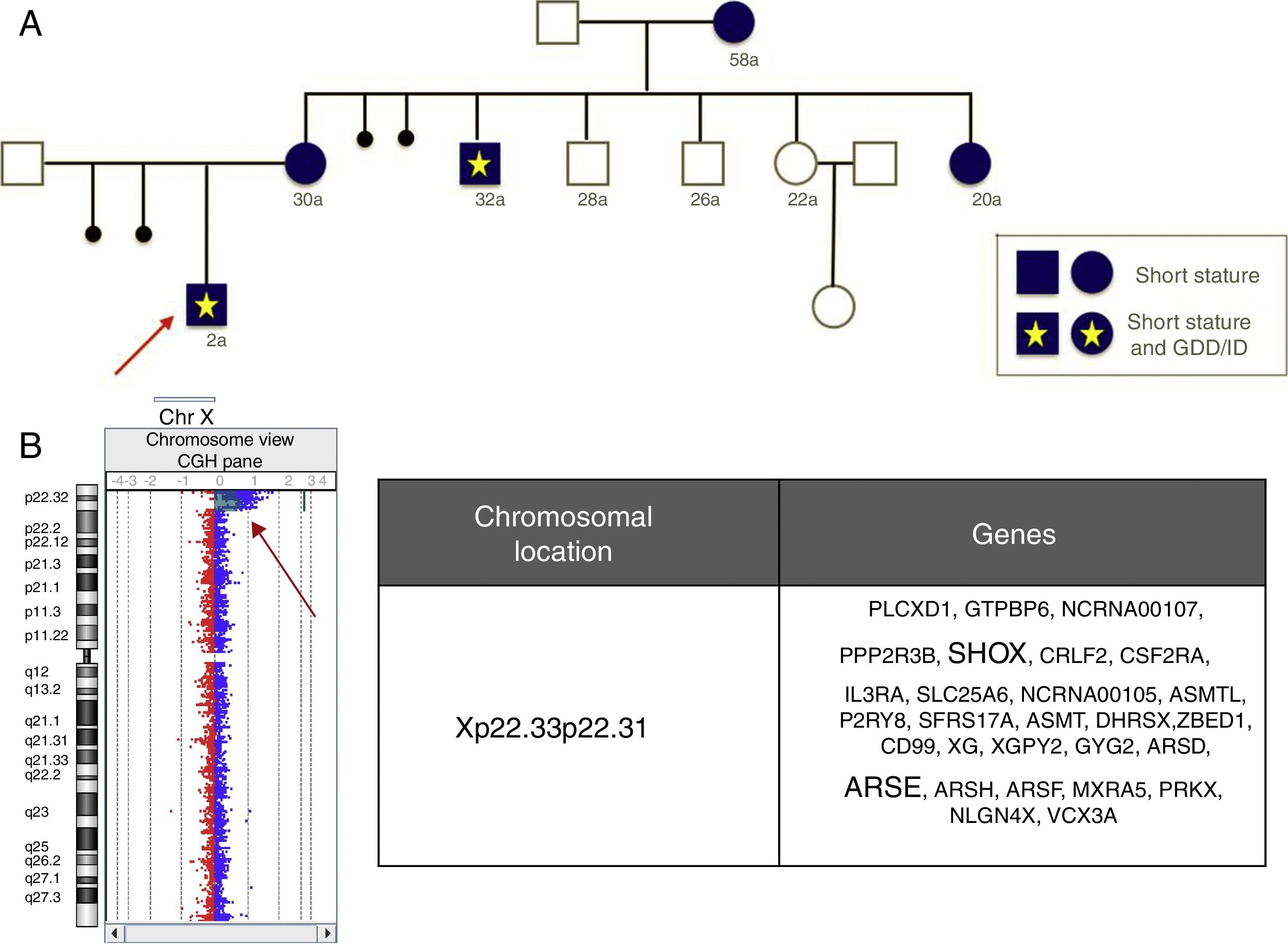
As an added value, aCGH allows a more accurate characterisation of genetic abnormalities compared to commercial FISH or MLPA kits, which may have significant implications for reproductive counselling. For example, Fig. 2 shows a deletion that affects the SHOX gene and other non-pseudoautosomal genes, such as ARSE, in female carriers with short stature and diseased males with a much more severe phenotype including short stature, chondrodysplasia punctata and GDD/ID.
 Pedigree of a family carrying a Xp22.33-p22.31 deletion. (B) Result of the analysis by aCGH, which evinced loss of the SHOX and ARSE genes, the main genes involved in the phenotypic abnormalities.")
The use of aCGH allows diagnosis of microdeletion syndromes with atypical breakpoints that may be undetectable by FISH, the most widely used diagnostic method. Specifically, our analysis of these series allowed us to detect a novel recurrent breakpoint in the Williams–Beuren syndrome critical region that, in cases of deletion, allows the partial hybridisation of the most widely used commercial FISH assays, resulting in false negatives.20
Comparative genomic hybridisation can identify gains and losses of genetic material more precisely than karyotype analysis, but it cannot detect balanced rearrangements (mainly translocations and inversions) that may in some cases be pathological. However, we must take into account that such rearrangements account for a very small proportion of clinically relevant genetic abnormalities.5,21
Comparative genomic cytogenetic arrays detect the presence of mosaicism when intermediate levels of fluorescence are produced, although determining the level of mosaicism may be difficult. Our findings were heterogeneous in this regard, for while the system readily detected abnormalities that were only present in 9.8% or 1.2% of analysed metaphase chromosomes (Fig. 1A and B), some mosaics in which the abnormalities were present in more than 40% of metaphase chromosomes caused such subtle changes in fluorescence that they were not detected by the Cytogenomics analytical software, although they were visible in the plot that it generated (Fig. 1C). A possible explanation is that different techniques analyse different cell populations: T cells from cell cultures stimulated with phytohaemagglutinin for karyotype analysis and FISH assays, compared to every type of nucleated blood cell in aCGH.
The differences in the detection rate based on the indication for testing may account for the disparity of the published results, with rates as extreme as the 7.6% reported by Shen et al.22 and the 35% reported by Aradhya et al.23 Both studies analysed the genome with a similar resolution (35kb) but differed as to the population under analysis: in the first study, the population was vaguely defined from a phenotypic standpoint, whereas the second study selected patients with phenotypes considered highly suggestive of the presence of CNVs. The large case series published in the literature that have used aCGH as the first-line diagnostic test, such as the series of 8300 patients published by Ahn et al.24 and the series of 8789 patients published by Shaffer et al.,6 identified a considerable number of pathogenic CNVs but provided little information about patient phenotypes. On the other hand, the case series that have documented patient phenotypes in detail have usually been small and focused on specific diseases, as occurred in the study published by Roselló et al.,25 whose sample consisted of 246 patients with ID. Our case series presents the results obtained using aCGH as the first-tier test in 1000 patients with a detailed phenotypic characterisation.
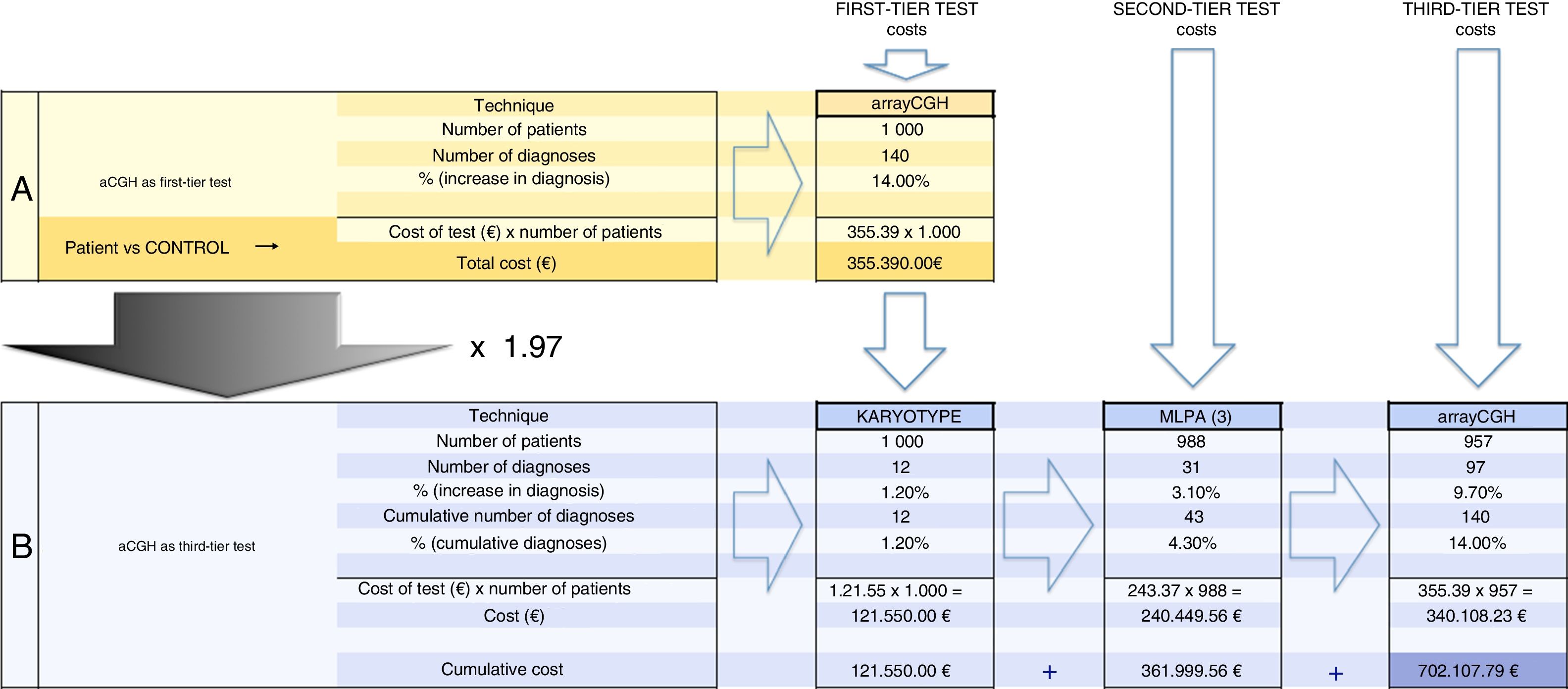
Thus, our study allowed us to reliably compare the effectiveness and efficiency of the approach of using aCGH as the first-tier diagnostic test for the detection of genetic imbalances compared to the conventional strategy of using karyotype analysis and MLPA with the possible addition of aCGH as a third-tier test (Fig. 2). Comparing published case series of patients evaluated by karyotype analysis, FISH, MLPA or aCGH poses challenges, as different studies analyse different populations. Our study allowed us to make an approximate assessment based on a single population, with the assumption that aCGH detects nearly all clinically relevant abnormalities. The traditional strategy of using karyotype analysis and the MLPA assays currently available for these diseases (2 kits for recurrent genomic imbalances and 1 kit for subtelomeric abnormalities) would have led to diagnosis in only 43 (30.7%) of the 140 patients with abnormalities detected by aCGH at an approximate cost of 326.000€ (1000 karyotype analyses+988 MLPA assays), to which we would have to add the cost of performing 957 aCGHs to reach a diagnosis in all 140 patients, for an overall cost of 702107€ (Fig. 3). The evaluation of the same patients using only aCGH would cost approximately 355.390€. In short, the traditional approach of using MLPA and karyotype analysis would have achieved less than one third of the diagnoses made in our sample, while achieving the detection of 100% of the abnormalities detectable by aCGH would increase the cost by a factor of 1.97.
 Calculation of the cost of using aCGH as the first-tier diagnostic test. (B) Calculation of the costs of using aCGH as the third-tier test after karyotype analysis and MLPA. Of the 140 cases diagnosed by means of aCGH, 12 would have been detected by karyotype analysis (of which 2 were aneuploidies and 10 genetic imbalances >6Mb); 31 would have been detected by MLPA using assays for assessment of subtelomeric and recurrent genetic imbalances, and the remaining 97 cases would have been diagnosed using aCGH in patients with normal results of karyotype analysis and MLPA. Costs16: karyotype 121.55€/test; MLPA 243.37€/3 MLPA assays; aCGH 355.39€/array. The traditional approach of karyotype analysis and MLPA (strategy B) would have achieved fewer than 1/3 of the diagnoses made in our sample (43/140). Adding performance of aCGH to diagnose 100% of abnormalities would increase the overall cost by a factor of 1.97 compared to using aCGH as the first and only diagnostic test (strategy A).")
Calculation of costs for each of the two approaches to diagnosis. (A) Calculation of the cost of using aCGH as the first-tier diagnostic test. (B) Calculation of the costs of using aCGH as the third-tier test after karyotype analysis and MLPA. Of the 140 cases diagnosed by means of aCGH, 12 would have been detected by karyotype analysis (of which 2 were aneuploidies and 10 genetic imbalances >6Mb); 31 would have been detected by MLPA using assays for assessment of subtelomeric and recurrent genetic imbalances, and the remaining 97 cases would have been diagnosed using aCGH in patients with normal results of karyotype analysis and MLPA. Costs16: karyotype 121.55€/test; MLPA 243.37€/3 MLPA assays; aCGH 355.39€/array.
The traditional approach of karyotype analysis and MLPA (strategy B) would have achieved fewer than 1/3 of the diagnoses made in our sample (43/140). Adding performance of aCGH to diagnose 100% of abnormalities would increase the overall cost by a factor of 1.97 compared to using aCGH as the first and only diagnostic test (strategy A).
Our findings illustrate the effectiveness and efficiency of using aCGH as the first-tier test for genetic diagnosis in patients with GDD/ID, CAs, ASD, short stature and epilepsy. These results and the cost–benefit analysis support the inclusion of this method in the Spanish public health system to supplement or substitute karyotype analysis, MLPA and FISH methods.
Comparative genomic hybridisation methods are constantly evolving, and new array formats have been recently developed that include backbone and targeted probes designed according to the ISCA Consortium standards and that guarantee coverage of a significant number of genes at the exon level. In this context, further studies are needed to reassess and compare the costs and benefits 8×60K and 4×180K aCGH platforms.
FundingThe study was partially funded by a grant from the Fondo de Investigaciones Sanitarias (Health Care Research Fund) of the Spanish Ministry of Health and Consumption (grant PS09/00632).
Conflicts of interestThe authors have no conflicts of interest to declare.
We want to thank our patients and their families.
Please cite this article as: Castells-Sarret N, Cueto-González AM, Borregan M, López-Grondona F, Miró R, Tizzano E, et al. Array CGH como primera opción en el diagnóstico genético: 1.000 casos y análisis de coste-beneficio. An Pediatr (Barc). 2018;89:3–11.
Previous presentations: part of this study has been included in the doctoral dissertation of Neus Castells Sarret, titled Array CGH como primera opción en el diagnóstico genético postnatal, defended on December 1, 2015 at the Hospital Universitario Vall d’Hebron, Universitat Autònoma de Barcelona. The study has also been presented as a poster titled 1.000 estudios de aCGH como técnica de primera línea: la experiencia del Hospital Vall d’Hebron at the XXVIII Congreso Nacional de la Asociación Española de Genética Humana (AEGH); May 13–15, 2015; Palma de Majorca, Spain. Part of this study was the subject of the article titled A novel recurrent breakpoint responsible for rearrangements in the Williams–Beuren region; Cytogenet Genome Res. 2015;146(3):181–186.
- Documento de manejo clínico del paciente pediátrico con infección por SARS-CoV-2.
(Actualizado el 26 de Noviembre de 2020) - Test de diagnóstico rápido en las consultas de Pediatría de Atención Primaria y Urgencias Pediátricas en la era COVID-19: más que una recomendación
(Actualizado el 21 de septiembre de 2020) - Consenso nacional sobre diagnóstico, estabilización y tratamiento del Síndrome Inflamatorio Multisistémico Pediátrico vinculado a SARS-CoV-2 (SIM-PedS).
(Actualizado el 27 de Julio de 2020) - Recomendaciones para el manejo del recién nacido en relación con la infección por SARS-CoV-2.
(Actualizado el 27 de Mayo de 2020) - Manejo del paciente pediátrico ante sospecha de infección por el nuevo coronavirus SARS-CoV-2 en atención primaria (COVID-19). AEPap-SEIP/AEP-SEPEAP.
(Actualizado el 27 de Abril de 2020) - Propuesta de adaptación de las recomendaciones de reanimación cardiopulmonar pediátrica avanzada a la infección por coronavirus.
(Actualizado el 11 de Abril de 2020)

- Laringitis aguda en neonato por VHS tipo 2
- Ingresos COVID-19: intentando comprender el impacto real de la infección en pacientes hospitalizados
- Úlcera de Lipschütz como posible manifestación del SARS-CoV-2
- Neurodesarrollo a los 2 años en cardiopatía congénita: utilidad pronóstica de los marcadores de daño cerebral

Anales de Pediatría sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas