

Congenital hyperinsulinism (CHI) is the most frequent cause of persistent hypoglycaemia in early childhood. It encompasses a heterogeneous group of genetic disorders whose overall incidence amounts to 1 in 30000 to 50000 live births, although the incidence can be as high as 1 in 2500 births in populations with a high rate of inbreeding.1
Although neonatal hypoglycaemia is the typical form of CHI, the age at onset and clinical picture are highly variable, and therefore CHI should be included in the differential diagnosis of non-ketotic hypoglycaemia at any age. It should be suspected in cases of persistent non-ketotic hypoglycaemia with abnormally high levels of insulin. The diagnosis of CHI is complicated by the lack of consensus on the biochemical criteria used to define it.2,3 Genetic testing identifies the causative mutation in only half of cases. Positron-emission tomography (PET) allows differentiation between focal and diffuse forms of CHI. The management consists of dietary measures and pharmacological treatment, and diazoxide is the first-line drug for treatment of CHI. Adequate glycaemic control is usually achieved in time. There are data suggesting that 50% of patients with CHI have neurologic sequelae secondary to unidentified episodes of hypoglycaemia.3,4
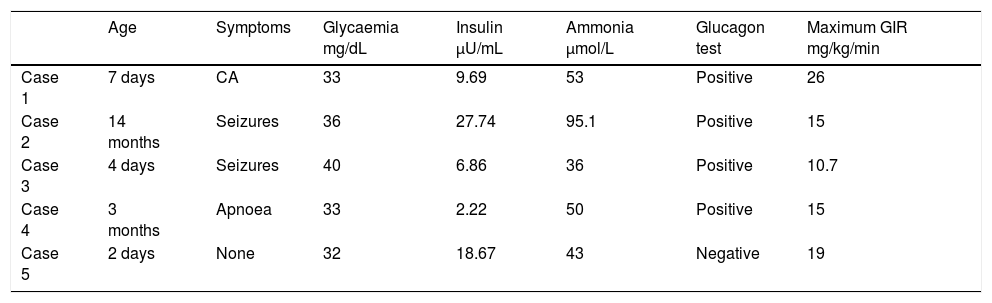
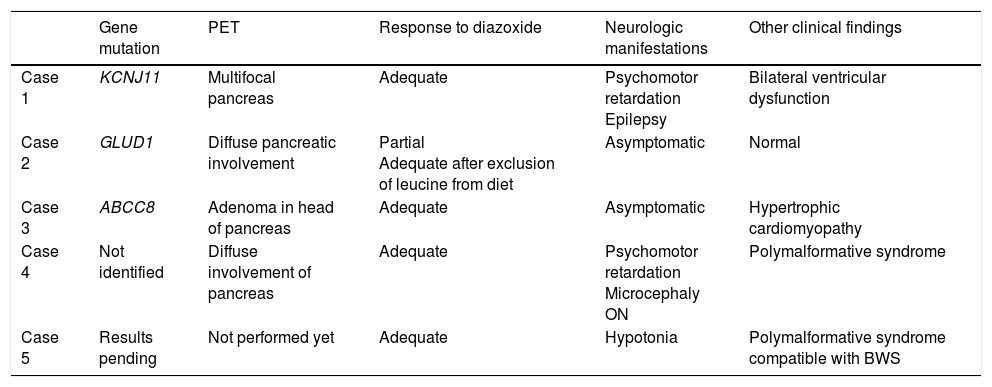
We present the cases of 5 patients with CHI diagnosed and treated in our hospital in the past 6 years. Tables 1 and 2 summarise the clinical features at diagnosis and during followup. The causative mutation was identified in 3 patients. One patient had a mutation in the KCNJ11 gene, which is associated with the most prevalent and severe form of CHI, and another had the second most frequent mutation (in the ABCC8 gene). The third patient had a novel mutation in the GLUD1 gene, changes in which are known to cause hyperinsulinism/hyperammonaemia syndrome. All 5 patients required continuous intravenous glucose infusion, whose dose could be tapered off after initiation of treatment with diazoxide. One patient received a diagnosis of hypertrophic cardiomyopathy (HCM). Four patients underwent an F-18 fluoro-L3,4-dihydroxyphenylalanine PET scan, which revealed focal involvement in only one of them. Two patients had psychomotor retardation, associated with polymalformative syndrome in one. A patient with a phenotype compatible with Beckwith-Wiedemann syndrome showed a favourable response to treatment starting at 6 months of age, and diazoxide was thereon tapered off.
Characteristics at the time of diagnosis.
| Age | Symptoms | Glycaemia mg/dL | Insulin μU/mL | Ammonia μmol/L | Glucagon test | Maximum GIR mg/kg/min | |
|---|---|---|---|---|---|---|---|
| Case 1 | 7 days | CA | 33 | 9.69 | 53 | Positive | 26 |
| Case 2 | 14 months | Seizures | 36 | 27.74 | 95.1 | Positive | 15 |
| Case 3 | 4 days | Seizures | 40 | 6.86 | 36 | Positive | 10.7 |
| Case 4 | 3 months | Apnoea | 33 | 2.22 | 50 | Positive | 15 |
| Case 5 | 2 days | None | 32 | 18.67 | 43 | Negative | 19 |
CA, cardiac arrest; GIR, glucose infusion rate.
Clinical characteristics.
| Gene mutation | PET | Response to diazoxide | Neurologic manifestations | Other clinical findings | |
|---|---|---|---|---|---|
| Case 1 | KCNJ11 | Multifocal pancreas | Adequate | Psychomotor retardation Epilepsy | Bilateral ventricular dysfunction |
| Case 2 | GLUD1 | Diffuse pancreatic involvement | Partial Adequate after exclusion of leucine from diet | Asymptomatic | Normal |
| Case 3 | ABCC8 | Adenoma in head of pancreas | Adequate | Asymptomatic | Hypertrophic cardiomyopathy |
| Case 4 | Not identified | Diffuse involvement of pancreas | Adequate | Psychomotor retardation Microcephaly ON | Polymalformative syndrome |
| Case 5 | Results pending | Not performed yet | Adequate | Hypotonia | Polymalformative syndrome compatible with BWS |
BWS, Beckwith-Wiedemann syndrome; ON, optic neuropathy; PET, positron emission tomography.
The incidence of CHI in our community exceeds the one described in the literature, as it amounts to 1 per 2400 live births. Our sample illustrates the clinical variability of CHI, which reflects the heterogeneity of the genetic causes of these disorders. In some mutations there is a clear genotype-phenotype correlation. If the mutation affects potassium channels, hyperinsulinism is severe, has an early onset, and is mostly refractory to pharmacological treatment. In contrast, mutations leading to changes in glutamate dehydrogenase cause late-onset CHI, which is less severe, and hyperammonaemia.3 The patient corresponding to case 2 had a mutation in this gene, with onset at 14 months and detection of hyperammonaemia, contrary to expectations, detected only on one occasion. He improved on a leucine-free diet, achieving greater stability in blood glucose levels.
Congenital hyperinsulinism is associated with cardiac abnormalities, mainly HCM and ventricular dysfunction. The prevalence of HCM seems increased in the most severe forms of CHI, reaching up to 65% in some case series.5 Some professionals recommend performance of an electrocardiogram and echocardiographic examination in all children with CHI.6 We performed electrocardiograms in all our patients, which detected severe HCM with mild ventricular outflow obstruction in only one patient, a problem that resolved gradually without treatment. We believe that detection of ventricular hypertrophy in a newborn with persistent hypoglycaemia is highly suggestive of CHI, and that awareness of this association could facilitate early diagnosis.
Congenital hyperinsulinism may be associated with several syndromes, and Beckwith-Wiedemann syndrome is the most frequent comorbidity out of the overgrowth disorders.4 In our series, 2 patients had polymalformative syndrome, of who 1 had a phenotype compatible with Beckwith-Wiedemann syndrome.
We detected a single case of focal pancreatic β-cell hypoplasia that could benefit from surgery in the future. Selective partial pancreatectomy in cases with focal hypoplasia would be sufficient for cure of CHI, avoiding the side effects of more aggressive surgical interventions.2
When it came to neurologic outcomes in our case series, CHI was far from being a benign disease. While most patients achieved adequate blood sugar control, neurodevelopmental impairment was common. The use of continuous glucose monitors could play a significant role in the followup of these patients.
In conclusion, we found considerable variability in the phenotypic expression of CHI. Genetic testing and PET are essential to determine the prognosis and the approach to treatment. The prevention of episodes of hypoglycaemia is essential to improve neurodevelopmental outcomes.
Please cite this article as: Reguera Bernardino J, Oulego Erroz I, Martínez Sáenz de Jubera J, Quiroga González R, Regueras Santos L. Heterogeneidad clínica y genética del hiperinsulinismo congénito. An Pediatr (Barc). 2018;89:58–59.

