
Cilia are evolutionarily conserved organelles found in most polarised cells of the body. They arise from basal bodies, extending from the cell surface to the extracellular space, and are made of proteins organised around a microtubular scaffold or axoneme. The position of cilia at the cell surface facilitates its function as a sensor and transmitter of information between the cell and the extracellular space.1–4
We present the cases of four patients that are phenotypically different but share a common feature, ciliary dysfunction.
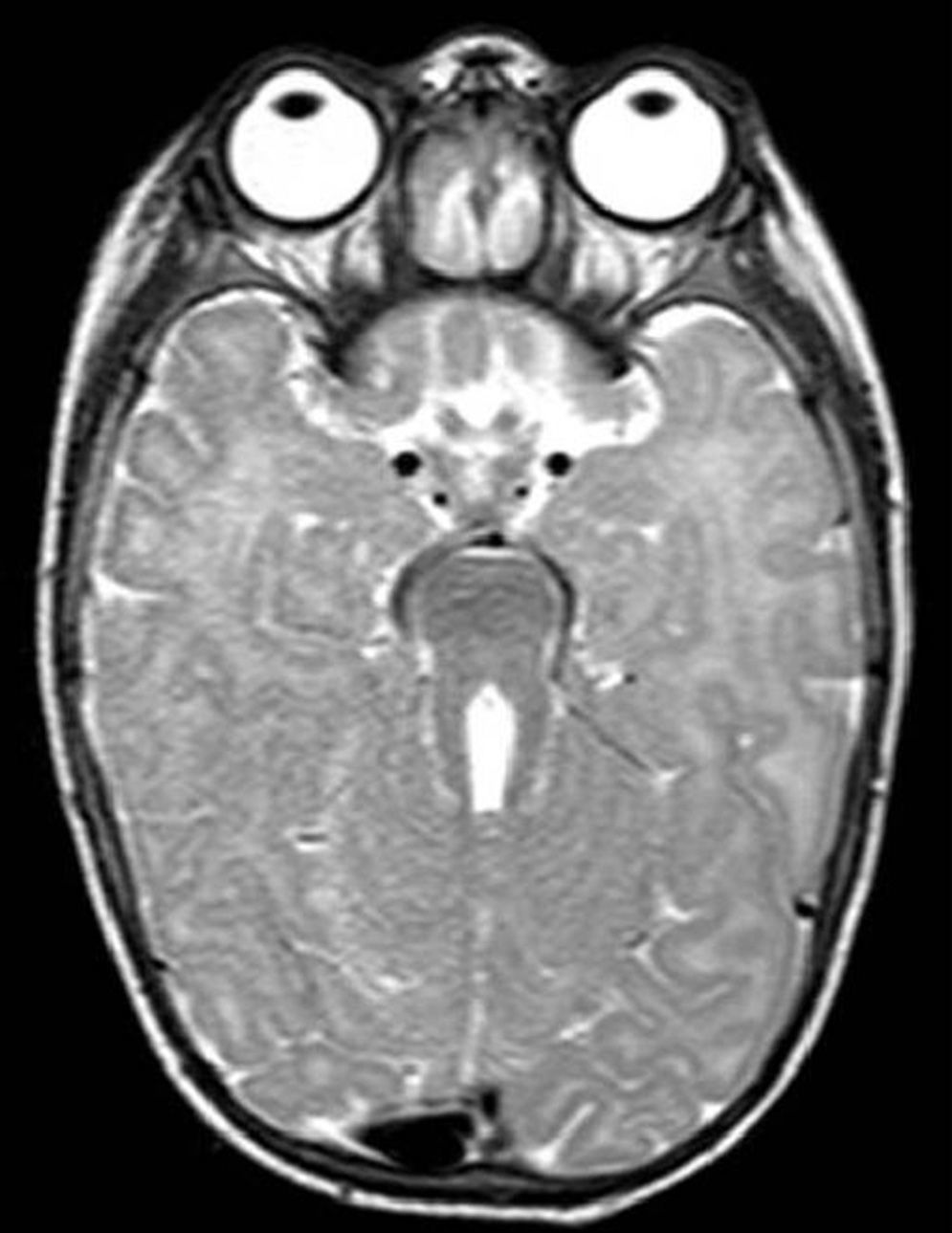
The first case corresponds to a 6-month-old female infant with psychomotor delay and axial hypotonia, vertical nystagmus and erratic eye movements. Cerebellar vermis hypoplasia had been suspected before birth. The postnatal brain MRI revealed the complete absence of the cerebellar vermis with abnormal cerebellar peduncles and a fourth ventricle with the “molar tooth” sign (Fig. 1). Joubert syndrome (OMIM 213300) was confirmed by the detection of the homozygous mutation c.2168G>A (pArg723Gln) in the AHI1 gene. The syndrome is characterised by the congenital malformation of the brainstem and agenesis or hypoplasia of the cerebellar vermis, causing hypotonia and ataxia, delayed motor development, nystagmus, and a tachypnoea/dyspnoea breathing pattern in the neonatal period. Cognitive deficits occur in varying degrees. It can be associated to retinal dystrophy, nephronophthisis and polydactyly, which occur in other ciliopathies. It follows an autosomal recessive inheritance, and mutations in different genes have been identified: AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B and CC2D2A.5 Bardet–Biedl syndrome, Meckel–Gruber syndrome, Leber congenital amaurosis and nephronophthisis share mutations in the CEP290 gene, demonstrating the genetic and clinical overlapping of syndromes associated to a primary ciliary dysfunction.6

The second patient was a newborn that developed respiratory distress requiring oxygen therapy, with no relevant perinatal history. The parents were consanguineous and the previously born sibling had been admitted to the hospital at 12 days of age for respiratory distress with lobar atelectasis. Radiological examination showed left upper lobe atelectasis and situs inversus. Primary ciliary dyskinesia (OMIM 244400) was suspected, a motor ciliopathy that includes Kartagener syndrome and is usually autosomal recessive. The literature has reported mutations in 11 genes: DNAI1, DNAH5, DNAH11, DNAI2, KTU, TXNDC3, LRRC50, RSPH9, RSPH4A, CCDC40 and CCDC39. The dysfunction of ciliated cells leads to chronic respiratory infections. It also affects the flagella of sperm cells and the cilia of the fallopian tubes.3,7 The development of the left-right axis in the embryo is a cilia-dependent mechanism. The dysfunction of nodal cilia leads to random placement of the internal organs, so 50% of these patients have situs inversus totalis.1,7
The third patient is a newborn with a prenatal history of right hydronephrosis. The patient had postaxial polydactyly in both hands and leukocoria in the left eye. Ultrasound examination showed a 50mm right kidney with grades III–IV hydronephrosis, dilation of the renal pelvis (30mm) and the entire ureter up to its distal end, with a prominent ureterocoele. He required decompression of the ureterocoele and early cataract surgery. The clinical picture was characteristic of Bardet–Biedl syndrome (OMIM 209900), a recessive sensory ciliopathy with great clinical variability: obesity, pigmentary retinopathy, postaxial polydactyly, polycystic kidneys, hypogonadism, cognitive impairment, diabetes mellitus and congenital heart disease. Its broad clinical spectrum derives from its genetic heterogeneity, with mutations identified in 12 genes (BBS1 through BBS12).2,3 The differential diagnosis includes Alström syndrome, which also manifests with obesity, diabetes mellitus, nephronophthisis, and pigmentary retinopathy.
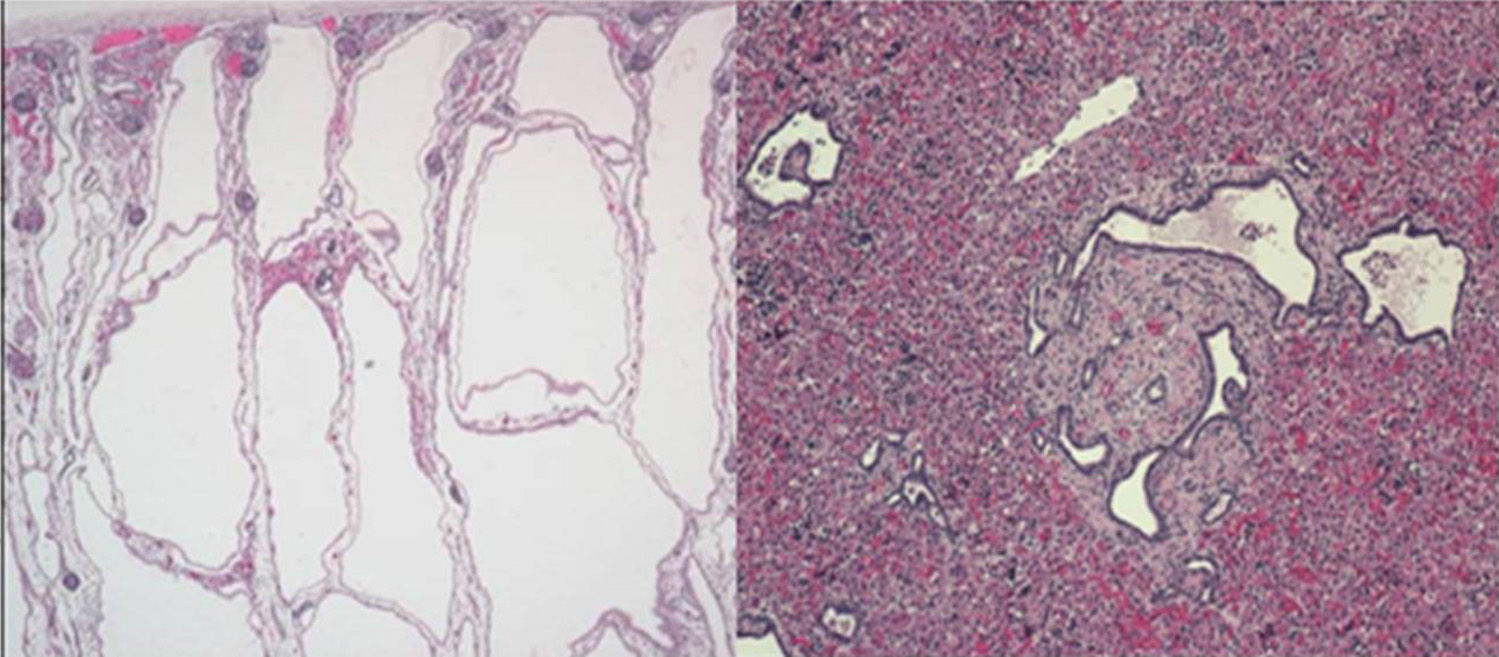
The last clinical case corresponded to a preterm newborn with oligohydramnios and bilateral nephromegaly detected before labour. The father had a history of hypertension and hypertransaminasemia. An emergency caesarean section was performed, and the newborn developed severe respiratory distress. The patient had a small bell-shaped chest with a distended abdomen, and masses could be felt on both sides upon palpation. Radiological examination revealed a pneumothorax unresponsive to drainage and pulmonary hypoplasia. The patient had anuria and hypotension. Abdominal ultrasonography showed microcysts in both kidneys and the liver, compatible with autosomal recessive polycystic kidney disease. The patient died 20h after birth. The autopsy confirmed the diagnosis of polycystic kidney disease (OMIM 263200) (Fig. 2). This is the most frequent ciliopathy in early childhood. It is characterised by the dilation of the renal collecting ducts, progressive renal cystic degeneration and liver fibrosis, and early mortality. It is caused by mutations in the PKHD1 gene that encodes polyductin, a protein that plays a role in the differentiation of the cells that line the collecting ducts.2,3

Ciliopathies comprise a group of genetically heterogeneous clinical entities due to the molecular complexity of the ciliary axoneme. They pose a challenge for research, as they play a part in various diseases and signalling pathways, and are involved in the pathogenesis of obesity, diabetes and oncogenesis.8–10
Please cite this article as: Faus Pérez A, Sanchis Calvo A, Codoñer Franch P, Camarasa Lillo N, Alcover Barrachina I. Ciliopatías: un viaje a través del cilio. An Pediatr (Barc). 2015;82:104–105.

