

Las braquidactilias constituyen un grupo de displasias óseas que afectan a las falanges y/o los metacarpianos/metatarsianos de manos y pies. Existen 5 tipos (A-E) y varios subtipos (A1-A4; E1-E3). La herencia es autosómica dominante con expresividad variable. Los casos de herencia recesiva son excepcionales.
La braquidactilia de tipo C (BDC) se caracteriza por un acortamiento de las falanges medias del segundo, tercer y quinto dedo, así como del primer metacarpiano, pudiendo observarse además desviación cubital del segundo dedo y polidactilia, con hipersegmentación muy característica de las falanges proximales o medias del segundo y tercer dedos. El cuarto dedo está menos afectado, siendo habitualmente el de mayor longitud1–3. Las falanges en «forma de ángel» (fig. 1 A), aunque características, no son patognomónicas de la BDC, pues también acontecen en la denominada «displasia epifisaria falángica en forma de ángel». Es plausible que esta y la BDC formen parte de un espectro clínico común1. Esta anomalía desaparece al completarse la osificación de los huesos de la mano, quedando como una braquidactilia simple1. Otras anomalías asociadas a la BDC son: talla baja con retraso de la edad ósea, deformidad de Madelung, displasia de caderas, pies valgos o equinovaros, hipodontia o ausencia de falanges medias en dedos de los pies2,3.
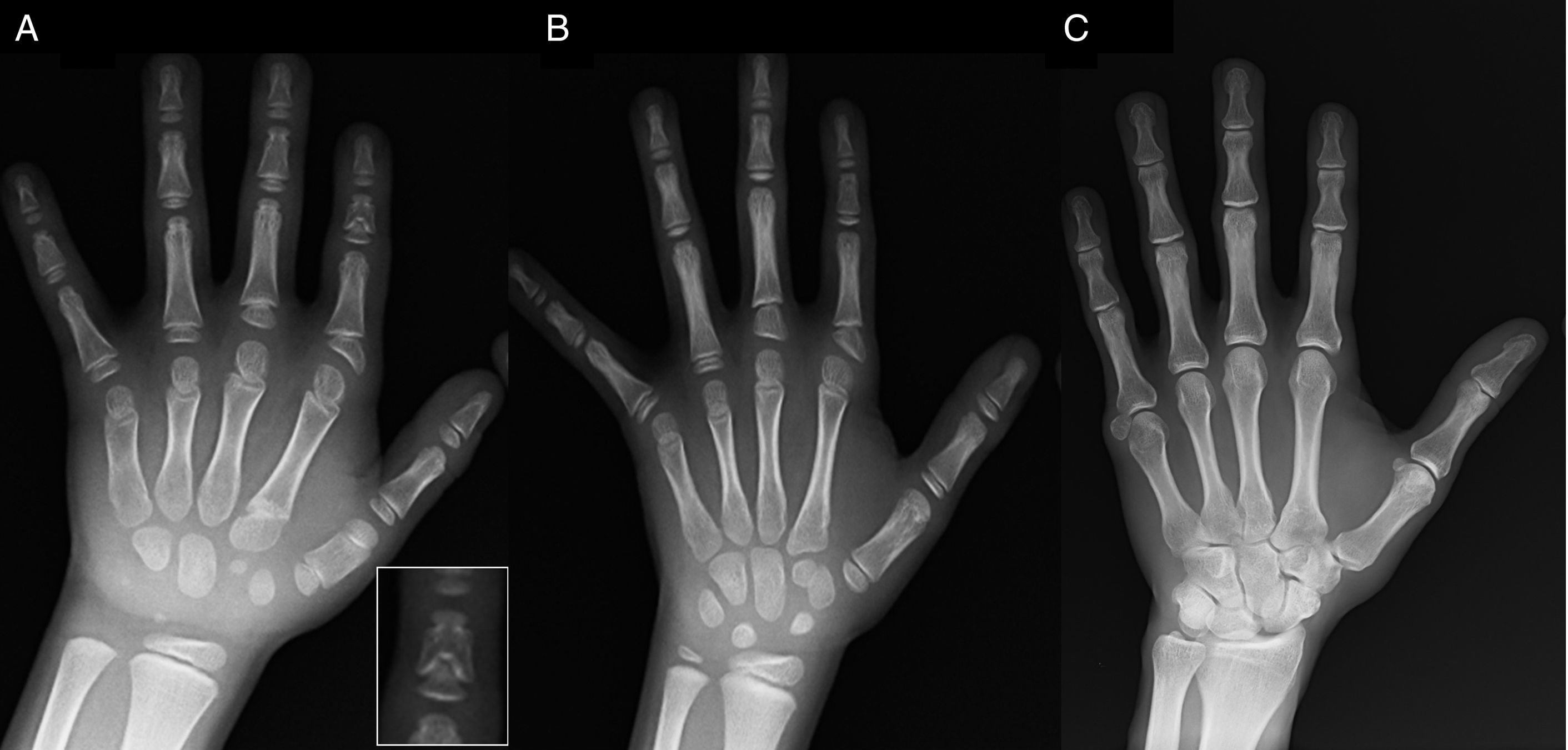
 Radiografía de la mano izquierda del probando (7 años de edad cronológica y 5 años de edad ósea) con: acortamiento de un primer metacarpiano anómalo (doble epífisis, proximal y distal) y de falanges medias del segundo, tercer y quinto dedos. El segundo dedo muestra una desviación cubital y el cuarto dedo es el menos afectado, siendo el más largo de la mano izquierda. Las epífisis proximales de las falanges proximales del segundo y tercer dedos son displásicas, y llama la atención la forma de ángel de la falange media del segundo dedo (detalle en A). B). Radiografía de la mano izquierda de la hermana del probando (5 años y medio de edad cronológica, sin retraso en la edad ósea) con: acortamiento de las falanges medias del segundo, tercer y quinto dedos, sin afectación del cuarto. En este caso, el primer metacarpiano es normal. Destaca la forma triangular de la epífisis proximal de la falange proximal del segundo dedo, similar a la del hermano, y la forma trapezoidal de la del tercer dedo. Al igual que su hermano, el segundo dedo presenta desviación cubital. C) Radiografía de la mano izquierda del padre del probando, donde solo se aprecia un resto óseo correspondiente a una hexadactilia postaxial intervenida en la infancia.")
A) Radiografía de la mano izquierda del probando (7 años de edad cronológica y 5 años de edad ósea) con: acortamiento de un primer metacarpiano anómalo (doble epífisis, proximal y distal) y de falanges medias del segundo, tercer y quinto dedos. El segundo dedo muestra una desviación cubital y el cuarto dedo es el menos afectado, siendo el más largo de la mano izquierda. Las epífisis proximales de las falanges proximales del segundo y tercer dedos son displásicas, y llama la atención la forma de ángel de la falange media del segundo dedo (detalle en A). B). Radiografía de la mano izquierda de la hermana del probando (5 años y medio de edad cronológica, sin retraso en la edad ósea) con: acortamiento de las falanges medias del segundo, tercer y quinto dedos, sin afectación del cuarto. En este caso, el primer metacarpiano es normal. Destaca la forma triangular de la epífisis proximal de la falange proximal del segundo dedo, similar a la del hermano, y la forma trapezoidal de la del tercer dedo. Al igual que su hermano, el segundo dedo presenta desviación cubital. C) Radiografía de la mano izquierda del padre del probando, donde solo se aprecia un resto óseo correspondiente a una hexadactilia postaxial intervenida en la infancia.
Presentamos a un varón de 7 años con signos radiológicos compatibles con BDC, remitido por talla baja (–1,8 SDS). El padre mostraba polidactilia postaxial unilateral, siendo bilateral en el tío paterno. La radiografía de mano y muñeca izquierdas evidenciaba retraso en la edad ósea de 2 años y anomalías que motivaron la realización de una serie ósea. Se detectaron alteraciones en las manos (fig. 1 A) y de forma muy sutil en los pies (leve displasia epifisaria en las falanges proximales de algunos dedos). El estudio radiológico de la hermana de 6 años mostró así mismo lesiones en manos similares a las del probando, pero menos marcadas (fig. 1 B). La radiografía de la mano del padre (fig. 1 C) solo mostró un resto óseo correspondiente a la hexadactilia postaxial intervenida en la infancia.
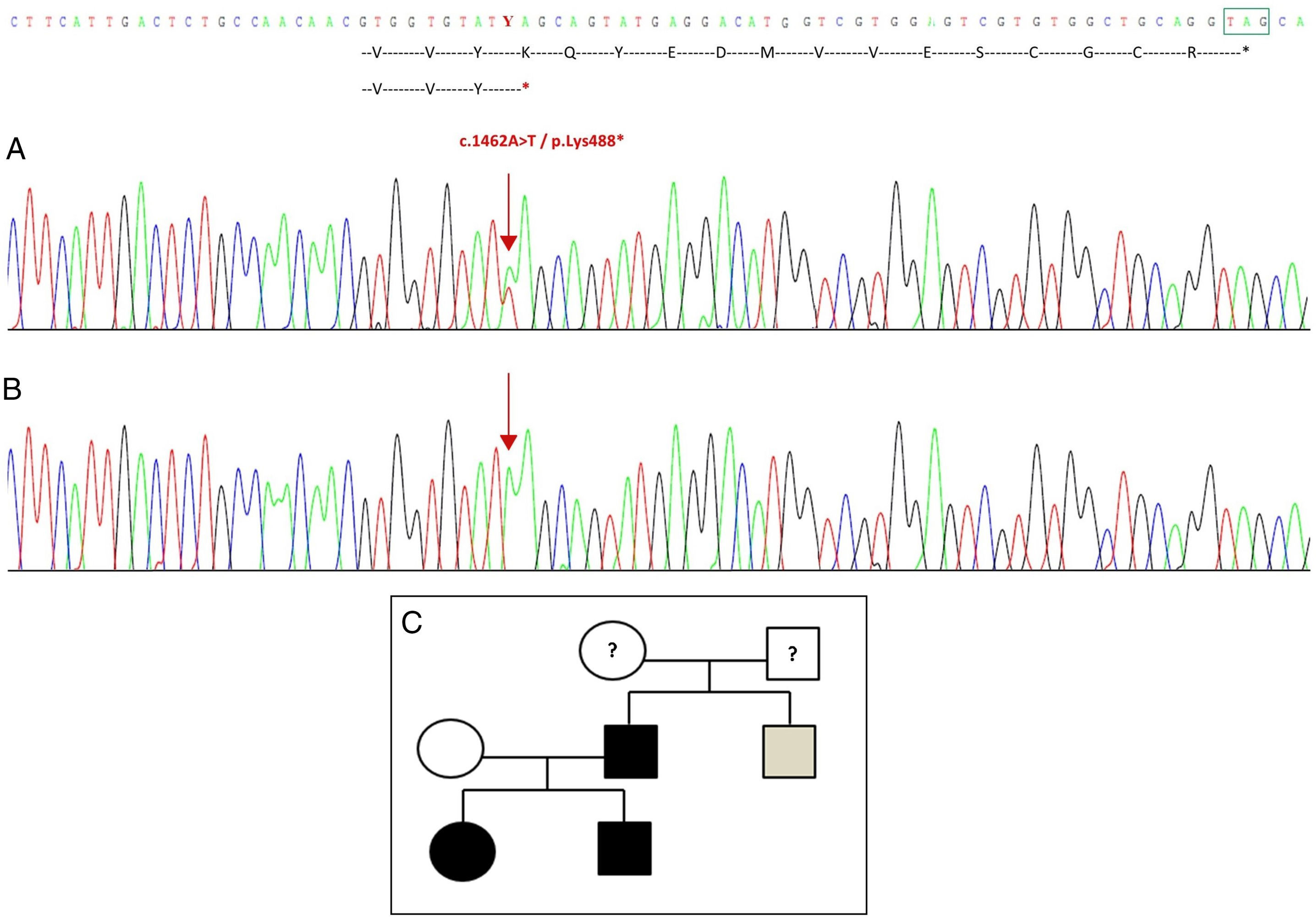
La secuenciación del gen GDF5 en el probando reveló un cambio de nucleótido (c.1462A>T) en heterocigosis en el exón 2, que conlleva un codón de terminación prematuro (p.Lys488*, mutación nonsense), dando lugar así a una proteína truncada de 14 aminoácidos menos que la silvestre (fig. 2 A). Esta mutación está presente en el padre y en la hermana del paciente, pero no en la madre sana (fig. 2 B). El pedigree se representa en la figura 2 C. Se trata de una nueva variante, probablemente patogénica y con patrón de herencia autosómico dominante.
 Secuenciación del exón 2 del probando donde se observa el cambio c.1462A>T que da lugar a la aparición de un codón stop prematuro y a una proteína truncada (p.Lys488*) (seha indicado el codón stop natural en cuadrado verde). B) Misma secuencia normal de la madre sana. C) Árbol genealógico familiar: en negro se muestra al padre y ambos hijos con mutación demostrada en GDF5. En blanco, la madre sana. En gris, se muestra un hermano del padre con polidactilia postaxial bilateral, probablemente afectado.")
Mutación novel hallada en GDF5. A) Secuenciación del exón 2 del probando donde se observa el cambio c.1462A>T que da lugar a la aparición de un codón stop prematuro y a una proteína truncada (p.Lys488*) (seha indicado el codón stop natural en cuadrado verde). B) Misma secuencia normal de la madre sana. C) Árbol genealógico familiar: en negro se muestra al padre y ambos hijos con mutación demostrada en GDF5. En blanco, la madre sana. En gris, se muestra un hermano del padre con polidactilia postaxial bilateral, probablemente afectado.
El growth differentiation factor 5 (GDF5) es un factor de crecimiento estrechamente relacionado con las proteínas morfogenéticas del hueso y perteneciente a la superfamilia de los transforming growth factor ß, que interviene en el desarrollo embrionario del esqueleto y las articulaciones4. El GDF5 es un gen hot-spot para enfermedades asociadas a malformaciones esqueléticas5. La mayoría de las mutaciones en homocigosis o heterocigosis compuesta se asocian a enfermedades graves: condrodisplasia de Grebe (OMIM#200700), displasia acromesomélica de Hunter-Thomson (OMIM#201250) o síndrome de Du Pan (OMIM#228900). En cambio, las mutaciones en heterocigosis se asocian a displasias esqueléticas más leves: sinfalangismo proximal 1B (OMIM#615298) y síndrome de sinostosis múltiple tipo 2 (OMIM#610017), en ambos casos con mutaciones missense de ganancia de función, y, con las braquidactilias A1 y A2, asociadas también con mutaciones missense, pero con pérdida de función5. La BDC está asociada a mutaciones en heterocigosis con pérdida de función, aunque también se han descrito 3 casos con un patrón recesivo6. La mayoría de las mutaciones asociadas a BDC se localizan en el propéptido y son de tipo frameshift; en cambio, las mutaciones localizadas en el dominio activo son en su mayoría missense y muy variables en sus manifestaciones clínicas4.
Nuestra familia presenta una mutación nonsense localizada en el dominio activo, que eliminaría los 14 últimos aminoácidos de la proteína. Es la segunda mutación nonsense que se describe afectando al dominio activo maduro3. La ya descrita es una mutación similar en el aminoácido anterior (p.Tyr487*/c.1461T>G) al de nuestra familia, lo que indica que ambas darían lugar a monómeros mutantes con haploinsuficiencia funcional de GDF5 causando así la BDC.
FinanciaciónEste trabajo ha sido parcialmente financiado por los proyectos PI13/00467 y PI13/01295, integrados en el plan Estatal de I+D+I 2013-2016 y cofinanciados por el ISCIII-Subdirección General de Evaluación y Fomento de la investigación y el Fondo Europeo de Desarrollo Regional (FEDER) y por el CIBER de Fisiopatología de la Obesidad y Nutrición (CIBEROBN). Instituto de Salud Carlos III, Madrid.

