

Las anomalías nefrourológicas congénitas (CAKUT) son alteraciones con una alta prevalencia en la población general; de ellas, las más frecuentes son las dilataciones de la vía urinaria. Suponen además la causa más importante de enfermedad renal crónica en la edad infantil.
En este artículo se hace especial énfasis en el papel del pediatra de Atención Primaria en la valoración y el seguimiento de los niños con CAKUT, fundamentalmente en lo que hace referencia a las infecciones urinarias asociadas, a la progresión hacia la insuficiencia renal y a su base genética.
The congenital abnormalities of kidney and urinary tract (CAKUT) are disorders with a high prevalence in the general population, with urinary tract dilations being the most frequent. CAKUT also account for the most important cause of chronic kidney disease in childhood.
This paper focuses on the role of the primary care paediatrician in the diagnosis, assessment, and follow-up of children with CAKUT, with special emphasis on the associated urinary tract infections, the progression toward chronic renal failure, and the genetic basis.
Las anomalías nefrourológicas congénitas se conocen en la literatura internacional como congenital abnormalities of kidney and urinary tract (CAKUT), por lo que este será el término que se empleará en este artículo. La finalidad del mismo no es revisar la sistemática de diagnóstico y tratamiento que debe de seguirse ante los diversos tipos de CAKUT, ya que habitualmente estos procedimientos se realizan en unidades hospitalarias de Nefrología y Urología Pediátricas, sino comentar aspectos actuales sobre esta patología de interés para el pediatra. En su conjunto, las CAKUT constituyen un grupo de enfermedades de gran relevancia en la práctica clínica por ser de alta prevalencia, por causar enfermedad renal crónica y por estar produciéndose en los últimos años importantes novedades y avances en el conocimiento de su patogenia y expresividad clínica.
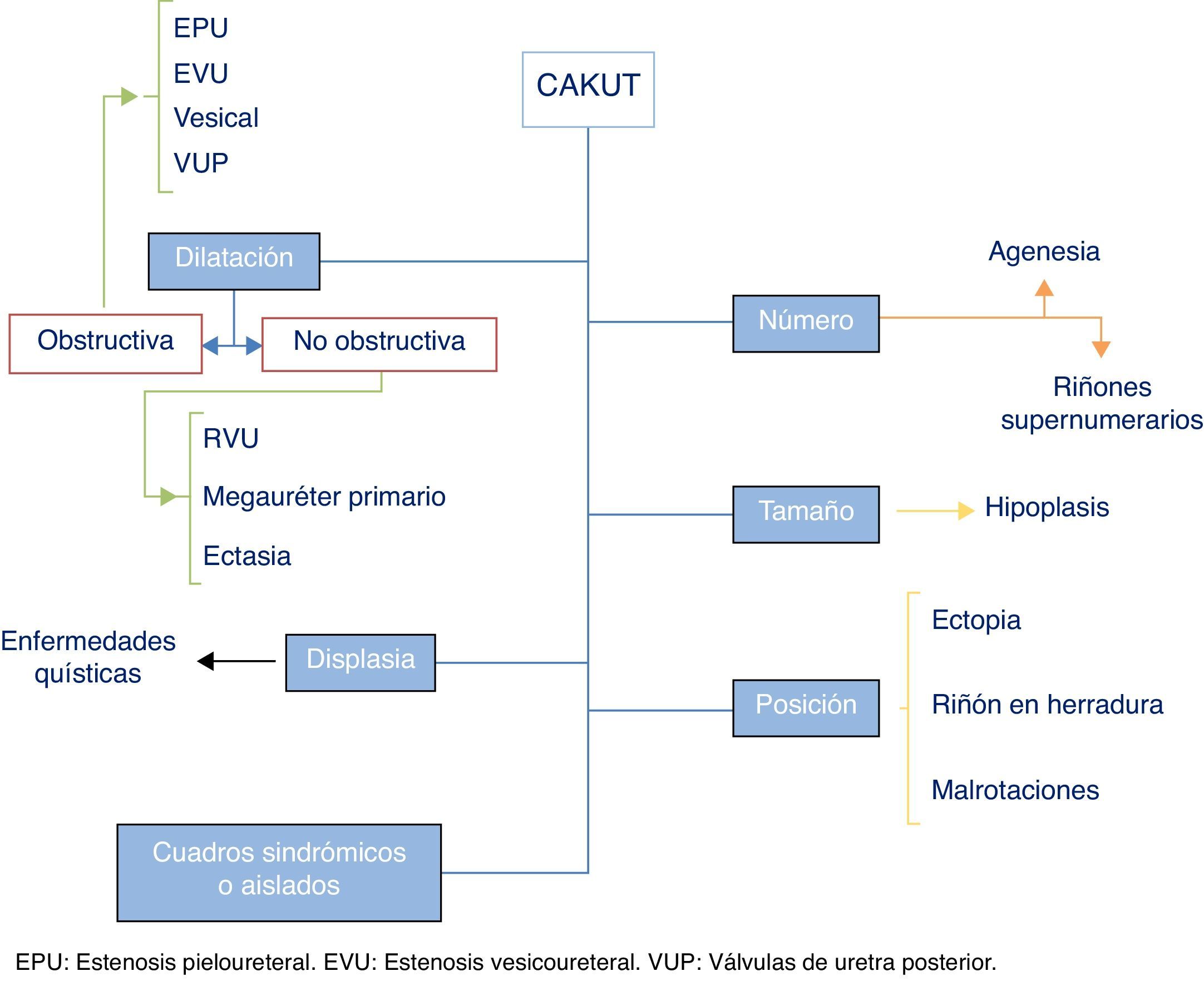
Se incluyen como CAKUT un gran número de entidades ocasionadas por el anormal desarrollo embriológico del aparato urinario, que comprenden alteraciones en el número, el tamaño y/o la posición de los riñones, dilatación obstructiva o no obstructiva de la vía urinaria y lesiones de displasia renal, incluyendo enfermedades quísticas. Pueden darse de forma aislada o en el contexto de un cuadro sindrómico. La figura 1 presenta gráficamente una clasificación general de los diversos tipos de CAKUT (es de notar que el doble sistema pieloureteral debe de considerarse una variante de la normalidad, a no ser que tenga otras anomalías asociadas). Algunas series incluyen dentro de este término a la poliquistosis renal, en sus formas autosómica dominante y autosómica recesiva, y al complejo nefronoptisis. Aunque en sentido estricto puede ser así, estas enfermedades requieren un planteamiento diagnóstico y terapéutico claramente diferenciado y no tienen muchas de las características clínicas y evolutivas que comparten en general las CAKUT, por lo que no se hará nueva mención a estas entidades en este artículo.
.")
En nuestro medio, las CAKUT se detectan generalmente antes de nacer, de forma que representan un tercio aproximadamente de las alteraciones ecográficas prenatales. Un 5-10/1.000 de los recién nacidos vivos tienen algún tipo de CAKUT, siendo así las alteraciones del desarrollo más habitualmente encontradas en humanos1. Del total de estas anomalías, aquellas que cursan con dilatación de la vía urinaria son las más frecuentes. Hay que señalar que así como las dilataciones de la vía urinaria se identifican fácilmente en el periodo prenatal, no es infrecuente que los riñones pequeños (hipoplasia renal) pasen inadvertidos en los exámenes ecográficos de la gestante. Es importante tener presente esta consideración a la hora de establecer las indicaciones clínicas de la ecografía renal en la vida postnatal.
Este artículo intentará ser de utilidad para el pediatra en relación con la respuesta a las siguientes preguntas acerca de CAKUT: ¿implican más riesgo de infecciones urinarias (IU) y cómo son estas?, ¿cuál es la evolución renal a largo plazo?, ¿qué conocemos sobre su base genética y qué orientación hay que dar a las familias sobre herencia y riesgo de recurrencia en otros hijos?
Infección urinariaEs bien conocido que las IU son más frecuentes en el sexo femenino, de forma que un 7-8% de niñas y un 2% de niños tienen alguna IU durante los primeros 8 años de vida2. Las IU febriles tienen la máxima incidencia en el primer año de vida en ambos sexos2 y esta mayor presencia de varones en el periodo neonatal y los primeros meses de la vida está relacionada con la mayor frecuencia de CAKUT en niños que en niñas3. Los lactantes con dilatación de la vía urinaria tienen mayor riesgo de presentar pielonefritis aguda durante el primer año de vida, de manera que la probabilidad de ser hospitalizados por esta causa es aproximadamente 12 veces mayor que en los niños sin hidronefrosis.
Aunque el uso de quimioprofilaxis continuada para la prevención de IU es controvertido, una evaluación de 21 estudios publicados entre 1990 y 2010 sobre la incidencia de IU en niños menores de 2 años con hidronefrosis prenatal concluyó que la quimioprofilaxis no disminuye el porcentaje de IU en los pacientes con hidronefrosis de bajo grado (hallazgos ecográficos de ectasia calicilial inexistente o mínima, y parénquima renal no adelgazado y/o diámetro anteroposterior de pelvis renal postnatal entre 4,0 y 14,9mm), en los que la IU se diagnosticó en un 2,5% aproximadamente, con independencia de que recibieran o no profilaxis, pero sí disminuye significativamente el riesgo de IU del 29 al 15% en pacientes con hidronefrosis de grados iii y iv y/o diámetro anteroposterior de pelvis mayor o igual a 15mm, aunque con un bajo grado de evidencia4. Así pues, los lactantes con hidronefrosis prenatal de alto grado pueden beneficiarse de quimioprofilaxis para reducir la probabilidad de presentar IU.
Otro aspecto que es importante señalar es que el Escherichia coli (E. coli) tiene menos protagonismo como agente productor de IU en los pacientes pediátricos con CAKUT que en la población general5, lo que es preciso tener en cuenta a la hora de instaurar medidas terapéuticas y/o profilácticas. Es asimismo necesario recordar que así como la IU, incluso si es recurrente, entraña un riesgo muy bajo de causar enfermedad renal crónica6, la asociación de pielonefritis aguda y uropatía obstructiva implica una alta probabilidad de daño renal permanente, a no ser que se actúe rápidamente con antibioterapia y solución de la obstrucción que permita el drenaje de la orina infectada en el riñón afecto.
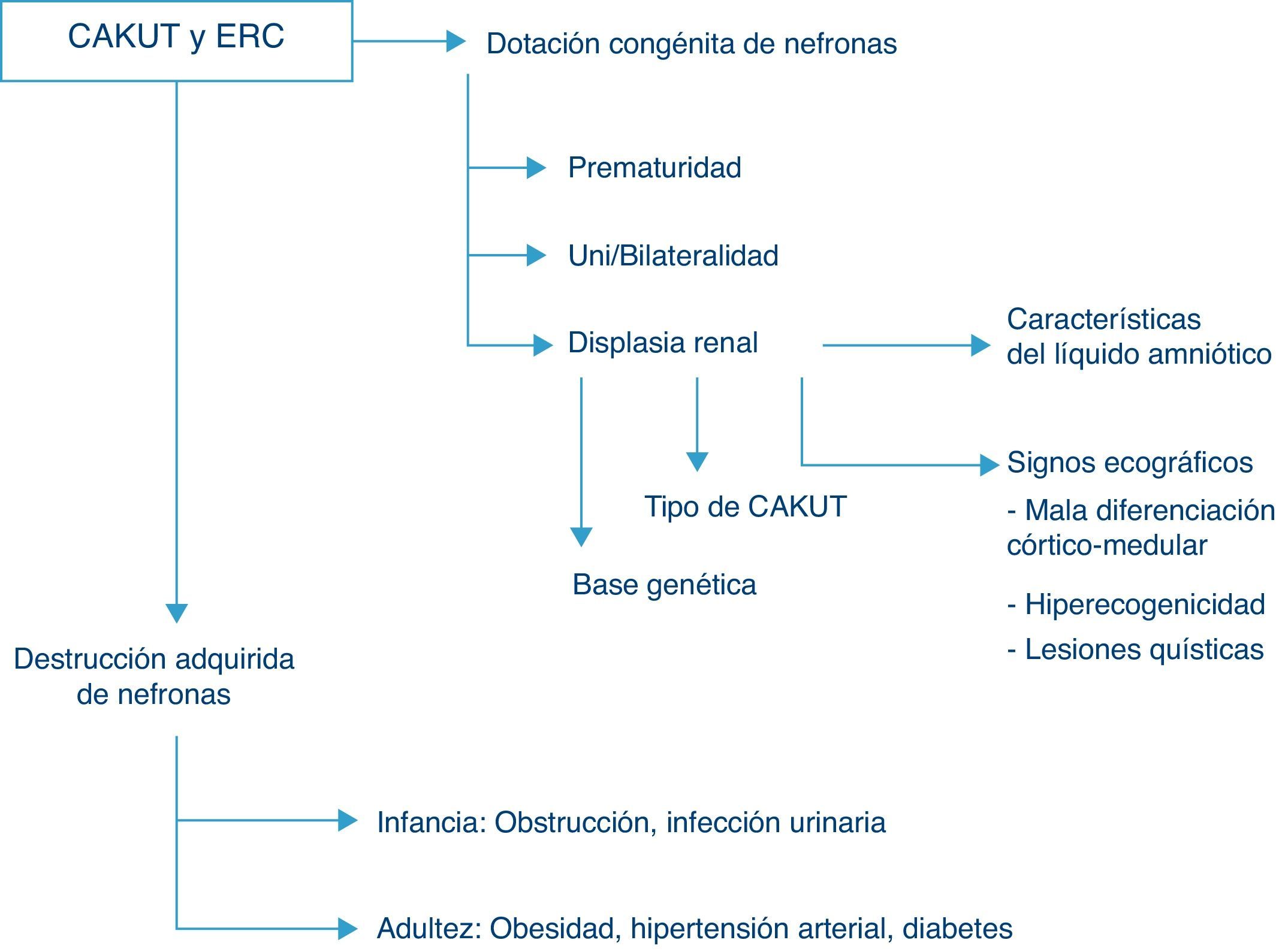
Evolución de la función renalLas CAKUT representan la causa porcentualmente más importante de enfermedad renal crónica en todas las series pediátricas, pudiendo originar si se incluyen las enfermedades quísticas hasta casi las 3 cuartas partes del total de los casos de insuficiencia renal crónica en niños. En el registro de la Asociación Española de Nefrología Pediátrica un 56% de los pacientes con fallo renal crónico en estadios 2 a 4 (no en diálisis) son debidos a anomalías nefrourológicas estructurales y este porcentaje es similar al observado en series pediátricas europeas y norteamericanas, en las que el porcentaje de enfermedad renal crónica debida a CAKUT se sitúa entre el 48 y el 59%7. El riesgo de enfermedad renal crónica depende del número congénito de nefronas funcionantes, y por lo tanto depende de la existencia de prematuridad, del grado de displasia renal y de la afectación unilateral o bilateral, así como de la destrucción adquirida de nefronas, fundamentalmente debida, en la edad infantil, a la concurrencia de IU de vías altas y/o a la persistencia de obstrucción (fig. 2).
 de las anomalías congénitas nefrourológicas (CAKUT). Véase el texto para explicación y detalles.")
Aunque el término displasia hace referencia a características anatomopatológicas del tejido que se trate, diversos datos clínicos pueden hacer sospechar su existencia, por ejemplo, los hallazgos ecográficos de hiperecogenicidad del parénquima renal, pérdida de la normal diferenciación córtico-medular o la detección de quistes renales. Prenatalmente, a las 20 semanas de gestación la orina fetal ya representa más del 90% del volumen total de líquido amniótico, de modo que el hallazgo de oligohidramnios más allá de esta edad gestacional es un excelente indicador de función renal fetal anormal8. En general, concentraciones de sodio y cloro mayores de 90 mEq/l y osmolalidad inferior a 210 mOsm/kg H2O en el líquido amniótico indican afectación tubular renal y pronóstico sombrío, aunque ninguna de estas determinaciones bioquímicas predice con seguridad clínica suficiente la función renal posnatal9. Además del daño renal congénito por displasia, la nefropatía asociada a las CAKUT es inicialmente de origen intersticial, por lo que, en caso de dar lugar a insuficiencia renal crónica, esta cursa con diuresis conservada e incluso con poliuria. La aparición de microalbuminuria se considera un marcador de daño glomerular, ya manifiesto cuando se detecta proteinuria mantenida, siempre que no sea de proteínas de bajo peso molecular.
La presencia de insuficiencia renal desde el nacimiento o desde las primeras semanas de vida es un dato de mal pronóstico con respecto a la función renal, aunque en algunos de estos niños la filtración glomerular, corregida por 1,73 m2, puede mejorar con el rápido crecimiento corporal del primer año. También es preciso tener en cuenta que la disminución de la función renal puede agravarse durante la pubertad al no crecer el riñón o los riñones afectados en la misma proporción que el resto del organismo. Del mismo modo, una estenosis relativa de la vía urinaria puede acentuarse durante el estirón puberal y ocasionar una uropatía obstructiva que comprometa la función renal del riñón dilatado. Hay que tener en mente estas circunstancias y posibilidades evolutivas al planificar el seguimiento clínico de estos pacientes cuando inician el periodo puberal.
Base genéticaLa patogenia de las diferentes entidades englobadas como CAKUT es compleja, en consonancia con el complicado mecanismo de formación y desarrollo embriológico del aparato urinario. Las CAKUT son más frecuentes en varones que en mujeres y existen casos de agregación familiar. Pueden formar parte de cuadros multiorgánicos en entidades de transmisión dominante o recesiva en las que se conoce que el defecto causal es monogénico, como el síndrome branquio-oto-renal, el síndrome de Kallmann, el síndrome de Fraser, el síndrome de Ehlers-Danlos y el síndrome de Townes-Brocks, entre otras.
Existen además formas de CAKUT de origen monogénico en las que el fenotipo renal es predominante o aislado. Entre los diversos genes involucrados destacan por su frecuencia los genes HNF1β y PAX2. Alteraciones del gen PAX2 causan hipodisplasia renal asociada a coloboma y sordera10. Además, se han relacionado mutaciones de este gen con hipoplasia y displasia renal aisladas y con displasia renal multiquística11. Los defectos del gen HNF1β que codifica el factor de transcripción del mismo nombre (factor nuclear de hepatocitos 1 β) se han mostrado en los últimos años como causantes de diversas alteraciones del desarrollo, habiéndose incluso sugerido recientemente el término de enfermedad HNF1β12. Este factor de transcripción está involucrado en la organogénesis de los riñones, la vía urinaria, el hígado y el páncreas, y se ha demostrado que mutaciones en este gen explican hasta un 10% aproximadamente de los casos de CAKUT13. La existencia de mutaciones subyacentes en el gen HNF1β responsables de CAKUT debería sospecharse ante lesiones renales bilaterales con hallazgos de quistes renales de origen no conocido, asociados a antecedentes familiares de diabetes, hipoplasia pancreática y alteraciones electrolíticas como hipomagnesemia e hiperuricemia.
Así pues, la evaluación de un paciente con CAKUT debería de incluir, además de los estudios relacionados directamente con su alteración estructural nefrourológica, la búsqueda específica de manifestaciones extrarrenales y la realización de una meticulosa historia familiar que ayuden a identificar la causa molecular subyacente y a facilitar el consejo genético a las familias afectas.
Puntos ClaveLos siguientes puntos clave deben de recordarse en el diagnóstico y seguimiento de un niño con CAKUT por parte de su pediatra:
- –
Bajo el término de CAKUT se engloban un grupo heterogéneo de entidades que son muy frecuentes, se asocian con IU y representan la causa más importante de enfermedad renal crónica en la edad pediátrica.
- –
En los casos en los que no se ha detectado prenatalmente, debe sospecharse CAKUT en lactantes varones con IU, especialmente ante pielonefritis que requieren hospitalización, y sean debidas a gérmenes distintos al E. coli. Es necesario tener en cuenta este hecho no solo para establecer la antibioterapia, sino en el seguimiento, pues en el contexto de una uropatía obstructiva las IU de vías altas entrañan alto riesgo de causar daño renal permanente, por lo que hay que vigilar estrechamente su evolución.
- –
Un correcto diagnóstico de CAKUT exige realizar una historia clínica detallada, incluyendo antecedentes familiares de problemas renales y de diabetes y la búsqueda de anomalías extrarrenales asociadas: coloboma, sordera, hipomagnesemia, hiperuricemia, etc.
- –
El seguimiento de estos pacientes debe de procurar evitar los factores que puedan agravar el daño renal congénito existente, con especial atención a la prevención y el tratamiento de pielonefritis agudas, a la erradicación de la obstrucción urinaria y a la promoción de hábitos de vida saludables. En este sentido, es muy importante la entidades, todas ellas que conllevan una situación de hiperfiltración mantenida de las nefronas funcionantes residuales, y con ello agravan el riesgo de glomeruloesclerosis. La aparición de microalbuminuria patológica y, desde luego, de proteinuria mantenida, es marcador de daño glomerular.
- –
La profilaxis antibiótica en niños con hidronefrosis solo ha demostrado disminuir el riesgo de IU en pacientes con hidronefrosis de alto grado.
- –
Durante el estirón puberal, la disminución de la función renal puede agravarse, por lo que, especialmente en casos de alteraciones nefrourológicas como hipoplasia renal o estenosis de la vía urinaria, se debe plantear de forma específica al médico de familia de Atención Primaria (que va a continuar la atención sanitaria) la situación clínica del paciente para asegurar un seguimiento específico en este periodo de la vida.
Los autores declaran no tener ningún conflicto de intereses.
Parcialmente presentado en la Reunión de la Sociedad de Pediatría de Asturias, Cantabria y Castilla y León, 10-11 abril del 2015.

