

Global developmental delay (GDD) and intellectual disability (ID) are frequent diseases that have a considerable social impact and a varied aetiology. Cytogenetic testing by comparative genomic hybridisation (CGH) array analysis has increased the number of aetiological diagnoses in our paediatric neurology clinic by more than 20%.1
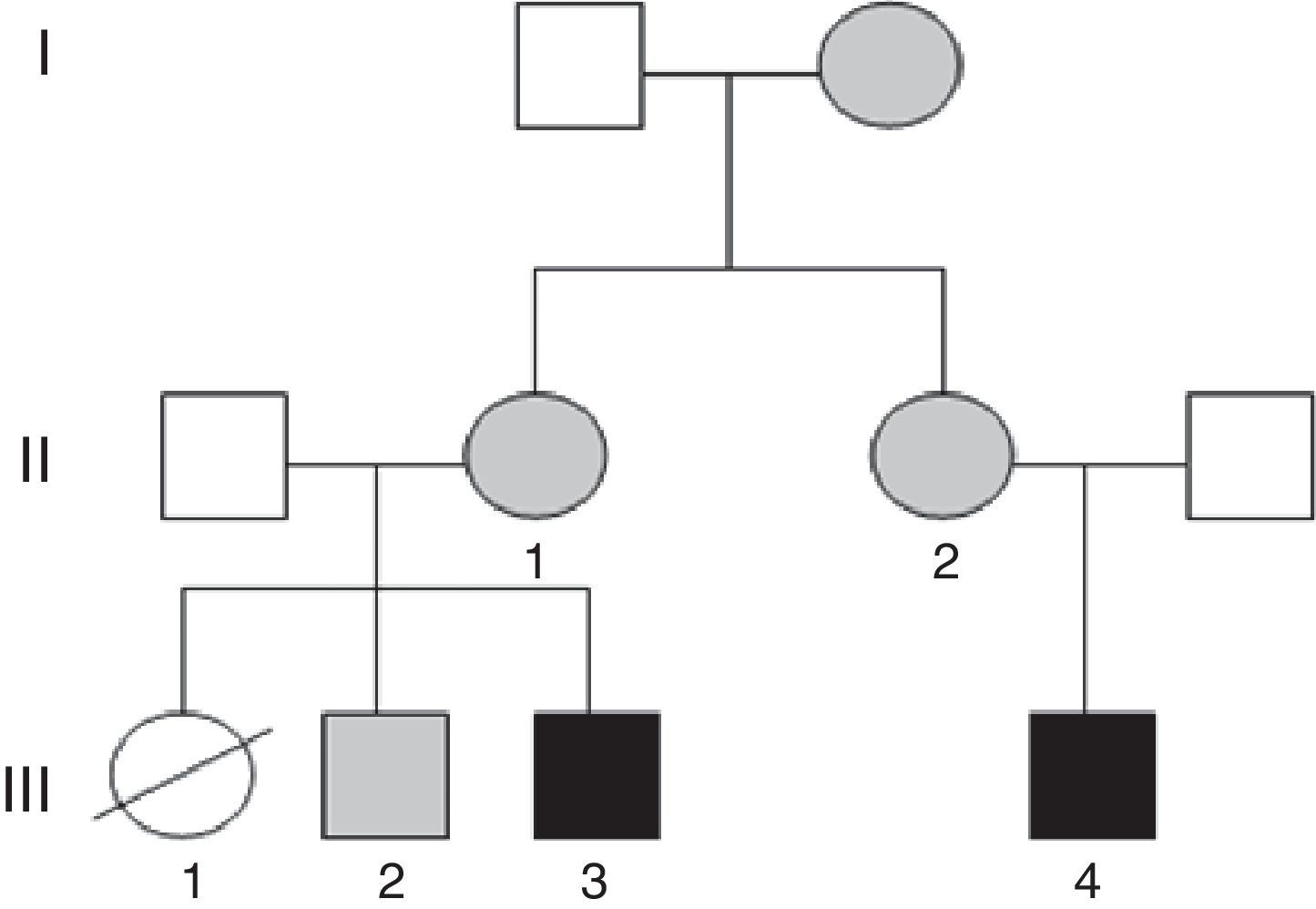
We present the clinical cases in a family with several members with ID in whom testing detected balanced and unbalanced chromosomal rearrangements secondary to a different chromosomal segregation process in each.
Case 1 (Fig. 1, individual III.4): boy aged 5 years, only child, with ID, oppositional behaviours, deficits in propositional language and no sphincter control. He had microcephaly from birth (head circumference, −4.21 SD), with height and weight at the 75th percentile, a dysmorphic phenotype, hexadactyly in both feet, and atrial septal defect that required surgery. Testing by CGH array detected a duplication at the 15q26.3 locus and a partial deletion of the 7p22 region.
.")
Haploinsufficiency of the MAD1L1, FTSJ2, NUDT1, SNX8, EIF3B and FAM20C genes in chromosome 7, whose dosage was abnormal in the patient, has been associated to heart malformations and ID.2
Increased dosage of the distal region of chromosome 15 has been associated with 15q overgrowth syndrome (tendency to macrosomia, polydactyly, ID and behavioural problems), described in more than 100 patients.3 Classically it has been associated with the IGF1R gene. However, a recent study described a family in which the duplication was not found in this gene, as occurred in case 1, and proposed that it was located in the LRRK1 gene instead (of which there were also 3 copies in case 1 in our series), giving rise to the described phenotype. This gene encodes a protein that is believed to be involved in the regulation of cell growth and proliferation.4
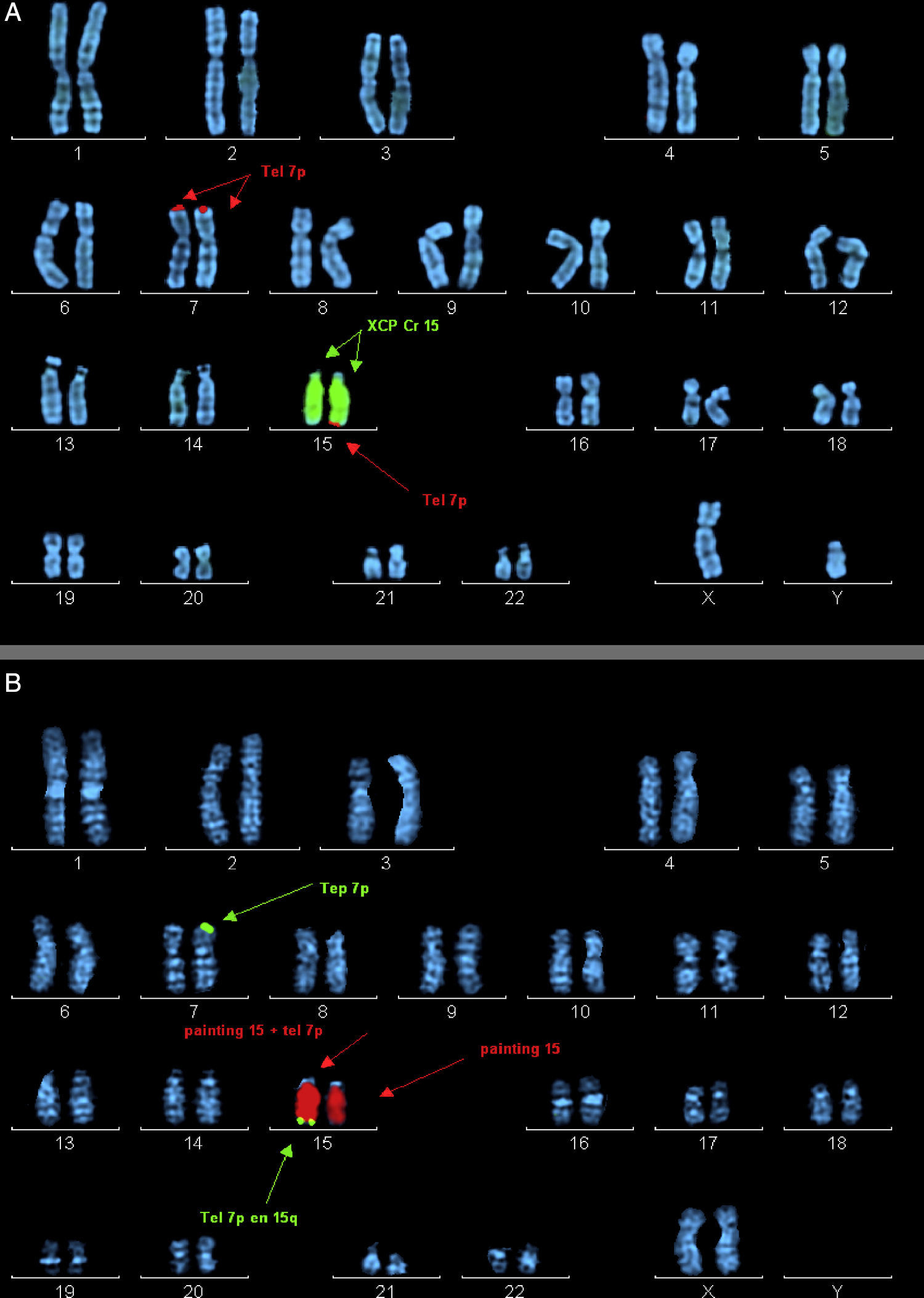
Case 2 (Fig. 1, individual III.3): boy aged 3 years, first cousin of case 1, whose older sister died at age 2 months after suffering from transposition of the great vessels, cleft lip and microcephaly (Fig. 1, individual III.1). The boy presented with GDD, macrocephaly, dysmorphic features, delayed fontanelle closure, joint hyperlaxity and clubfoot. Array analysis detected a deletion in region 15q26.3 and duplication of the 7p22.3p22.1 locus (Fig. 2).
. (A) Case 2 proband. Labelled with probe XCP Cr15 (marked with central arrows) and telomere probe 7p (marked by top and bottom arrows). (B) Mother of case 2. Labelled with probe XCP Cr15 (marked by central arrows) and telomere probe 7p (marked by top and bottom arrows).")
Ordered metaphase chromosomes labelled by means of fluorescence in situ hybridisation (FISH). (A) Case 2 proband. Labelled with probe XCP Cr15 (marked with central arrows) and telomere probe 7p (marked by top and bottom arrows). (B) Mother of case 2. Labelled with probe XCP Cr15 (marked by central arrows) and telomere probe 7p (marked by top and bottom arrows).
As for partial trisomy of the 5.1Mb distal segment of the short arm of chromosome 7, which alters the dosage of more than 40 RefSeq genes, there are more than 40 patients with similar duplications described in the literature. They are characterised by a specific facial dysmorphism (macrocephaly, delayed fontanelle closure, hypertelorism and low-set ears) in addition to ID, language disorders, hypotonia, low birth weight, cryptorchidism and heart malformations. A recent study has attributed macrocephaly to the duplication of RNF216, which was found in case 2 (III.3).5
On the other hand, the terminal deletion of the long arm of chromosome 15, of approximately 2.2Mb, impacts more than 10 RefSeq genes. To present, 12 cases with distal deletions have been described in the literature, manifesting with pre- and postnatal growth retardation, heart malformations, developmental delays and dysmorphic features.6
The mothers of both subjects (who are sisters) had the karyotype 46, XX t(7;15) (p22;q26), and there was a healthy sibling of case 2 with karyotype 46, XY t(7;15) (p22;q26) and normal array results (Fig. 1, individual III.2). Subsequently, we found out that the maternal grandmother was also a carrier of the balanced translocation.
It is essential that all members of the family receive genetic counselling, given the high probability that the sister (III.1) of case 2 (III.3) and the cousin of case 1 (III.4) had inherited an unbalanced translocation. Heart disease, which was found in the patient, is associated both to deletions and duplications in the short arm of chromosome 7; however, duplications manifest with macrocephaly, which was present in her brother (case 2, III.3), so it is more likely that she had a deletion in chromosome 7 and a duplication in chromosome 15, as did her cousin (case 1, III.4); both of them had microcephaly, which is frequent in genetic neurodevelopmental disorders.
Individuals that carry more than one variant of the pathological number of copies have more severe phenotypes. Due to this fact and the variable expression of many of these chromosomal disorders, it is difficult to establish genotype–phenotype correlations in these patients.
On the other hand, we ought to highlight recent findings obtained through massive sequencing of the breakpoints of balanced as well as unbalanced translocations, with chromosome rearrangements at multiple breakpoints with inversions or insertions that cannot be detected by other techniques that break or fuse the genes located in these regions, and its relevance to clinical practice.7
Thus, multidisciplinary teams should be formed to better interpret the results of genetic tests, with clinicians and geneticists working in close collaboration. Doing this can not only achieve an aetiological diagnosis, which allows the optimisation of management and genetic counselling of families, but also prevent the performance of unnecessary tests.
Please cite this article as: Caballero Pérez V, López-Pisón FJ, Miramar Gallart MD, González Álvarez A, García Jiménez MC. Repercusión clínica de la traslocación t(7;15) (p22;q26) en varios miembros de una misma familia. An Pediatr (Barc). 2017;87:113–115.

