
A study of epilepsy, according to the age at onset of the crisis and its causes, monitored by a Paediatric Neurology Unit over a period of three years.
Patients and methodsHistorical cohorts study was conducted by reviewing the Paediatric Neurology medical records database of epileptic children followed-up from 1 January 2008 to 31 December 2010.
ResultsA total of 4595 children were attended during the study period. The diagnosis of epilepsy was established in 605 (13.17%): 277 (45.79%) symptomatic, 156 (25.79%) idiopathic, and 172 (28.43%) with cryptogenic epilepsy. Absence epilepsy and benign childhood epilepsy with centro-temporal spikes are the idiopathic epileptic syndromes most prevalent, and the most prevalent symptomatic epilepsies are prenatal encephalopathies. More than one-quarter (26.12%) of epilepsies began in the first year of life, and 67.72% were symptomatic. Refractory epilepsy was observed in 25.29%, 42.46% with cognitive impairment, 26.45% with motor involvement, and 9.92% with an autism spectrum disorder, being more frequent at an earlier age of onset.
ConclusionsThe absence of a universally accepted classification of epileptic syndromes makes tasks like this difficult, starting with the terminology. A useful classification would be aetiological, with two groups: a large group with established aetiology, or very likely genetic syndromes, and another with no established cause. The age of onset of epilepsy in each aetiological group helps in the prognosis, which is worsened by refractoriness and associated neurodevelopmental disorders, and are generally worse at an earlier onset and in certain aetiologies.
Estudio de las epilepsias según la edad de inicio de las crisis y la etiología, de los pacientes controlados en una unidad de neuropediatría durante 3 años.
Pacientes y métodosEstudio de cohortes históricas. Revisión de historias de niños con epilepsia de la base de datos de neuropediatría controlados del 1 de enero de 2008 al 31 de diciembre de 2010.
ResultadosDe 4.595 niños atendidos en el periodo, se estableció el diagnóstico de epilepsia en 605 (13,17%), siendo 277 (45,79%) epilepsias sintomáticas, 156 (25,79%) idiopáticas y 172 (28,43%) criptogénicas. La epilepsia de ausencias y la epilepsia benigna de la infancia con paroxismos centrotemporales son los síndromes epilépticos idiopáticos con mayor prevalencia, y las encefalopatías prenatales las epilepsias sintomáticas más prevalentes. El 26,12% iniciaron su epilepsia el primer año, siendo sintomáticas el 67,72%. Se han considerado refractarias el 25,29% de las epilepsias; el 42,46% asocia déficit cognitivo, el 26,45% afectación motora y el 9,92% trastorno del espectro autista, siendo más frecuentes a menor edad de inicio.
ConclusionesLa ausencia de una clasificación universalmente aceptada de los síndromes epilépticos dificulta trabajos como este, empezando por la terminología. Una clasificación útil es la etiológica, con 2 grupos: un gran grupo con las etiologías establecidas o síndromes genéticos muy probables, y otro de casos sin causa establecida. La edad de inicio de la epilepsia en cada grupo etiológico añade orientación pronóstica. El pronóstico de la epilepsia lo ensombrecen la refractariedad y las alteraciones asociadas del neurodesarrollo, siendo peor en general a más precoz inicio y en etiologías concretas.
Epilepsy is one of the most frequent neurologic disorders in childhood, with an estimated prevalence of 3.4–11.3 cases per 1000 inhabitants.1–3
Epilepsy syndromes are age-dependent, and their characteristics vary based on the stage of brain maturation, with certain disorders presenting predominantly in specific age groups.
The prognosis of epilepsy depends mainly on its aetiology. Another key factor is the age at the first seizure (which depends on the aetiology), with early ages generally associated with poorer outcomes.4–7 Epilepsy with onset in the first year of life usually has a poor prognosis that worsens the earlier the onset, is frequently refractory to treatment, and is associated with neurodevelopmental disorders,8,9 although there are forms of epilepsy in infants that have a favourable prognosis.5,10
We conducted a study of cases of epilepsy and epilepsy syndrome by age of onset and aetiology followed up at a regional reference paediatric neurology unit during a three-year period. We analysed aetiologic and prognostic differences in epilepsy by age of onset in the patients that received care in our unit during the period under study.
Materials and methodsThe population under study consisted of all patients aged more than 1 month with a diagnosis of epilepsy assessed (for the first time or in follow-up visits) at the Unit of Paediatric Neurology of the Hospital Miguel Servet of Zaragoza over a three-year period (from January 1, 2008 to December 31, 2010). The services provided by this unit since its creation in 1990 have been documented in an electronic database, that includes records of all the relevant data for each patient11,12 that are updated when there are clinically relevant changes, new test results or changes in treatment.
We conducted a retrospective cohort study by reviewing the medical records of the patients included in the sample.
We have defined epilepsy as a history of at least two spontaneous epileptic seizures. The exclusion criteria included neonatal convulsions in the absence of subsequent epilepsy, isolated afebrile seizures, febrile seizures, and other acute provoked or symptomatic seizures.
We have defined symptomatic epilepsy as epilepsy secondary to a brain abnormality and that manifests with seizures in addition to other neurologic manifestations. We developed our own classification scheme into aetiologic groups to facilitate our analysis: prenatal encephalopathies, perinatal encephalopathies, postnatal encephalopathies, metabolic and degenerative encephalopathies, mesial temporal sclerosis, neurocutaneous disorders, vascular malformations, cavernomas, intracranial tumours and other.
We defined idiopathic epilepsy as seizures that are not caused by a brain abnormality and constitute the main manifestation of disease in the absence of other associated neurologic signs and symptoms, with the assumption that these are genetic and age-dependent. We defined cryptogenic epilepsy as epilepsy that cannot be classified as either idiopathic or symptomatic due to insufficient clinical and outcome data. The term cryptogenic was used based on its etymology: cryptic genesis.13
We assessed prognosis taking into account treatment effectiveness (refractory disease) and the impact on neurologic function. Adhering to the 2010 definition of the International League Against Epilepsy (ILAE),14 we defined refractory epilepsy as failure of adequate trials of two tolerated and appropriately chosen and used antiepileptic drug schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom. Subsequently, we assigned each patient one or several neurological functioning diagnosis of the following: neurologically normal (if no neurologic abnormalities other than epilepsy were identified), cognitive disorder, autism spectrum disorder and motor impairment.
We used the following tests in our statistical analysis: the chi square test, the nonparametric Mann–Whitney U test and the Kruskal–Wallis test with post hoc tests with the Bonferroni correction. We uploaded the data and analysed it using STATA for Windows version 14.0 (StataCorp LP, Texas, USA). We defined statistical significance as a p-value of less than 0.05.
ResultsAt the time of the study, the database of the unit had records for 15808 patients. During the period under study, 4595 patients had received care in the unit. Out of the total, 1654 had sought care for seizures (35.99%) and 605 received a diagnosis of epilepsy (13.17% of the total patients and 36.58% of patients with seizures). Epilepsy was categorised as symptomatic in 277 patients (45.79%), idiopathic in 156 (25.78%) and cryptogenic in 172 (28.43%). In the three years under study, 184 new cases of epilepsy were diagnosed, while the rest of assessments corresponded to patients in follow-up. Of all included patients, 53.22% were male and 46.78% female. The mean duration of follow-up was 6.21 years overall, and 8.13 for symptomatic epilepsy, 4.66 for idiopathic epilepsy, and 4.54 for cryptogenic epilepsy.
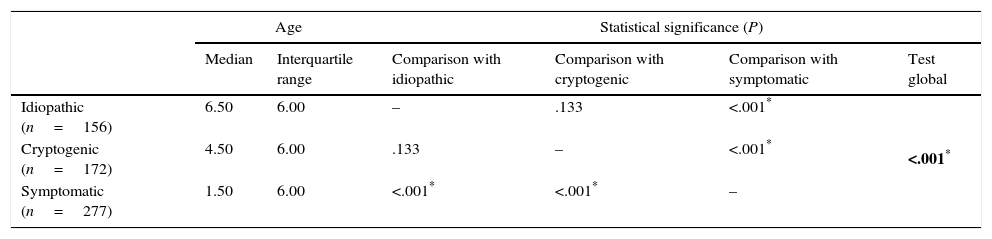
The mean age of onset of epilepsy was 4.78 years for the overall sample, 3.53 for idiopathic epilepsy and 5.43 for cryptogenic epilepsy.
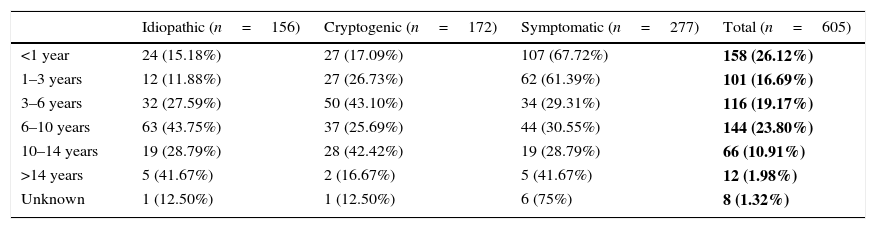
Table 1 shows the age of onset of epilepsy (in age bands) by aetiology. In our sample, 26.12% of patients had the first seizure before age 1 year. The most prevalent aetiology in children with early onset was symptomatic (67.09% of children with onset before 1 year and 61.39% of children with onset at age 1–3 years). Idiopathic epilepsy predominated in children with onset between 6 and 10 years of age (43.75%), and cryptogenic epilepsy in children with onset between 3 and 6 years of age (43.10%). Table 2 analyses the differences between the three aetiological groups by age of onset.
Age of onset groups by aetiology of epilepsy.
| Idiopathic (n=156) | Cryptogenic (n=172) | Symptomatic (n=277) | Total (n=605) | |
|---|---|---|---|---|
| <1 year | 24 (15.18%) | 27 (17.09%) | 107 (67.72%) | 158 (26.12%) |
| 1–3 years | 12 (11.88%) | 27 (26.73%) | 62 (61.39%) | 101 (16.69%) |
| 3–6 years | 32 (27.59%) | 50 (43.10%) | 34 (29.31%) | 116 (19.17%) |
| 6–10 years | 63 (43.75%) | 37 (25.69%) | 44 (30.55%) | 144 (23.80%) |
| 10–14 years | 19 (28.79%) | 28 (42.42%) | 19 (28.79%) | 66 (10.91%) |
| >14 years | 5 (41.67%) | 2 (16.67%) | 5 (41.67%) | 12 (1.98%) |
| Unknown | 1 (12.50%) | 1 (12.50%) | 6 (75%) | 8 (1.32%) |
The absolute frequencies and percentages (over the total sample) of epilepsies in each age group are shown in bold.
Comparison of the age of onset of epilepsy by aetiology (Kruskal–Wallis test with post hoc tests with Bonferroni correction).
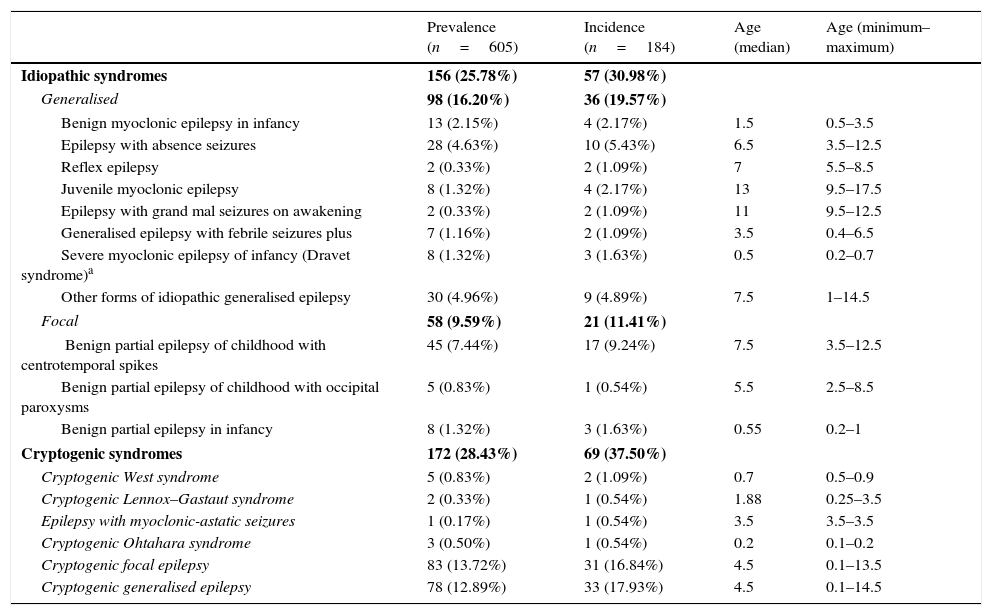
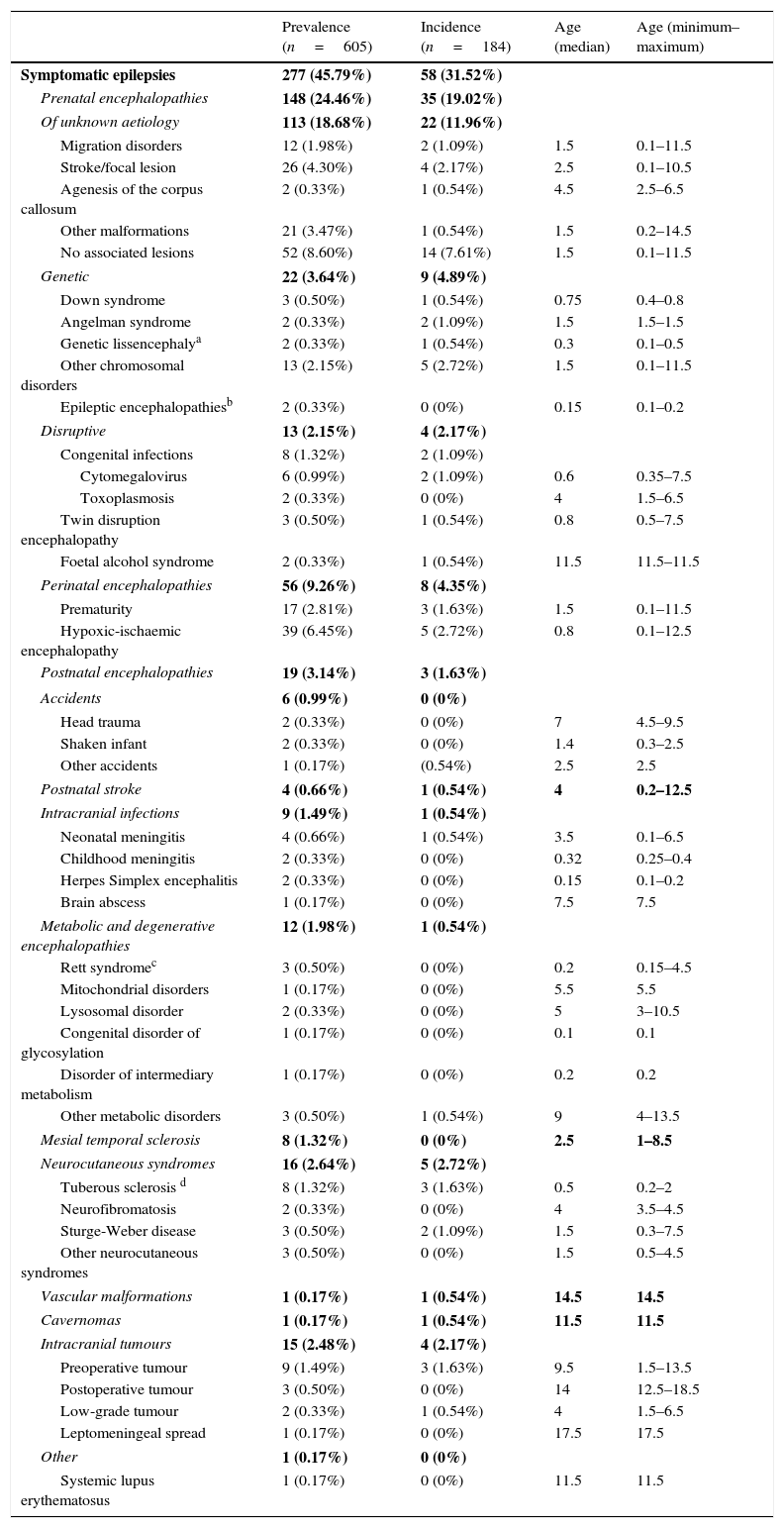
Table 3 summarises the incidence, prevalence and age of onset of idiopathic and cryptogenic epilepsy syndromes. Table 4 summarises the incidence, prevalence and age of onset of symptomatic epilepsies by aetiology.
Prevalence, incidence and age of onset (in years) of idiopathic and cryptogenic epilepsy syndromes.
| Prevalence (n=605) | Incidence (n=184) | Age (median) | Age (minimum–maximum) | |
|---|---|---|---|---|
| Idiopathic syndromes | 156 (25.78%) | 57 (30.98%) | ||
| Generalised | 98 (16.20%) | 36 (19.57%) | ||
| Benign myoclonic epilepsy in infancy | 13 (2.15%) | 4 (2.17%) | 1.5 | 0.5–3.5 |
| Epilepsy with absence seizures | 28 (4.63%) | 10 (5.43%) | 6.5 | 3.5–12.5 |
| Reflex epilepsy | 2 (0.33%) | 2 (1.09%) | 7 | 5.5–8.5 |
| Juvenile myoclonic epilepsy | 8 (1.32%) | 4 (2.17%) | 13 | 9.5–17.5 |
| Epilepsy with grand mal seizures on awakening | 2 (0.33%) | 2 (1.09%) | 11 | 9.5–12.5 |
| Generalised epilepsy with febrile seizures plus | 7 (1.16%) | 2 (1.09%) | 3.5 | 0.4–6.5 |
| Severe myoclonic epilepsy of infancy (Dravet syndrome)a | 8 (1.32%) | 3 (1.63%) | 0.5 | 0.2–0.7 |
| Other forms of idiopathic generalised epilepsy | 30 (4.96%) | 9 (4.89%) | 7.5 | 1–14.5 |
| Focal | 58 (9.59%) | 21 (11.41%) | ||
| Benign partial epilepsy of childhood with centrotemporal spikes | 45 (7.44%) | 17 (9.24%) | 7.5 | 3.5–12.5 |
| Benign partial epilepsy of childhood with occipital paroxysms | 5 (0.83%) | 1 (0.54%) | 5.5 | 2.5–8.5 |
| Benign partial epilepsy in infancy | 8 (1.32%) | 3 (1.63%) | 0.55 | 0.2–1 |
| Cryptogenic syndromes | 172 (28.43%) | 69 (37.50%) | ||
| Cryptogenic West syndrome | 5 (0.83%) | 2 (1.09%) | 0.7 | 0.5–0.9 |
| Cryptogenic Lennox–Gastaut syndrome | 2 (0.33%) | 1 (0.54%) | 1.88 | 0.25–3.5 |
| Epilepsy with myoclonic-astatic seizures | 1 (0.17%) | 1 (0.54%) | 3.5 | 3.5–3.5 |
| Cryptogenic Ohtahara syndrome | 3 (0.50%) | 1 (0.54%) | 0.2 | 0.1–0.2 |
| Cryptogenic focal epilepsy | 83 (13.72%) | 31 (16.84%) | 4.5 | 0.1–13.5 |
| Cryptogenic generalised epilepsy | 78 (12.89%) | 33 (17.93%) | 4.5 | 0.1–14.5 |
An additional genetic test for mutations in the SCN1A gene was performed in 34 patients and was positive in 8, who are the patients with a diagnosis of Dravet syndrome.
The cumulative data for the idiopathic and cryptogenic syndromes, and within the idiopathic, of generalised and focal syndromes, are shown in bold.
Prevalence, incidence and age of onset (in years) of symptomatic epilepsy syndromes.
| Prevalence (n=605) | Incidence (n=184) | Age (median) | Age (minimum–maximum) | |
|---|---|---|---|---|
| Symptomatic epilepsies | 277 (45.79%) | 58 (31.52%) | ||
| Prenatal encephalopathies | 148 (24.46%) | 35 (19.02%) | ||
| Of unknown aetiology | 113 (18.68%) | 22 (11.96%) | ||
| Migration disorders | 12 (1.98%) | 2 (1.09%) | 1.5 | 0.1–11.5 |
| Stroke/focal lesion | 26 (4.30%) | 4 (2.17%) | 2.5 | 0.1–10.5 |
| Agenesis of the corpus callosum | 2 (0.33%) | 1 (0.54%) | 4.5 | 2.5–6.5 |
| Other malformations | 21 (3.47%) | 1 (0.54%) | 1.5 | 0.2–14.5 |
| No associated lesions | 52 (8.60%) | 14 (7.61%) | 1.5 | 0.1–11.5 |
| Genetic | 22 (3.64%) | 9 (4.89%) | ||
| Down syndrome | 3 (0.50%) | 1 (0.54%) | 0.75 | 0.4–0.8 |
| Angelman syndrome | 2 (0.33%) | 2 (1.09%) | 1.5 | 1.5–1.5 |
| Genetic lissencephalya | 2 (0.33%) | 1 (0.54%) | 0.3 | 0.1–0.5 |
| Other chromosomal disorders | 13 (2.15%) | 5 (2.72%) | 1.5 | 0.1–11.5 |
| Epileptic encephalopathiesb | 2 (0.33%) | 0 (0%) | 0.15 | 0.1–0.2 |
| Disruptive | 13 (2.15%) | 4 (2.17%) | ||
| Congenital infections | 8 (1.32%) | 2 (1.09%) | ||
| Cytomegalovirus | 6 (0.99%) | 2 (1.09%) | 0.6 | 0.35–7.5 |
| Toxoplasmosis | 2 (0.33%) | 0 (0%) | 4 | 1.5–6.5 |
| Twin disruption encephalopathy | 3 (0.50%) | 1 (0.54%) | 0.8 | 0.5–7.5 |
| Foetal alcohol syndrome | 2 (0.33%) | 1 (0.54%) | 11.5 | 11.5–11.5 |
| Perinatal encephalopathies | 56 (9.26%) | 8 (4.35%) | ||
| Prematurity | 17 (2.81%) | 3 (1.63%) | 1.5 | 0.1–11.5 |
| Hypoxic-ischaemic encephalopathy | 39 (6.45%) | 5 (2.72%) | 0.8 | 0.1–12.5 |
| Postnatal encephalopathies | 19 (3.14%) | 3 (1.63%) | ||
| Accidents | 6 (0.99%) | 0 (0%) | ||
| Head trauma | 2 (0.33%) | 0 (0%) | 7 | 4.5–9.5 |
| Shaken infant | 2 (0.33%) | 0 (0%) | 1.4 | 0.3–2.5 |
| Other accidents | 1 (0.17%) | (0.54%) | 2.5 | 2.5 |
| Postnatal stroke | 4 (0.66%) | 1 (0.54%) | 4 | 0.2–12.5 |
| Intracranial infections | 9 (1.49%) | 1 (0.54%) | ||
| Neonatal meningitis | 4 (0.66%) | 1 (0.54%) | 3.5 | 0.1–6.5 |
| Childhood meningitis | 2 (0.33%) | 0 (0%) | 0.32 | 0.25–0.4 |
| Herpes Simplex encephalitis | 2 (0.33%) | 0 (0%) | 0.15 | 0.1–0.2 |
| Brain abscess | 1 (0.17%) | 0 (0%) | 7.5 | 7.5 |
| Metabolic and degenerative encephalopathies | 12 (1.98%) | 1 (0.54%) | ||
| Rett syndromec | 3 (0.50%) | 0 (0%) | 0.2 | 0.15–4.5 |
| Mitochondrial disorders | 1 (0.17%) | 0 (0%) | 5.5 | 5.5 |
| Lysosomal disorder | 2 (0.33%) | 0 (0%) | 5 | 3–10.5 |
| Congenital disorder of glycosylation | 1 (0.17%) | 0 (0%) | 0.1 | 0.1 |
| Disorder of intermediary metabolism | 1 (0.17%) | 0 (0%) | 0.2 | 0.2 |
| Other metabolic disorders | 3 (0.50%) | 1 (0.54%) | 9 | 4–13.5 |
| Mesial temporal sclerosis | 8 (1.32%) | 0 (0%) | 2.5 | 1–8.5 |
| Neurocutaneous syndromes | 16 (2.64%) | 5 (2.72%) | ||
| Tuberous sclerosis d | 8 (1.32%) | 3 (1.63%) | 0.5 | 0.2–2 |
| Neurofibromatosis | 2 (0.33%) | 0 (0%) | 4 | 3.5–4.5 |
| Sturge-Weber disease | 3 (0.50%) | 2 (1.09%) | 1.5 | 0.3–7.5 |
| Other neurocutaneous syndromes | 3 (0.50%) | 0 (0%) | 1.5 | 0.5–4.5 |
| Vascular malformations | 1 (0.17%) | 1 (0.54%) | 14.5 | 14.5 |
| Cavernomas | 1 (0.17%) | 1 (0.54%) | 11.5 | 11.5 |
| Intracranial tumours | 15 (2.48%) | 4 (2.17%) | ||
| Preoperative tumour | 9 (1.49%) | 3 (1.63%) | 9.5 | 1.5–13.5 |
| Postoperative tumour | 3 (0.50%) | 0 (0%) | 14 | 12.5–18.5 |
| Low-grade tumour | 2 (0.33%) | 1 (0.54%) | 4 | 1.5–6.5 |
| Leptomeningeal spread | 1 (0.17%) | 0 (0%) | 17.5 | 17.5 |
| Other | 1 (0.17%) | 0 (0%) | ||
| Systemic lupus erythematosus | 1 (0.17%) | 0 (0%) | 11.5 | 11.5 |
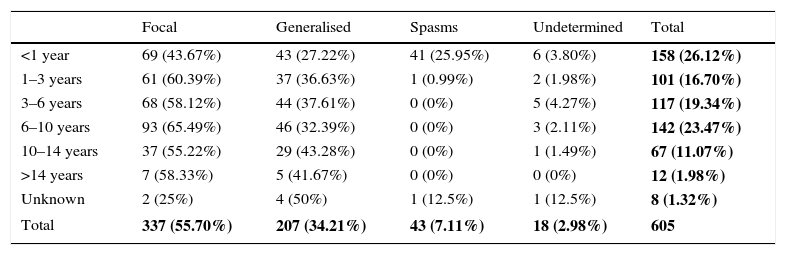
Table 5 shows the clinical presentation of epileptic seizures by age of onset of epilepsy. Focal seizures were more common (55.70%) than generalised seizures (34.21%) in all age groups. Epilepsy presented as infantile spasms in 25.95% of infants aged less than 1 year. Table 6 contrasts the age of onset of epilepsy with the type of seizure.
Type of epileptic seizure by onset age group.
| Focal | Generalised | Spasms | Undetermined | Total | |
|---|---|---|---|---|---|
| <1 year | 69 (43.67%) | 43 (27.22%) | 41 (25.95%) | 6 (3.80%) | 158 (26.12%) |
| 1–3 years | 61 (60.39%) | 37 (36.63%) | 1 (0.99%) | 2 (1.98%) | 101 (16.70%) |
| 3–6 years | 68 (58.12%) | 44 (37.61%) | 0 (0%) | 5 (4.27%) | 117 (19.34%) |
| 6–10 years | 93 (65.49%) | 46 (32.39%) | 0 (0%) | 3 (2.11%) | 142 (23.47%) |
| 10–14 years | 37 (55.22%) | 29 (43.28%) | 0 (0%) | 1 (1.49%) | 67 (11.07%) |
| >14 years | 7 (58.33%) | 5 (41.67%) | 0 (0%) | 0 (0%) | 12 (1.98%) |
| Unknown | 2 (25%) | 4 (50%) | 1 (12.5%) | 1 (12.5%) | 8 (1.32%) |
| Total | 337 (55.70%) | 207 (34.21%) | 43 (7.11%) | 18 (2.98%) | 605 |
The overall data for each different type of epileptic seizures relative to the total sample are shown in bold.
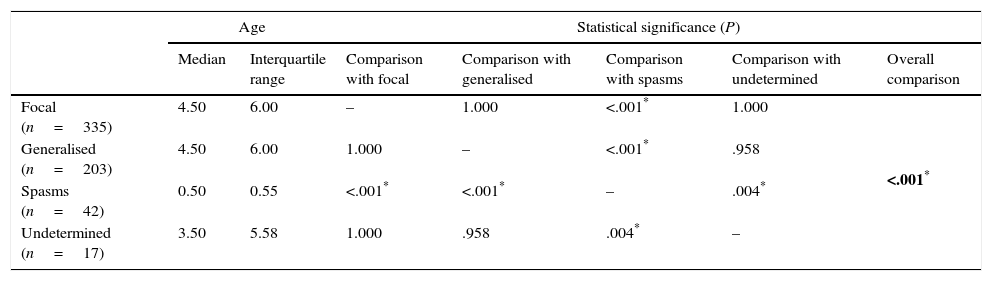
Comparison of age of onset of epilepsy by type of seizure (Kruskal–Wallis test with Bonferroni correction).
| Age | Statistical significance (P) | ||||||
|---|---|---|---|---|---|---|---|
| Median | Interquartile range | Comparison with focal | Comparison with generalised | Comparison with spasms | Comparison with undetermined | Overall comparison | |
| Focal (n=335) | 4.50 | 6.00 | – | 1.000 | <.001* | 1.000 | <.001* |
| Generalised (n=203) | 4.50 | 6.00 | 1.000 | – | <.001* | .958 | |
| Spasms (n=42) | 0.50 | 0.55 | <.001* | <.001* | – | .004* | |
| Undetermined (n=17) | 3.50 | 5.58 | 1.000 | .958 | .004* | – | |
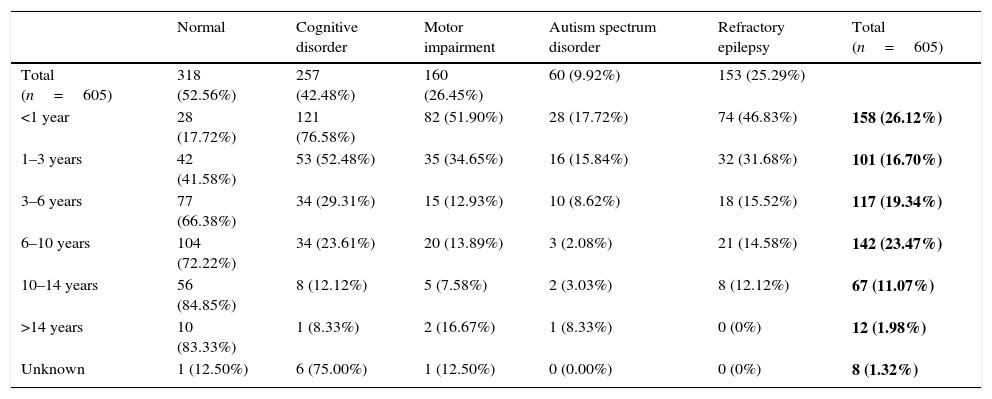
Table 7 summarises the prevalence of neurologic function disorders and refractory disease by age group. The mean duration of follow-up of refractory epilepsies was 8.87 years. The median age of onset was significantly lower in patients with refractory epilepsy (1.5 years) compared to patients that responded well to treatment (4.5 years). Of the total sample, 25.29% had refractory epilepsy, and only 52.56% did not have associated neurologic disorders. The younger the age of onset, the greater the percentage of refractory disease and of neurologic disorders: out of the patients with onset before 1 year of age, 46.83% had refractory epilepsy and only 17.72% did not have associated neurologic disorders.
Functional neurological disorders and refractory epilepsy by age of onset of epilepsy.
| Normal | Cognitive disorder | Motor impairment | Autism spectrum disorder | Refractory epilepsy | Total (n=605) | |
|---|---|---|---|---|---|---|
| Total (n=605) | 318 (52.56%) | 257 (42.48%) | 160 (26.45%) | 60 (9.92%) | 153 (25.29%) | |
| <1 year | 28 (17.72%) | 121 (76.58%) | 82 (51.90%) | 28 (17.72%) | 74 (46.83%) | 158 (26.12%) |
| 1–3 years | 42 (41.58%) | 53 (52.48%) | 35 (34.65%) | 16 (15.84%) | 32 (31.68%) | 101 (16.70%) |
| 3–6 years | 77 (66.38%) | 34 (29.31%) | 15 (12.93%) | 10 (8.62%) | 18 (15.52%) | 117 (19.34%) |
| 6–10 years | 104 (72.22%) | 34 (23.61%) | 20 (13.89%) | 3 (2.08%) | 21 (14.58%) | 142 (23.47%) |
| 10–14 years | 56 (84.85%) | 8 (12.12%) | 5 (7.58%) | 2 (3.03%) | 8 (12.12%) | 67 (11.07%) |
| >14 years | 10 (83.33%) | 1 (8.33%) | 2 (16.67%) | 1 (8.33%) | 0 (0%) | 12 (1.98%) |
| Unknown | 1 (12.50%) | 6 (75.00%) | 1 (12.50%) | 0 (0.00%) | 0 (0%) | 8 (1.32%) |
The absolute frequencies and percentages (of the total sample) of epilepsies in each age group are shown in bold.
Table 8 compares the outcome characteristics by age of onset.
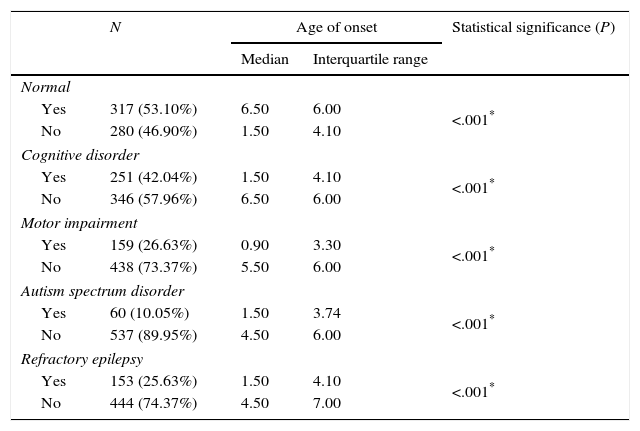
Functional neurological disorders and refractory disease by age of onset of epilepsy (Mann–Whitney U test).
| N | Age of onset | Statistical significance (P) | ||
|---|---|---|---|---|
| Median | Interquartile range | |||
| Normal | ||||
| Yes | 317 (53.10%) | 6.50 | 6.00 | <.001* |
| No | 280 (46.90%) | 1.50 | 4.10 | |
| Cognitive disorder | ||||
| Yes | 251 (42.04%) | 1.50 | 4.10 | <.001* |
| No | 346 (57.96%) | 6.50 | 6.00 | |
| Motor impairment | ||||
| Yes | 159 (26.63%) | 0.90 | 3.30 | <.001* |
| No | 438 (73.37%) | 5.50 | 6.00 | |
| Autism spectrum disorder | ||||
| Yes | 60 (10.05%) | 1.50 | 3.74 | <.001* |
| No | 537 (89.95%) | 4.50 | 6.00 | |
| Refractory epilepsy | ||||
| Yes | 153 (25.63%) | 1.50 | 4.10 | <.001* |
| No | 444 (74.37%) | 4.50 | 7.00 | |
After excluding patients with malignant intracranial tumours and brain malformations, 14 patients were considered eligible for surgery to treat refractory epilepsy, and five finally underwent surgical intervention: three patients with cortical dysplasia, one with mesial sclerosis and one with a low-grade tumour. One of the patients with cortical dysplasia experienced a significant decrease in the frequency of seizures, and in the other four the seizures disappeared after the intervention (although one of the patients with cortical dysplasia experienced a relapse that was refractory to treatment two years later).
DiscussionThe classification of epilepsy syndrome remains unresolved since 1989,15 as the classification proposed by the ILAE in 201016 has not been widely accepted, which poses challenges to the definition and delimitation of syndromes, and to the comparison of case data. This new proposal by the ILAE was met with considerable criticism by epilepsy specialists,17–19 as rather than offering a new classification it seemed to offer a new terminology. Thus, the classification does not accept the subdivision into generalised and focal epilepsy, although it does maintain the division between epileptic seizures of either type, while eliminating the concepts of idiopathic, symptomatic and presumed symptomatic epilepsy. It includes electroclinical syndromes (distributed by age of onset), clinically distinctive constellations (syndromes characterised by an association with specific lesions, such as mesial temporal lobe epilepsy with hippocampal sclerosis), structural/metabolic epilepsies, and a group of epilepsies of unknown cause (which the Commission currently believes would correspond to a third of all cases of epilepsy).16,17
Epilepsy syndromes are age-dependent and their clinical and electroencephalographic characteristics vary based on the stage of brain maturation,7,20,21 as demonstrated in this study. In our sample, the onset of 26.12% of the total cases of epilepsy occurred in the first year of life, and 67.72% were symptomatic. Furthermore, the median age of onset of symptomatic epilepsies was 1.5 years, significantly lower than the age of onset in cryptogenic and idiopathic epilepsies (4.5 and 6.5 years, respectively).
The incidence of some epilepsy syndromes peaks at certain ages. In our sample, West syndrome, Ohtahara syndrome, Dravet syndrome or benign partial epilepsy in infancy had onsets in the first months of life, while absence seizures, benign childhood epilepsy with centrotemporal spikes and other forms of idiopathic generalised epilepsy appear in school-aged children. In the group of symptomatic epilepsies, some aetiologies are associated with onset in the first year of life, such as genetic lissencephaly, Down syndrome, hypoxic-ischaemic encephalopathy or tuberous sclerosis, while others are associated with onset at later ages.
In our study, focal seizures (43.67%) predominated over generalised seizures (27.22%) in children with onset before 1 year, and we ought to highlight that the initial presentation in this group was in the form of epileptic spasms in 25.95%.
Despite the advances made in the field of epilepsy in recent years and the development of new antiepileptic drugs, a large number of cases are refractory to treatment, with an estimated reported incidence of 6–24%,22–25 and an incidence of 25.29% in our study. One of the factors that are associated with unresponsiveness to treatment is the age of onset, with refractory epilepsy occurring more frequently in cases with onset at earlier ages (first two to three years of life).26,27 In our study, the median age of onset was considerably lower in patients with refractory epilepsy than in patients that responded to treatment (1.5 versus 4.5 years). Out of all cases of epilepsy with onset before age 1 year, 46.83% were considered refractory to treatment, a percentage that was considerably higher than in other age groups.
Several publications have emphasised that the cognitive sequelae of epilepsy become more significant the lower the age of onset.5,28,29 In our study, only 17.72% of patients with epilepsy with onset before age 1 year were neurologically normal, while 76.58% had cognitive deficits, 51.90% motor impairment and 17.72% autism spectrum disorder. In the group of patients with onset between ages 1 and 3 years, 41.58% were neurologically normal.
It seems that the factor that has the strongest impact on cognitive development in the early stages of life is the aetiology of the epilepsy.10 Thus, epilepsies with onset in the first year of life usually have a symptomatic aetiology, which can be of a diverse nature (genetic, infectious, metabolic, perinatal complications, etc.).5,8 In our study, 67.09% of epilepsies with onset in the first year of life and 61.39% of epilepsies with onset between age 1 and 3 years were symptomatic (with prenatal and perinatal encephalopathies being the most prevalent aetiologies). Therefore, these patients usually have neurodevelopmental comorbidities. However, there are some epilepsies with early onset that have a favourable prognosis. Benign partial epilepsy in infancy usually has an excellent prognosis with seizures resolving spontaneously in some cases,30,31 and the eight patients in our study that had this form of epilepsy responded well to treatment, while only one had cognitive impairment. The prognosis of benign myoclonic epilepsy in infancy is generally good, with good seizure control and normal neurologic development in 60–80% of cases, although the rest may be associated with behavioural and learning disorders and seizures later in life.20,32,33 For this reason, the “benign” qualifier may not be appropriate for this syndrome, and the ILAE even considered eliminating it, but the 2010 classification still maintained it.32–34 None of the thirteen patients that had this entity in our study was unresponsive to treatment, and three (23%) had some type of neurologic comorbidity.
In our sample, 66.67% of the patients with onset between 1 and 3 months of age (18 cases, nearly 3% of the total) had symptomatic epilepsy. Fifty percent were refractory to treatment and 83.33% had neurologic comorbidities. The aetiologies in this very-early-onset group are very diverse, so a diagnostic and treatment strategy must be established for the purpose of making an early diagnosis and avoid uncertainty, and to identify potentially treatable cases, such as those secondary to hereditary metabolic diseases. Advances in genetics, especially in the development of gene panels for early-onset epileptic encephalopathies and exome studies, will make early diagnosis possible.
Most of the epilepsies that develop in school-aged children have a genetic basis, while epilepsies with underlying structural abnormalities are less frequent.7 In our sample, epilepsy cases with onset between ages 3 and 10 years amounted to 42.98% of the total. Among them, 36.54% were idiopathic, 30% symptomatic and 33.46% cryptogenic. Well-defined syndromes usually appear during this period, for instance, epilepsy with absence seizures and benign childhood epilepsy with centrotemporal spikes,7 which were the most prevalent epileptic syndromes in this age group in our study. We observed that the peak incidence of epilepsy with absence seizures occurred between 7 and 8 years of age, while the peak incidence of benign childhood epilepsy with centrotemporal spikes occurred between 9 and 10 years. The epilepsies that have onset in this age group tend to be benign, and most have favourable outcomes9; in our sample, 15% of epilepsy cases with onset between ages 3 and 10 years were refractory, and 70% had no neurologic comorbidities.
According to various publications, epilepsy affects 1.5–2% of adolescents (19% of the cases of epilepsy in all age groups).35,36 In our study, the cases of epilepsy with onset between ages 10 and 14 years amounted to 10.91% of our sample, and those with onset at age more than 14 years to 1.98%. In this stage of development, the brain has already matured, so the clinical manifestations do not differ greatly from those found in adults.36 There is a predominance of generalised tonic-clonic seizures, absence seizures and simple and complex partial seizures. According to the literature, the epilepsy syndromes that develop most frequently in this age group are idiopathic generalised epilepsies, such as juvenile absence epilepsy, juvenile myoclonic epilepsy and epilepsy with generalised seizures on awakening36 (although in clinical practice it is sometimes difficult to distinguish these syndromes, as all of them may manifest with myoclonic and/or absence and/or tonic-clonic seizures37). In our study, 30.77% of epilepsy cases with onset at age ten or more were idiopathic, 30.77% symptomatic and 38.46% cryptogenic. We preferred to classify the epilepsy in several patients as cryptogenic when its clinical and/or outcome characteristics did not perfectly fit a well-defined idiopathic syndrome. We ought to highlight the benign course of epilepsies with onset in this age group, as a significant proportion of them tend to resolve without future sequelae, which reflects their non-lesional nature, as most of them have a genetic basis.7 In our series, only 11.98% of cases of epilepsy with onset between 10 and 14 years were refractory to treatment, and none were refractory in patients aged more than 14, while 85% were free of neurologic comorbidities.
The age of onset is a decisive factor in the approach to childhood epilepsy; in older children, neuroimaging tests and electroencephalography may be sufficient, but infants may require extensive workups (metabolic and genetic).20 We did not identify any cases with an autoimmune aetiology in our case series, but this is an emerging cause (as are all neuroimmune conditions) of refractory epilepsy, and they need to be investigated for their adequate, albeit complex, diagnosis and management. In cases with early onset, epilepsy is due much more frequently to a severe brain abnormality, responding poorly to treatment and carrying a poor neurologic and neurodevelopmental prognosis.5 In rare cases, it may be due to hereditary metabolic diseases, which will be unresponsive to antiepileptic therapy, and some of which may require specific treatment (with vitamin supplementation or a ketogenic diet). Considering the significant concern regarding its prognosis, the risk of recurrence (since they often involve a genetic defect), and the options, although infrequent, for specific treatments that they can respond to, we need to establish a diagnostic and therapeutic protocol to facilitate, whenever possible, early treatment and identification of the aetiology, and which contemplates treatment with vitamins.
In patients of any age with refractory epilepsy, searching for potentially operable lesions is a must, as resection of the lesion may be curative. Therefore, in cases of focal epilepsy the need for neuroimaging and functional neuroimaging tests must be emphasised, and surgery must be performed as early as possible to prevent the negative impact of seizures and medication.38,39
The absence of a universally accepted classification of epilepsy syndromes16,40 poses challenges to studies like the one presented here, starting with the terminology used. Dravet syndrome and other epileptic encephalopathies with an identified mutation, such as the patients in our sample that had mutations in the STXBP1 and CDKL5 genes, could be classified both as idiopathic (genetically determined) and as symptomatic (encephalopathy of genetic causes with neurodevelopmental impairment that may not be secondary to epilepsy). Nondisruptive prenatal epilepsy syndromes, neurocutaneous syndromes, metabolic and degenerative disorders and many cases of vascular malformation, cavernomas, brain tumour and mesial temporal sclerosis have a genetic basis. Obviously, all epilepsies are symptomatic, as they all have a cause, whether genetic or acquired.
An aetiological classification of epilepsy may be useful: one broad group including established aetiologies or probable genetic syndromes, and another of cases with no known cause. The age of onset in each aetiological group can further guide the prognosis.
The prognosis of epilepsy worsens with pharmacoresistance and the presence of neurodevelopmental abnormalities, and it is generally poorer the younger the age of onset and for specific aetiologic groups.
As noted above, epilepsy is age-dependent, so it is very important that we get to know the characteristics of different epilepsy syndromes in each age group for the purposes of identification, appropriate management without the use of unnecessary diagnostic tests, initiation of the most suitable antiepileptic treatment, and offering a prognosis to the families of epileptic children.
Conflict of interestsThe authors have no conflict of interests to declare.
Please cite this article as: Ochoa-Gómez L, López-Pisón J, Lapresta Moros C, Fuertes Rodrigo C, Fernando Martínez R, Samper-Villagrasa P, et al. Estudio de las epilepsias según la edad de inicio, controladas durante 3 años en una unidad de neuropediatría de referencia regional. An Pediatr (Barc). 2017;86:11–19.
