

Haematopoietic stem cell transplantation (HSCT) involves implanting cellular elements capable of generating a new and healthy haematopoietic system. Reduced intensity conditioning (RIC) consists of an immunosuppressive treatment to facilitate a progressive implant with lower morbidity. This type of conditioning can also lead to myelosuppression, which is potentially reversible over time.
Reduced intensity conditioning enables HSCT to be performed on patients with genetic diseases for whom added comorbidity is undesirable due to the high doses of chemotherapy that accompanies conventional myeloablative regimens.
Patients and methodsAn analysis was performed on the outcomes of 68 paediatric patients with genetic diseases who underwent HSCT with RIC between 2005 and 2013 in the of Paediatric Haematopoietic Stem Cell Transplantation Units that are part of the Spanish Working Group for Bone Marrow Transplantation in Children.
A multicentre study was conducted including 68 patients, of whom 43 had Primary Immunodeficiency, 21 with congenital haematological diseases, and 4 with metabolic diseases.
ResultsFifty (73.5%) of the 68 patients were still alive. The overall survival (OS) at nine years was 0.74. Twenty-three (33.8%) had some event during the course of the HSCT, with an event-free survival rate of 0.66. The OS in patients with haematological diseases was 0.81, being 0.7 in primary immunodeficiencies, and 0.4 in metabolic diseases. No significant difference was observed between the 3 groups of diseases. As regards the source of haematopoietic progenitors, there was an OS rate of 0.74 in patients transplanted with peripheral blood, 0.70 with bone marrow, and 0.70 and with cord blood, with no statistically significant differences.
ConclusionsFavourable results have been obtained in HSCT with reduced intensity conditioning in genetic diseases. It should be noted that the risks and benefits of the RIC in patients with metabolic diseases need to be assessed on an individual basis.
El trasplante de progenitores hematopoyéticos (TPH) consiste en implantar elementos celulares capaces de generar un sistema hematopoyético nuevo y sano. El régimen de intensidad reducida (RIR) consiste en un tratamiento predominantemente inmunosupresor, para facilitar un implante progresivo con menor morbilidad. Este tipo de acondicionamiento puede también provocar mielosupresión, aunque potencialmente reversible en el tiempo.
El acondicionamiento RIR permite aplicar TPH a pacientes con enfermedad genética en los que no es deseable añadir comorbilidad por las altas dosis de quimioterapia que conlleva el régimen mieloablativo convencional.
Pacientes y métodosSe analiza la evolución de 68 pacientes pediátricos con enfermedades genéticas que entre los años 2005-2013 se han sometido a un TPH con RIR en las Unidades pediátricas de Trasplante Hematopoyético de los hospitales españoles integrantes del Grupo Español para Trasplante de Médula Ósea en niños.
Se trata de un estudio multicéntrico que incluye a 68 pacientes, de los cuales 43 presentan inmunodeficiencia primaria, 21 presentan hemopatía congénita y 4 están afectados de metabolopatía.
ResultadosCincuenta de los 68 pacientes se encuentran vivos (73,5%). La supervivencia global (SG) a 9 años es de 0,74. Veintitrés (33,8%) han presentado en el transcurso del TPH algún evento. Supervivencia libre de evento de 0,66. La SG en los pacientes con hemopatía es de 0,81; en las inmunodeficiencias primarias es de 0,70 y en las metabolopatías es de 0,4. No se observa diferencia significativa entre los 3 grupos de enfermedades. Respecto a la fuente de progenitores hematopoyéticos, la SG en los pacientes trasplantados con sangre periférica es de 0,74; con médula ósea es de 0,70 y con la sangre de cordón umbilical es de 0,70. No se observa tampoco diferencia estadística significativa.
ConclusionesEn nuestro trabajo, de ámbito nacional, hemos evidenciado unos resultados favorables en TPH con régimen de intensidad reducida en las enfermedades genéticas. Cabe destacar que las metabolopatías requieren una consideración individualizada para sopesar en cada paciente los riesgos y beneficios que comporta el RIR.
Haematopoietic stem cell transplantation (HSCT) involves the infusion of cellular elements with the potential of re-establishing healthy haematopoietic function. Haematopoietic stem cells (HSCs) are multipotent cells capable of self-renewal and differentiation and proliferation into different cellular lineages. The term conditioning regimen refers to the treatment administered to patients prior to the infusion of HSCs.
Reduced intensity conditioning (RIC) consists of a predominantly immunosuppressive conditioning regimen aimed at facilitating progressive engraftment and reducing regimen-related morbidity. This type of conditioning can also produce myelosuppression, although its effects may be reversible. Once donor cells are transplanted, mixed chimerism usually results (coexistence of donor and recipient cells in the same bone marrow environment), which may stabilise or evolve to complete chimerism or graft failure.
Reduced-intensity conditioning regimens allow the use of HSCT in patients with genetic disorders in whom exposure to conventional myeloablative regimens is not advisable on account of the additional morbidity that may result from high-dose chemotherapy.
According to the Center of International Blood and Marrow Research,1 RIC regimens are based on the use of immunosuppressive drugs at high doses, especially fludarabine (Flu) and antilymphocyte globulin, combined with lower doses of alkylating agents (bulsulfan, melphalan, treosulfan [Tre], cyclophosphamide or thiotepa [TT]). Total body irradiation (TBI) is used to achieve haematopoietic tissue ablation and immunosuppression.
Reduced-intensity conditioning regimens are those that include:
- -
TBI≤500cGy as a single fraction or ≤800cGy in fractionated doses.
- -
Bulsulfan: total dose≤9mg/kg.
- -
Melphalan: total dose≤140mg/m2.
- -
TT: total dose≤10mg/kg.
- -
Cyclophosphamide: total dose≤120mg/kg.
- -
Tre: total dose≤42g/m2.
- -
Flu: total dose≤160mg/m2.
Engraftment or haematopoietic reconstitution is defined as recovery of neutrophil counts of 0.5×109cells/L or higher and platelet counts of 20×109/L or higher in the peripheral blood (PB) of the recipient.
Immune reconstitution after HSCT is a slow process, and normal levels of circulating T cells may not be achieved until 6–12 months post transplantation. Therefore, morbidity in the first year after HSCT may be high.2
In most children, the indication for HSCT is malignant disease, although in this study we focused on HSCT for treatment of genetic diseases, where the use of RIC is of greater interest.
Thalassaemia major is the most severe form of thalassaemia. It manifests with transfusion-dependent dyserythropoietic anaemia, splenomegaly, bone deformities and haemosiderosis. At present, HSCT is the only available curative treatment. There is still little experience with matched unrelated donor HSCT following RIC, and the main problem described in relation to this type of conditioning is graft rejection in multiply transfused patients.3,4
Sickle-cell disease, which manifests with vaso-occlusive crises, is another blood disorder for which allogeneic HSCT is the sole available curative treatment, reserved for patients with severe presentations.5
Fanconi anaemia is a hereditary disease characterised by pancytopaenia, physical abnormalities and increased risk of malignant disease. These patients have chromosomal instability and are more sensitive to chemotherapy and radiation therapy. Despite supportive care, the disease is invariably fatal once bone marrow failure or malignancy occurs. Allogeneic HSCT is the only curative treatment currently available.6,7
Primary immunodeficiency diseases (PIDDs) are a heterogeneous group of congenital defects that affect different mechanisms essential to the immune response. These patients are characterised by a greater susceptibility to infection, autoimmune disease and malignancy. Haematopoietic stem cell transplantation is the only curative treatment available for a broad range of PIDDs.8,9
Metabolic diseases are managed with a multidisciplinary approach, and HSCT is a treatment option for some of them. The main problems described in relation to HSCT in these patients are graft failure with autologous reconstitution and the unavailability of related histocompatible donors.10 Favourable outcomes have been achieved with transplantation of HSCs from umbilical cord blood (UCB).11
We present the outcomes of HSCT following reduced-intensity conditioning in patients with genetic diseases reported by the Grupo Español para el Trasplante de Médula Ósea en Niños (Spanish Working Group on Bone Marrow Transplantation in Children [GETMON]).
Patients and methodsWe conducted a multicentre, observational, retrospective-prospective, descriptive and analytical study.
The individuals included in the study were 68 paediatric patients (aged 0–18 years) with genetic diseases that underwent HSCT following reduced-intensity conditioning between 2005 and 2013 in the paediatric haematopoietic stem cell transplantation units of the Spanish hospitals members of the GETMON. The researchers of participating centres filled out data collection forms. The legal guardians of the patients signed their informed consent for performance of HSCT and associated research.
In the statistical analysis, we obtained overall survival (OS) and event-free survival (EFS) curves using the Kaplan–Meier method and compared survival by means of the log rank test. We defined statistical significance as a p-value of 0.05 or less. We analysed the data with the SPSS software (Statistical Package for Social Sciences) version 22.0 for Windows.
We defined event as any adverse event occurring after HSCT, such as death or graft failure. We defined OS as the time elapsed between performance of HSCT and the most recent follow-up. We defined EFS as the time elapsed between performance of HSCT and the occurrence of any event.
ResultsPatient characteristicsOf the 68 patients, 40 (59%) were male and 28 (41%) female. The median age at diagnosis was 21 months (range, 10 days–180 months). The median age at transplantation was 46 months (range, 3–190 months).
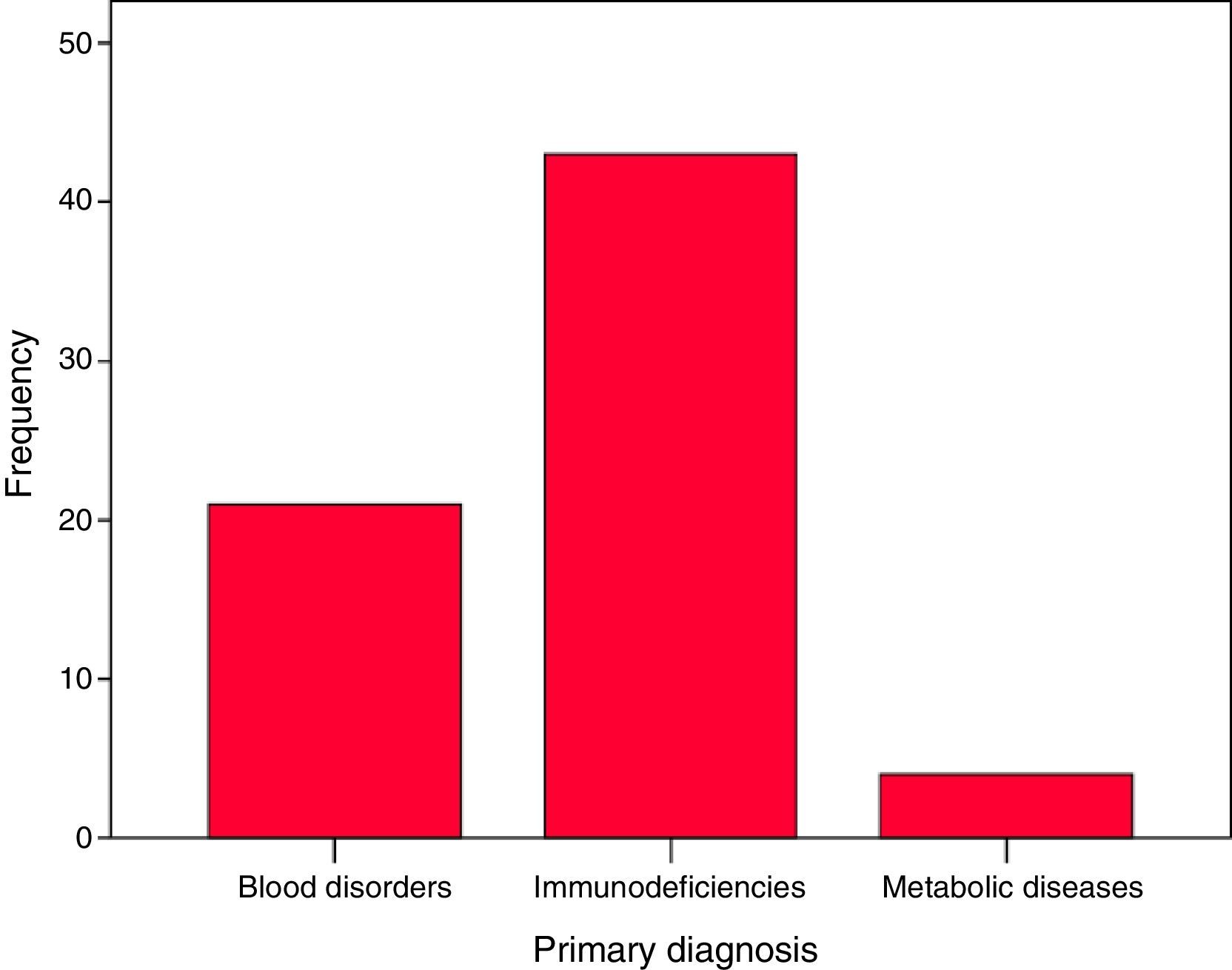
Forty-three patients had PIDDs: severe combined immunodeficiency (22), familial haemophagocytic lymphohistiocytosis (7), X-linked hyper IgM syndrome (3), Omenn syndrome (2), Wiskott-Aldrich syndrome (2), MHC class II deficiency (2), IPEX syndrome (2), Griscelli syndrome (1), leucocyte adhesion deficiency type 1 (1) and undetermined severe PIDD (1). Twenty-one patients had congenital blood disorders: Fanconi anaemia (12), major thalassaemia (7), sickle-cell anaemia (1) and pyruvate kinase deficiency (1). Four patients had metabolic diseases: mucopolysaccharidosis (2), fucosidosis (1) and globoid cell leukodystrophy or Krabbe disease (1). Of the total patients, 63.2% had PIDDs, 30.9% blood disorders and 7% metabolic diseases (Fig. 1).
Transplant characteristics, inherited blood disorders to 30.9% (n=21) and inherited metabolic diseases to 7% (n=4).")
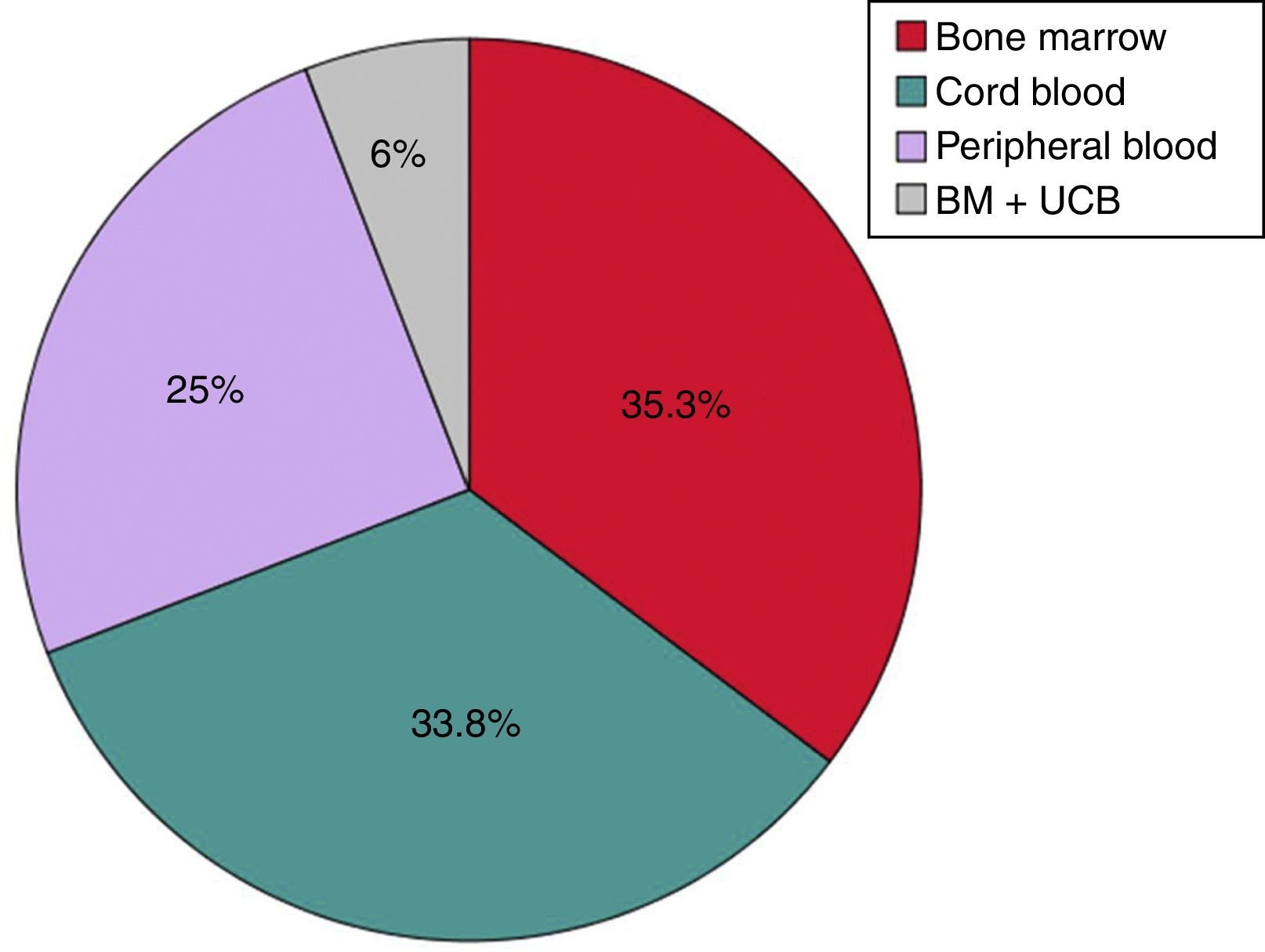
The source of HSCs for transplantation was bone marrow (BM) in 24 patients (35.3%), UCB in 23 (33.8%), PB in 17 (25%) and a combination of UCB and BM in 4 (6%) (Fig. 2).

As for the type of donor, 47 transplants (69.2%) were from an unrelated donor (UD), 25 of who were HLA-mismatched and 22 HLA-identical (allele match at the HLA-A, B, C, DR [8/8] and DQ [10/10] loci). Twenty-one received the graft from a related donor (31%): an HLA-identical sibling in 13, and an HLA-mismatched related donor (parent) in 8. Based on HLA identity, there were 35 transplants from HLA-identical donors (51.5%) and 33 from HLA-mismatched donors (48.5%).
The pre-transplantation conditioning regimen was most frequently based on the use of Flu, specifically in 65 of the 68 patients. The regimens used were Flu+melphalan (33); Flu+cyclophosphamide+TBI (13); Flu+Tre+TT (7); Flu+Tre+cyclophosphamide (6); Flu+bulsulfan+TT (7); Flu+melphalan+TBI (1) and bulsulfan+cyclophosphamide (1). In all cases, the maximum doses were within the defined limits for RIC.
Fifty-seven patients were also treated with monoclonal antibodies. Forty (58.8%) received antithymocyte globulin (ATG) for a total dose of 6–10mg/kg, and seventeen (25%) alemtuzumab (anti-CD52 antibody) for a total dose of 1mg/kg.
T-cell depletion was performed in 14 patients: 6 who received a PB graft from a parent, 6 who received a PB graft from an HLA-identical unrelated donor, 1 who received a PB graft from an HLA-identical sibling, and 1 who received a BM graft from an HLA-identical unrelated donor.
Prophylactic treatment for the prevention of graft-versus-host disease (GVHD) consisted of cyclosporine alone in 29 patients, cyclosporine+mycophelonic acid in 26, and cyclosporine+methotrexate+mycophelonic acid in 10. Only 3 patients did not receive pharmacologic prophylaxis; these patients had been infused with PB cells from their HLA-identical mothers, and the only prophylactic measure taken to prevent GVHD was T-cell depletion prior to HSCT.
When it came to the cell dose infused for HSCT, the 24 patients with BM transplants received a mean (±SD) of 19.12×108/kg±36.8×108/kg nucleated cells and 11.51×106/kg±14.68×106/kg CD34+ cells. The mean cell dose in the 23 patients who received UCB transplants was 3.51×108/kg±5.1×108/kg nucleated cells, with 1.3×106/kg±2.12×106/kg CD34+ cells. The mean number of CD34+ cells infused in the 17 patients that received PB grafts was 15.86×106/kg±17.83×106/kg. Four patients received a combination of UCB and BM cells from an HLA-identical sibling, with infusion of a mean of 12.82×108/kg±21.2×108/kg total cells, including 4.43×106/kg±2.04×106/kg CD34+ cells.
Assessment of transplantation outcomesNeutrophil engraftment occurred in 65 patients (95.6%). These patients reached neutrophil counts of 0.5×109/L or greater in a median of 17.4 days. Platelet engraftment occurred in 61 patients (89.7%), who achieved platelet counts of 20×109/L or greater a median of 24 days after transplantation.
Chimerism analysis revealed complete chimerism (100% donor-derived cells) in T lymphocytes and granulocytes between 30 and 60 days post HSCT in 38 patients (55.9%). Twenty-six patients (38.2%) exhibited mixed chimerism, and four patients (5.9%) primary graft failure with autologous bone marrow recovery.
Out of the 60 patients that could be evaluated 6 or more months post HTSC, chimerism analysis detected complete donor chimerism in T lymphocytes and granulocytes in 32 patients (47.1%). Twenty-one patients (30.9%) had stable mixed chimerism, 2 (2.9%) had primary graft failure and 5 (10.2%) who initially had mixed chimerism experienced secondary graft failure.
When it came to GVHD, 24 patients did not develop acute GVHD (35.3%), 33 developed grade I-II acute GVHD (48.5%) and 11 grade III-IV acute GVHD (16.2%).
Of the 64 patients that could be evaluated for chronic GVHD 100 days post transplantation, 55 did not have chronic GVHD (80.9%), 4 had limited chronic GVHD and 5 extensive chronic GVHD.
In the 18 patients who died, the main causes of death, isolated or combined, were: GVHD, sepsis, multiple organ failure, primary or secondary graft failure, pulmonary haemorrhage, brain haemorrhage and infection by cytomegalovirus.
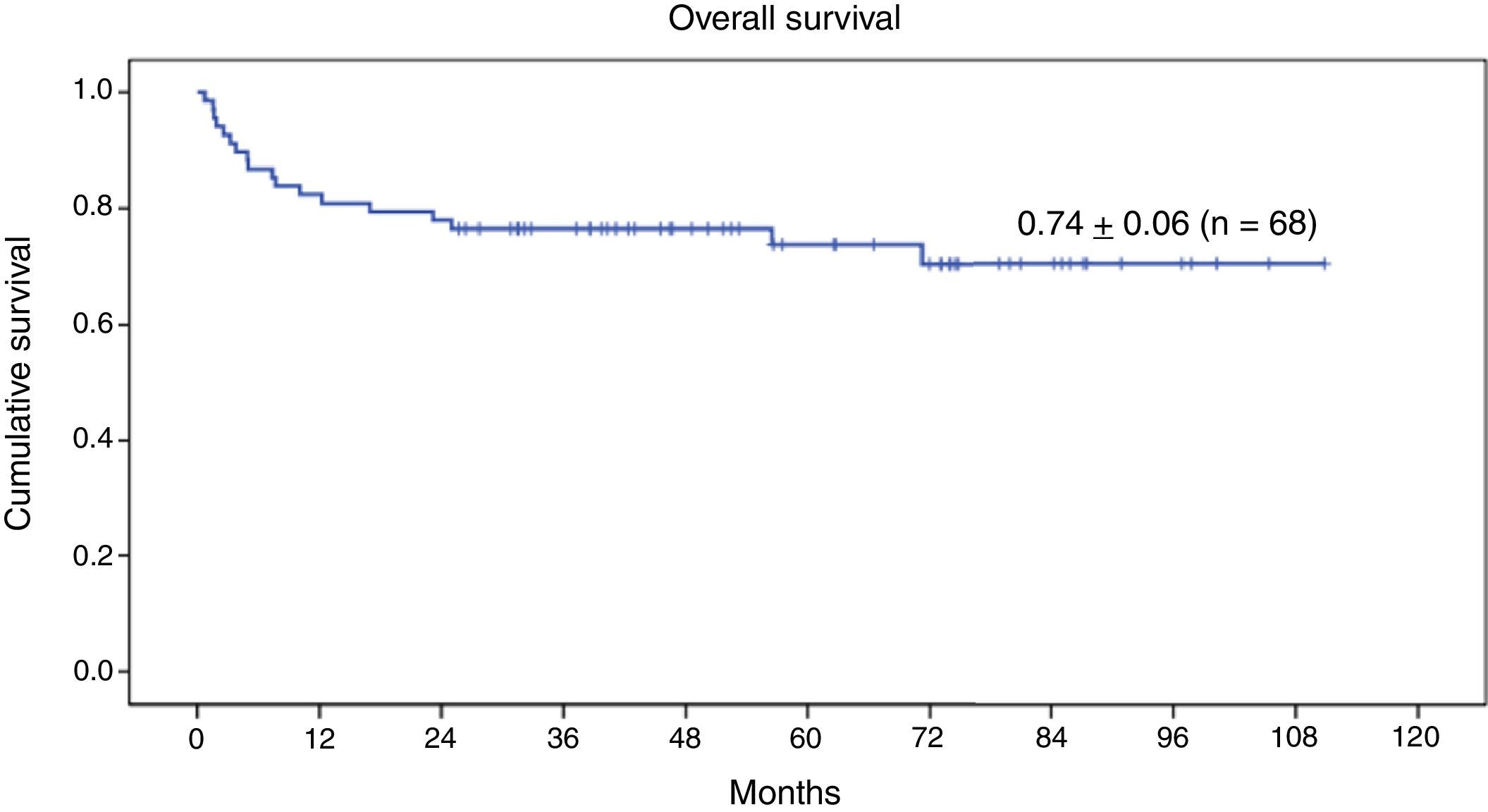
Survival analysisAt the time of this writing, 50 of the 68 patients remain alive (73.5%) (Fig. 3).
 in 68 patients with genetic disease treated with HSCT following reduced-intensity conditioning.")
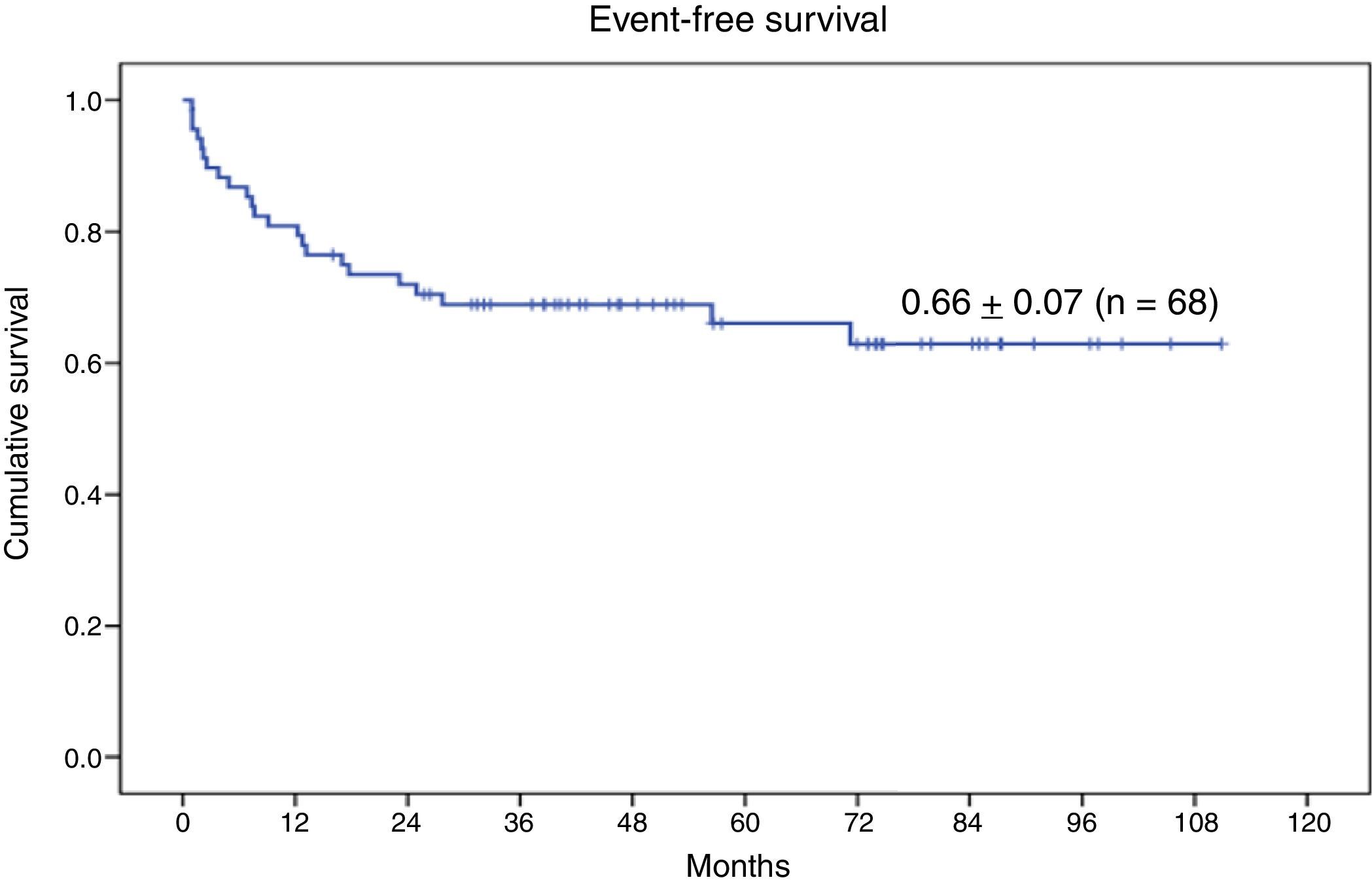
The 9-year OS for the entire series of 68 patients with genetic disease treated with HSCT after reduced-intensity conditioning was 0.74±0.06 (Fig. 3). Twenty-three patients (33.8%) experienced events after transplantation. The 9-year EFS was 0.66±0.07 (Fig. 4).

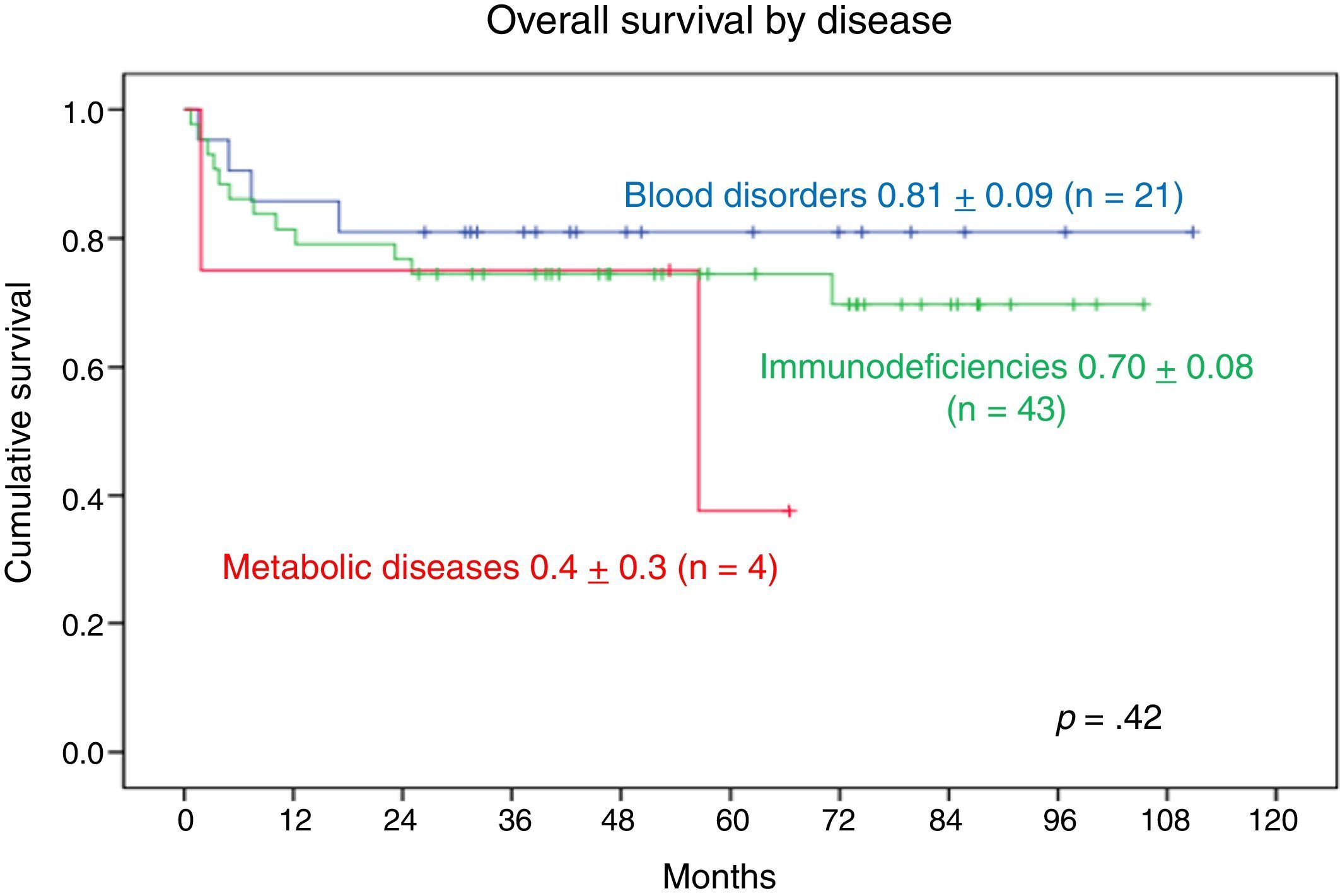
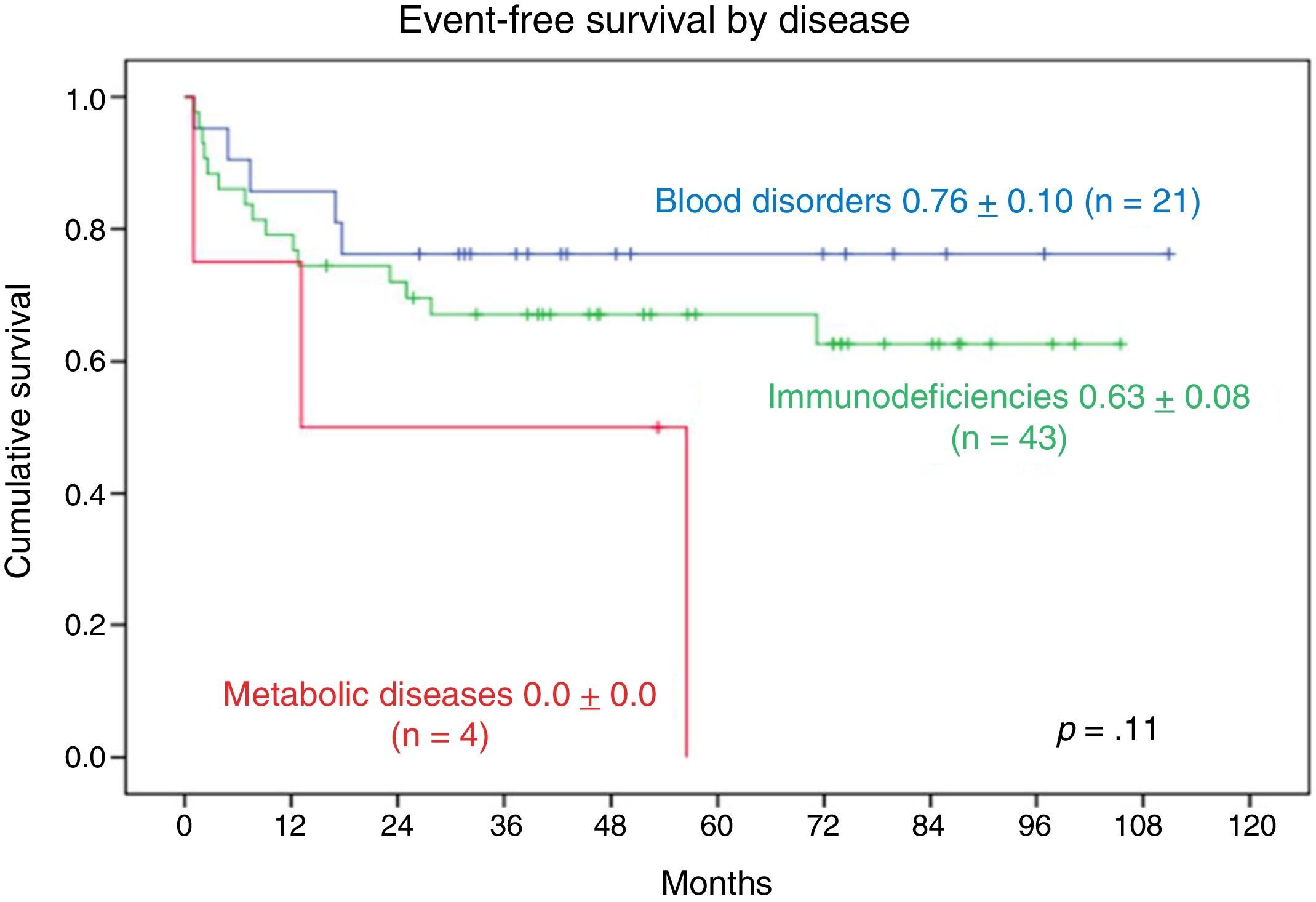
Comparing the 3 main disease groups, we found that 17 of the 21 patients with blood disorders and 31 out of the 43 patients with PIDD remained alive, compared to only 2 of the 4 patients with metabolic diseases.
The 9-year OS was 0.81±0.09 in patients with blood disorders and 0.70±0.08 in patients with PIDDs. The 5-year OS in patients with metabolic diseases was 0.4±0.3. We found no statistically significant differences (Fig. 5).

The 9-year EFS was 0.76±0.10 in patients with blood disorders and 0.63±0.08 in patients with PIDDs. The 2-year EFS in patients with metabolic diseases was 0.0±0.0. We found no statistically significant differences (Fig. 6).

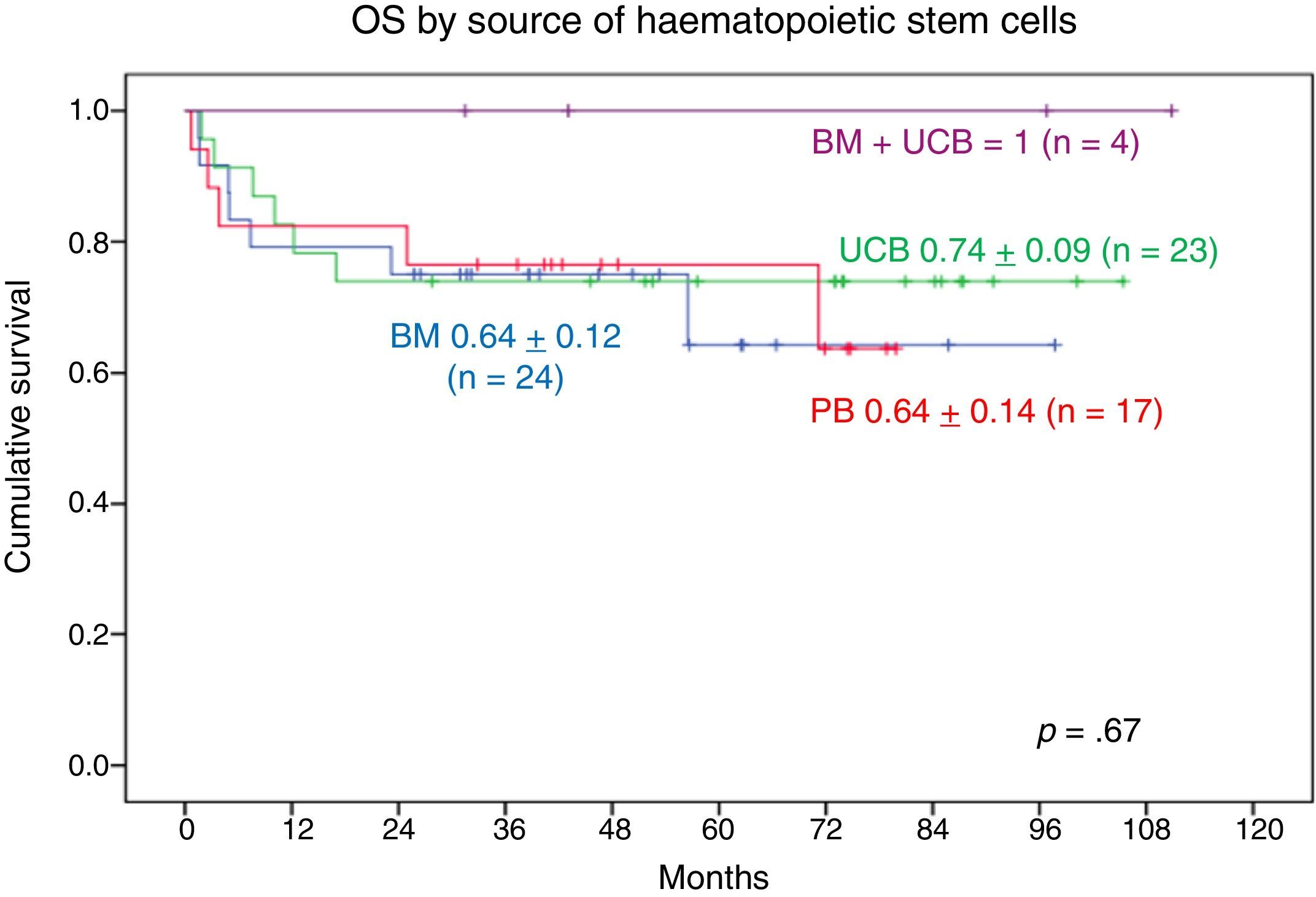
When it came to the graft source, the 9-year OS was 1 in patients who received UCB+BM grafts and 0.70±0.11 in patients who received UCB grafts, the 8-year OS in patients who received BM grafts was 0.70±0.10 and the 7-year OS in patients who received PB grafts was 0.74±0.13. We found no statistically significant differences (Fig. 7).

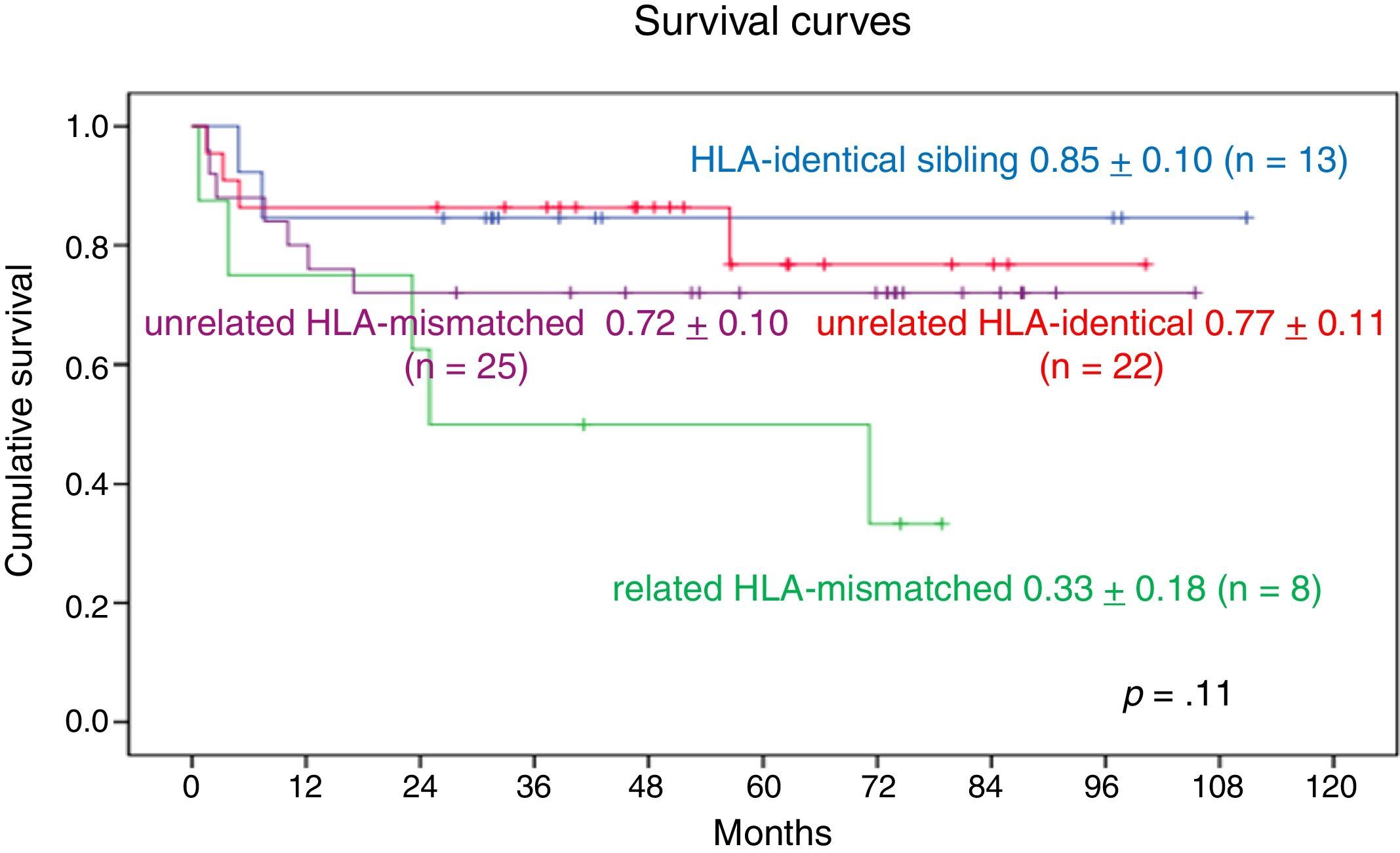
When we analysed the impact of related vs unrelated donors and HLA matching, we found that 9-year OS in patients who received grafts from HLA-identical unrelated donors was 0.77±0.11, compared to 0.72±0.1 in patients with grafts from HLA-mismatched unrelated donors. As for patients who received grafts from related donors, the 9-year OS in those with grafts from HLA-identical donors was 0.85±0.10 and the 7-year survival in those with grafts from HLA-mismatched donors was 0.33±0.18. We found no statistically significant differences (Fig. 8).
Discussion
We present the results obtained from the analysis of the data for 68 paediatric patients with genetic diseases treated with HSCT following RIC recorded by the GETMON. Considering the low prevalence in children of the genetic diseases under study, we managed to obtain a relatively large sample of patients.
Patients with genetic diseases receive the diagnosis at a median age of 21 months and undergo HSCT at a median age of 46 months. This suggests that these patients undergo transplantation in the first years of life to attempt a cure as soon as a suitable HSC donor is found after the diagnosis.
When it comes to HSC infusion, we ought to highlight that the total number of nucleated and CD34+ cells infused when the graft source is UCB is lower, although it is also important to consider that UCB stem cells have a higher proliferative and colony-forming potential compared to cells obtained from BM or PB, which may compensate for their lower absolute number. When the UCB donor is an HLA-identical sibling, BM can also be harvested to deliver a higher cell dose closer to the recommended amount, a strategy associated with excellent outcomes in our study.
Donor neutrophil engraftment after HSCT with a RIC was greater than 90% in the 3 groups of patients.
The published literature on paediatric HSCT with RIC is scarce. Most of the research published before 2007 corresponded to single-centre studies with small and heterogeneous samples of patients, variable conditioning regimens and a short duration of follow-up.
Bacigalupo12 published a study in 35 patients that underwent allogeneic HSCT in Jerusalem. The patients had non-malignant and autoimmune diseases. The conditioning regimen was Flu+bulsulfan+ATG in all. At 2 years of follow-up, the OA was 95% and the EFS was 80%.
Shenoy et al.13 presented a study in 16 patients treated with Flu+melphalan+alemtuzumab. They had non-malignant and autoimmune diseases. The graft sources were BM, PB or UCB, from related or unrelated donors. The outcomes were good in all the surviving patients (75%), with resolution or improvement of the underlying disease.
Based on the graft source, we found the best outcomes in terms of OS in the combination of UCB and BM cells from an HLA-identical sibling, for which the 9-year OS was 1. In 2014, Parikh et al.14 published a study in 22 patients with genetic diseases who received UCB grafts from unrelated donors, most of them HLA-mismatched. The RIC protocol consisted of Flu+melphalan+thiotepa and alemtuzumab. In this study, the OS at 31 months of follow-up was 68.2%. In our series, the 9-year OS in patients that received UCB grafts from unrelated donors was 70%, outcomes that were better than those reported by Parikh et al.
When it came to related versus unrelated donors and HLA matching, the best outcomes in terms of OS, as would be expected in patients with genetic disease, were obtained with grafts from HLA-identical related donors.
As for HSCT in patients with thalassaemia major, Lisini et al.15 recently published an interesting study presenting the cases of 60 patients. The RIC regimen consisted of Tre+Flu+TT. Twenty patients received grafts from HLA-identical siblings and 40 from unrelated donors. The 5-year OS and EFS were 93% and 84%, respectively. Our study corroborated the safety and effectiveness of the protocol of the Italian group with conditioning based on the use of Tre for patients with thalassaemia undergoing allogeneic HSCT, with an OS and an EFS of 100%.
In patients with Fanconi anaemia, the cyclophosphamide+Flu regimen has proven safe and efficacious. The use of RIC is recommended in these patients due to their higher sensitivity to toxic agents.
The Great Ormond Street Hospital8 group published a case series of 113 patients with PIDD that had undergone allogeneic HSCT with RIC between 1998 and 2006. The conditioning regimen in most of the patients consisted of Flu+melphalan+alemtuzumab. The OS was 82%, with a median duration of follow-up of 2.9 years. In our study, the OS observed with a similar conditioning regimen was 70%, with a median duration of follow-up of 5.2 years.
In patients with metabolic diseases, the literature describes the occurrence of mixed chimerism and graft failure, consistent with the findings of our small case series. In these diseases, HSCT must be considered on a case-to-case basis, weighing the risks and benefits of reduced-intensity conditioning, as there is little data on the use of this approach and further research is required.16,17
Conflict of interestThe authors have no conflicts of interest to declare.
Please cite this article as: López-Granados L, Torrent M, Sastre A, Gonzalez-Vicent M, de Heredia CD, Argilés B, et al. Trasplante de progenitores hematopoyéticos con intensidad reducida en enfermedades genéticas. Experiencia del grupo GETMON (Grupo Español de Trasplante de Médula Ósea en Niños). An Pediatr (Barc). 2018;88:196–203.

