

Pseudoachondroplasia (PSACH; OMIM #177170) is a rare autosomal dominant osteochondrodysplasia with an estimated prevalence of 1 in 30000 individuals. It manifests with a wide-based gait (“waddling gait”) from onset of walking and delay in linear growth starting at age 2 years that progresses to a marked short stature with shortened limbs, brachydactyly and early-onset osteoarthritis. Some relevant aspects of its presentation are normal birth length and the absence of facial abnormalities and intellectual disability. Pseudoachondroplasia is due to mutations in the COMP gene, located at 19p13.11, which encodes cartilage oligomeric matrix protein (COMP).1 These mutations result in abnormal folding of the COMP protein and its accumulation in the endoplasmic reticulum of metaphyseal chondrocytes, leading to oxidative stress and cell death, with loss of chondrocytes in the growth plate. Autosomal dominant multiple epiphyseal dysplasia (MED) is also associated with mutations in the COMP gene, although shows greater genetic heterogeneity and may be associated with mutations in the MATN3, COL9A1, COL9A2 or COL9A3 genes.
We present one de novo case and one familial case of pseudoachondroplasia.
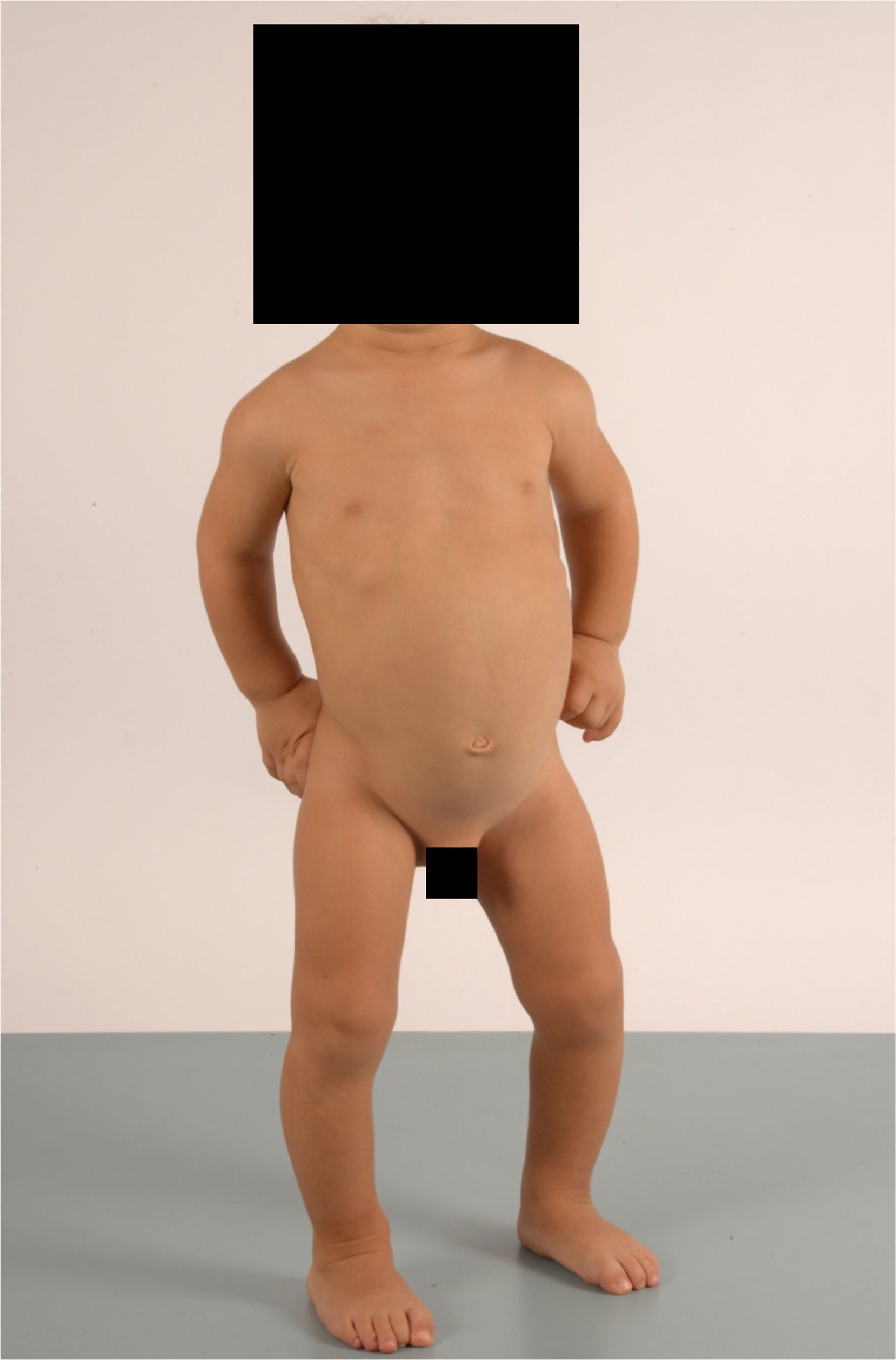
Case 1 corresponds to a boy aged 2 years referred to the outpatient paediatrics clinic for assessment of a limp present from onset of walking. He had been born at 41 weeks’ gestation with a weight of 3510g (z-score, 0.07), length of 53cm (z-score, 1.29) and head circumference of 36cm (z-score, 0.38). Case 2 corresponds to a girl aged 4 years referred for evaluation of poor linear growth starting at age 2 years. She had been born at 42 weeks’ gestation with a weight of 3360g (z-score, −0.29), length of 50cm (z-score, −0.38) and head circumference of 33cm (z-score, −1.5). The findings of prenatal ultrasound examinations had been normal in both patients, and neither had a relevant family history. At the time of diagnosis, case 1 presented with a weigh of 11.9kg (z-score, −0.91) and a height of 82.5cm (z-score, −2.36) and case 2 with a weight of 14.5kg (z-score, −1.32) and a height of 92.2cm (z-score, −3.44). Physical examination revealed short stature, rhizomelia of the upper extremities, brachydactyly, genu varum, lumbar lordosis and mild scoliosis (Fig. 1) in both patients. Other salient features were ligamentous laxity and “waddling gait.”
 with short stature, brachydactyly with rhizomelia and genu varum.")
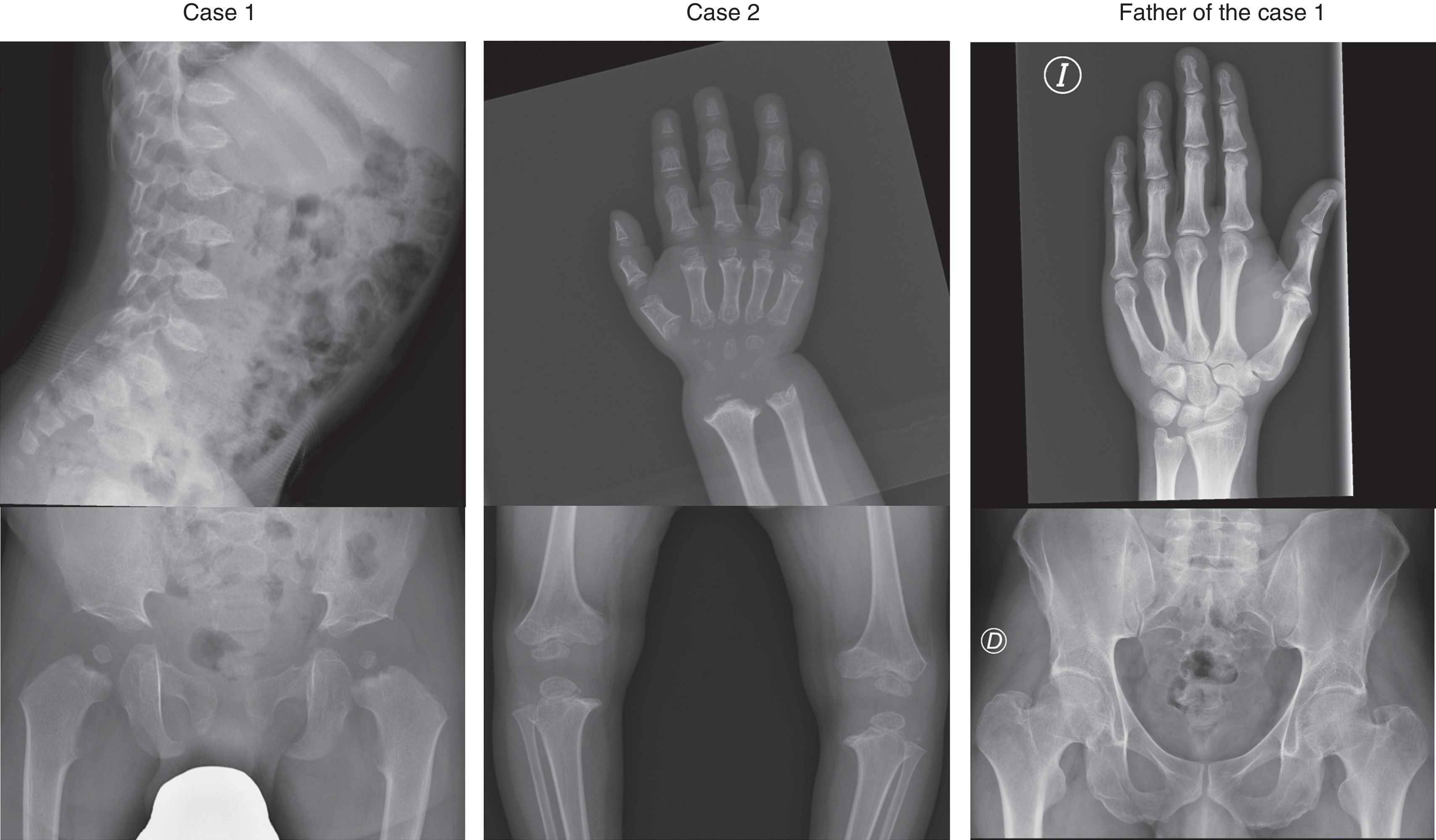
The skeletal survey revealed delayed ossification of the epiphyses and metaphyses and irregularities in the long bones, femoral head and acetabulum (Fig. 2). Hand radiographs showed shortened carpals and metacarpals, phalanges with cone-shaped epiphyses and irregular metaphyses. Another salient feature was anterior beaking of the vertebral bodies. The results of blood tests were normal. The differential diagnosis included PSACH, achondroplasia, MED and mucopolysaccharidosis type IV.

Molecular analysis of the exons and introns in the COMP gene region (NM_000095.2) in case 1 detected a heterozygous mutation in exon 14 (c.1552G>C) resulting in an amino acid substitution (p.Asp518His) in domain T38 of the protein, a mutation previously described in association with PSACH by Deere et al.2 In case 2, molecular analysis found a heterozygous mutation in exon 9 (c.868G>T) that resulted in an amino acid substitution (p.Asp290Tyr) in domain T31. This mutation had not been described before, but 2 other mutations in the same codon (p.Asp290Gly and p.Asp290Asn) had been previously described in association with PSACH.3,4 Four in silico predictions suggested that the mutation was pathogenic. The parents of both patients underwent gene sequencing, with positive results in the father of case 1, who had the same mutation as his son. This father had a height of 165cm (z-score, −1.95 based on the family context) and leg length discrepancy (1.5cm), with normal features in the skeletal survey save for a slightly narrow pelvis and lumbar hyperlordosis.
The father of case 1 posed a diagnostic challenge, as he did not have clinical or radiographic features compatible with PSACH or MED, yet carried that pathogenic mutation in the COMP gene already described in association with PSACH.3 Although there is a previous report of a case with possible incomplete penetrance,5 penetrance in this disease is considered to be complete, so a more likely explanation would be that phenotypic expression is variable with potential for mild phenotypes or somatic mosaicism, a possibility that could not be ruled out.
The mean adult height is 120cm in men and 116cm in women. The most significant complication is joint pain, with onset in childhood, due to precocious osteoarthritis in the knees, hips and spine. These patients may present with odontoid hypoplasia. Early treatment with antioxidants and anti-inflammatory agents and using antisense oligonucleotide technology has been associated with improvement in mouse models.6 Patients with PSACH require followup in a specialised clinic, should avoid intense physical activity, and require analgesia, physical therapy, corrective surgery in some cases, and psychosocial support in every case.
Please cite this article as: Casas-Alba D, Fernández López A, Gean Molins E, Suero Toledano P, Martínez-Monseny A. Seudoacondroplasia: descripción de un caso de novo y otro familiar. An Pediatr (Barc). 2018;89:60–61.

